
Abstract
Gene mutations and gene copy number variants are associated with autism spectrum disorders (ASDs). Affected gene products are often part of signaling networks implicated in synapse formation and/or function leading to alterations in the excitation/inhibition (E/I) balance. Although the network of parvalbumin (PV)-expressing interneurons has gained particular attention in ASD, little is known on PV’s putative role with respect to ASD. Genetic mouse models represent powerful translational tools for studying the role of genetic and neurobiological factors underlying ASD. Here, we report that PV knockout mice (PV−/−) display behavioral phenotypes with relevance to all three core symptoms present in human ASD patients: abnormal reciprocal social interactions, impairments in communication and repetitive and stereotyped patterns of behavior. PV-depleted mice also showed several signs of ASD-associated comorbidities, such as reduced pain sensitivity and startle responses yet increased seizure susceptibility, whereas no evidence for behavioral phenotypes with relevance to anxiety, depression and schizophrenia was obtained. Reduced social interactions and communication were also observed in heterozygous (PV+/−) mice characterized by lower PV expression levels, indicating that merely a decrease in PV levels might be sufficient to elicit core ASD-like deficits. Structural magnetic resonance imaging measurements in PV−/− and PV+/− mice further revealed ASD-associated developmental neuroanatomical changes, including transient cortical hypertrophy and cerebellar hypoplasia. Electrophysiological experiments finally demonstrated that the E/I balance in these mice is altered by modification of both inhibitory and excitatory synaptic transmission. On the basis of the reported changes in PV expression patterns in several, mostly genetic rodent models of ASD, we propose that in these models downregulation of PV might represent one of the points of convergence, thus providing a common link between apparently unrelated ASD-associated synapse structure/function phenotypes.
Similar content being viewed by others

Introduction
Autism spectrum disorders (ASDs) comprise a series of related neurodevelopmental disorders characterized by deficits in social interaction, reduced/impaired communication and restricted and stereotyped behavior.1 Anxiety, motor and sensory impairments, reduced nociception, increased seizure susceptibility and intellectual disability are common comorbidities.1 Recent data indicate that ~1/100 children displays symptoms or mild signs related to ASD.1 Results from many studies indicate a strong genetic component, and a plethora of gene mutations and/or copy number variants have been identified in ASD patients.2 Most ASD candidate gene products are involved in activity-dependent neuronal signaling and are (i) implicated in synapse formation/maintenance (for example, neurexins and neuroligins), (ii) proteins of synaptic membranes (for example, kainate-type glutamate receptor GluR6), (iii) scaffolding proteins in the postsynaptic density (for example, Shank1/2/3) or (iv) proteins involved in signaling pathways relaying information from the synapse to the nucleus.3, 4 At the functional level, these mutations are thought to finally translate into changes in the excitation/inhibition (E/I) balance.3, 4
Alterations in any ASD candidate gene only accounts for a minority of ASD cases, suggesting that they might be part of convergent molecular pathways. Genome-wide association studies5, 6 and transcriptomic co-expression network analyses7 have identified ASD risk gene networks. The top connections in one of the neuron-specific modules (‘Grey60’) contain the genes KCNC1, SCN1B, PVALB and HAPLN4,6 all highly expressed in fast-spiking parvalbumin (PV)-expressing (PV+) cortical interneurons.8 As the top module M12 (ref. 7) strongly overlaps with the PVALB+ interneuron module9 and most transcripts, including PVALB and DLX1, are downregulated in ASD brains, these two genes and/or gene products were proposed as ASD-related candidates.7 To our knowledge, no mutations in the human PVALB gene have been reported up to now, neither in ASD nor other neurodevelopmental disorders. However, decreased PV expression (protein and messenger RNA) and/or loss of PV+ neurons were reported not only in ASD patients,10, 11 but also in other neurodevelopmental disorders, such as schizophrenia and bipolar disorder.12 Initially, the decrease in the number and/or density of PV-immunoreactive (PV+) neurons was assumed to be the result of a loss/decrease of this neuron subpopulation in affected patients,10 but the weaker in situ hybridization signals observed in five out of eight ASD children were viewed as PVALB messenger RNA downregulation.11 Also others postulated PV downregulation (not PV+-neuron loss) as an adaptive/homeostatic mechanism.12, 13
Genetic mouse models are powerful translational tools for studying the role of those genes in the etiology of ASD.14, 15, 16, 17 Such mutations cause effects at various levels, from cell biology and morphology to electrophysiology and behavior. The network of PV+ neurons was analyzed in many studies, including not only genetic models of ASD focusing on the impact of Cntnap2,18 Met,19 Fmr1,20 Nlgn3R451C (ref. 21) and Nrp2,22 but also environmental ASD models, with the valproic acid model probably being the most prominent one.21 Most of these studies reported ‘a reduction in the number of PV+ neurons’ by ~20–25% (for details, see Supplementary Table 1). Constitutive PV−/− mice enabled us to directly address the role of PV in this ASD-linked neuronal subpopulation. Importantly, there is no evidence of a loss of ‘PV+’ neurons in PV−/− mice in various brain regions, including hippocampus23 and cortex.24 Yet the absence of the Ca2+ buffer PV profoundly affects synaptic transmission; PV prevents/delays short-term facilitation25, 26, 27 and reduces gamma power of associated hippocampal oscillations.23 PV affects neuron-firing timing and consequently influences the desynchronization of neuronal networks in the cortex,28 decreases spiking rhythmicity in striatal fast-spiking interneurons (FSIs)27 and modulates burst-firing properties in the thalamic reticular nucleus.29 These results from PV−/− mice indicate an activity-dependent increase in inhibition in the absence of PV, thus shifting the E/I balance towards increased inhibition, as reported in the neuroligin-3 knock-in ASD mouse model NL3R451C.30
The decreased PV levels and/or density of PV+ neurons reported in several pathological conditions, including ASD, led us to conduct a comprehensive set of behavioral phenotyping assays in PV−/− mice, using well-established paradigms with high relevance and sensitivity for the diagnostic and associated symptoms of ASD.14, 15, 16, 17 PV−/− mice not only display typical ASD-like phenotypes with relevance to all human autism core symptoms, that is, impaired social interactions, communication deficits and rigid/repetitive behaviors, but also reduced nociception and startle responses, as well as increased seizure susceptibility, viewed as ASD-associated comorbidities. At the neuroanatomical level, PV−/− mice show transient cerebral hypertrophy and cerebellar hypoplasia as juveniles at postnatal day (PND) 20, resembling morphological changes previously reported in ASD patients.31, 32 Finally, the absence of PV not only affects inhibitory synaptic transmission, but also excitatory transmission onto PV+ FSI is impaired, resulting in an altered E/I balance. Results obtained in PV+/− mice with a reduction of brain PV levels by ~60–70% further indicate that merely a downregulation of PV might be sufficient to elicit core ASD-like deficits.
Materials and methods
Animals
Wild-type PV+/+, heterozygous PV+/− and homozygous PV−/− mice (all littermates from heterozygous breedings, unless otherwise stated) used in the experiments were male (unless specified otherwise). They were all raised on a C57BL/6J background (backcrossed to C57BL/6J for >10 generations, as described previously33). Animals were genotyped by PCR.24 All animal experiments were carried out in accordance with local animal care legislation and to current Swiss law and EC Council Directive of 24/11/86 (86/609/EEC). Prior to and following experimentation, all animals were group housed and maintained on a 12-h light/dark cycle, with ad libitum access to standard rodent chow and water.
Reciprocal social interaction
To measure reciprocal social interaction behavior, pairs of juvenile mice were allowed to socially interact at PND25±1 for 5 min after one mouse of the pair being habituated to the test environment for 1 min. Same-sex/same-genotype pairs consisting of non-littermates were used. To enhance the level of social motivation, juvenile mice were socially isolated for 24 h prior to testing. Testing was performed in a clean Makrolon Type III cage with fresh bedding and a metal lid under dim red light. Behavior was recorded using a video camera placed 30 cm away from the cage. Reciprocal social interactions were tested between 0800 and 1800 hours during the light phase of the 12:12 h light/dark cycle.
Social interactions were scored by an experienced observer blind to the experimental conditions using the Noldus The Observer XT 10.0 software (Noldus Information Technology, Wageningen, The Netherlands). Parameters of social behaviors included facial sniffing (sniffing the nose and snout region of the partner), anogenital sniffing (sniffing the anogenital region of the partner), following (walking straight behind the partner, keeping pace with the one ahead), push past (squeezing between the wall and the partner), crawling under/over (pushing the head underneath the partner’s body or crawling over or under the partner’s body), social grooming (grooming the partner) and being socially inactive while having social contact (lying flat or standing still while maintaining close physical contact with the partner; for details see Yang et al.17 and Terranova and Laviola34). All social behaviors were analyzed for frequency of occurrence (that is, number of bouts) and duration in 1-min time bins. In addition to social behaviors, nonsocial behaviors were measured and included rearing (number of times an animal reared on its hind legs), grooming (number of bouts of face, body and genital grooming movements) and digging (number of bouts of digging in the bedding, pushing and kicking it around).
Ultrasonic vocalizations
Isolation-induced ultrasonic vocalizations in mouse pups
To induce ultrasonic vocalizations (USV) in pups, they were isolated from their mother and littermates on PND8 for 10 min at room temperature (22–24 °C). As described previously,35, 36 pups were removed individually from the nest at random and gently placed into an isolation container (10 × 8 × 7 cm; open surface) made of glass, containing clean bedding material. The isolation container was surrounded by a sound-attenuating box (20 × 20 × 20 cm) made of Styrofoam (thickness of walls: 4 cm). USV emission was monitored by an UltraSoundGate Condenser CM 16 Microphone (Avisoft Bioacoustics, Berlin, Germany) placed in the roof of the sound-attenuating box, 20 cm above the floor. After the 10-min isolation period, body weight and body temperature were determined. Body weight was measured using a palm scale (PS6-250; My Weigh Europe, Hückelhoven, Germany). For body temperature determination a Testo 110 thermometer (Testo, Lenzkirch, Germany) was used. In addition, the following somatosensory reflexes were determined: surface righting and vertical screen holding. To measure the righting reflex, pups were placed on their back on a flat, hard surface, and a stopwatch was used to measure the time that it took to right themselves on all four paws (maximum: 30 s). To measure vertical screen holding, pups were placed on a wire mesh screen (8 × 11 cm) that was slanted vertically at an angle of 90°. Length of time the pup was able to stay on the grid at an angle of 90° was measured using a stopwatch (maximum: 30 s). Then, pups were marked with a paw tattoo for identification, using nontoxic animal tattoo ink (Ketchum permanent Tattoo Inks green paste, Ketchum Manufacturing, Brockville, ON, Canada). The ink was inserted subcutaneously through a 30-gauge hypodermic needle tip into the center of the paw. Pup isolation, determination of somatosensory reflexes and paw marking occurred between 0800 and 1800 hours during the light phase of the 12:12 h light/dark cycle. Prior to each test, behavioral equipment was cleaned using a 0.1% acetic acid solution, followed by water and dried with paper towels.
Interaction-induced USV in juvenile mice
To induce USV in juvenile mice, pairs of juvenile mice were allowed to socially interact at PND25±1 for 5 min after one mouse of the pair being habituated to the test environment for 1 min. Same-sex/same-genotype pairs consisting of non-littermates were used. To enhance the level of social motivation, juvenile mice were socially isolated for 24 h prior to testing. Testing was performed in a Makrolon Type III cage with clean bedding and metal lid under dim red light. USV emission was monitored by an UltraSoundGate Condenser CM 16 Microphone (Avisoft Bioacoustics) placed 15 cm above the cage lid. Reciprocal social interactions were tested between 0800 and 1800 hours during the light phase of the 12:12 h light/dark cycle. Clean cages and lids were used for reciprocal social interactions.
Female-induced USV in adult male mice
To induce USV in adult male mice, male mice at PND~120 were exposed to a wild-type female for 5 min after being habituated to the test environment for 1 min. Testing was performed in a Makrolon Type III cage with clean bedding and a metal lid under dim red light. USV emission was monitored by an UltraSoundGate Condenser CM 16 Microphone (Avisoft Bioacoustics) placed 15 cm above the cage lid. Female exposure occurred between 0800 and 1800 hours during the light phase of the 12:12 h light/dark cycle. Clean cages and lids were used for all female exposures.
Acoustical recording
UltraSoundGate Condenser CM 16 Microphones (Avisoft Bioacoustics) used for USV recordings were connected via an UltraSoundGate 416H USB audio device (Avisoft Bioacoustics) to a personal computer, where acoustic data were recorded with a sampling rate of 250 000 Hz in 16-bit format by Avisoft RECORDER USGH (Avisoft Bioacoustics). The microphones are sensitive to frequencies of 15–180 kHz with a flat frequency response (±6 dB) between 25 and 140 kHz.
Acoustical analysis
For acoustical analysis, recordings were transferred to Avisoft SASLab Pro (version 4.50; Avisoft Bioacoustics) and a fast Fourier transform was conducted (512 fast Fourier transform length, 100% frame, Hamming window and 75% time window overlap). Correspondingly, the spectrograms were produced at 488 Hz of frequency resolution and 0.512 ms of time resolution. Call detection of isolation-induced USV emitted by pups was provided by an automatic amplitude threshold-based algorithm and a hold-time mechanism (hold time: 10 ms). As no USV were detected below 30 kHz, a high-pass filter of 30 kHz was used to reduce background noise outside the relevant frequency band to 0 dB. The accuracy of call detection by the software was verified manually by an experienced user. When necessary, missed calls were marked by hand to be included in the automatic parameter analysis. Interaction-induced and female-induced USV were analyzed interactively by an experienced user to avoid false positives due to high levels of background noise resulting from the animals’ movements. Total number of USV was calculated for the entire session and in 60-s time bins to visualize the time course of the USV response. Additional parameters, based on previous USV studies,35, 36 included peak frequency and peak amplitude, that is, loudness, which were derived from the average spectrum of the entire call, were determined automatically. Peak amplitude was defined as the point with the highest energy within the spectrum. Peak frequency was defined as the frequency at the location of the peak amplitude within the spectrum. In addition, the extent of frequency modulation, the difference between the lowest and the highest peak frequency within each call, was measured automatically. Temporal parameters included latency to start calling, total calling time and call duration. The experienced user was blind to the experimental conditions.
Repetitive and stereotyped patterns of behavior
T-maze reversal learning paradigm
Male PV+/+ (n=9) and PV−/− (n=8) mice were tested for restrictive, repetitive behavior using a T-maze reversal paradigm.37 The mice were first habituated to the maze (entry arm: 35 × 10 cm, T-arms, 40 × 10 cm each, as described previously37) over 5 days immediately prior to the experiment. An appetitive reward (in this case ~200 μl of malted chocolate milk) was placed in a shallow container in one of the short arms of the T-maze and the mouse was trained to retrieve it in 10 consecutive, daily trials until it had reached the criterion, in this case at least 70% of the mice had to enter the correct arm and at least partially consume the reward in at least 8/10 trials on three consecutive days. If a mouse failed to enter either arm, entered the wrong arm or entered the correct arm but failed to consume the reward, this was scored as an incorrect response. The mouse was allowed 3 min to complete each trial. In order that the mouse was not guided by olfactory clues, a little of the reward was placed outside of each short arm of the maze for the duration of the trials. Once a mouse had reached acquisition, the reward was swapped into the opposite short arm and 10 daily trials were repeated until the criterion had once again been attained or until the mouse would fail to achieve reversal by 20 days (in which case, if the mouse did not achieve 8/10 correct trials on day 18 post reversal, the mouse was discontinued in the experiment). The mice were scored for the number of correct trials/day during the acquisition and reversal phases, and each group was scored for the percentage of mice achieving acquisition or reversal. Mice failing to achieve initial acquisition did not proceed to the reversal stage.
Water-maze reversal learning paradigm
Place navigation testing in the Morris water maze (MWM) was performed as described previously38 in a round swim tank of 150 cm diameter. The goal platform (14 × 14 cm) was hidden 0.5 cm below the water surface. Animals performed 30 trials (max. duration 120 s), six per day with inter-trial intervals of 30–60 min. During the first 18 trials (acquisition), the hidden platform was kept in the same position and then moved to the opposite quadrant for the remaining 12 trials (reversal). The first trial of the reversal phase served as probe trial to test for spatial retention. Trials were tracked using Noldus EthoVision 2.6 (Noldus Information Technology) and transferred to Wintrack 2.4 software (http://www.dpwolfer.ch/wintrack) for analysis.
Radial-maze working-memory task
The radial-maze working-memory procedure39 was implemented and adapted as described previously.40 The apparatus was constructed of gray polyvinyl chloride. Eight arms (7 × 38 cm) with clear Perspex sidewalls (5 cm high) extended from an octagonal center platform (diameter 18.5 cm, distance platform center to end of arm 47 cm). It was placed 38 cm above the floor in a dimly lit room (4 × 40 W bulbs, 12 lux) rich in salient extra-maze cues (same room as for MWM place navigation task). Small cereal pellets (ca. 6 mg) were placed as baits in small metal cups (diameter 3 cm, 1 cm deep) at the end of each arm, in such a way that the mouse could not see them without completely entering the arm. A reversed box of clear Perspex served to confine the mouse on the center platform before each test session during which the mice were allowed to move freely on the maze. Mice were gradually reduced to and maintained at 85% of their free-feeding body weight using a premeasured amount of chow each day. Water was available ad libitum. Mice performed one trial per day lasting maximally 10 min or until the animal had collected all pellets. They began with two habituation sessions during which they were accustomed to collecting pellets from the maze that were distributed all over the maze. During the following 10 training trials each cup was baited only with one pellet. Behavioral measures: consumption of each pellet was recorded by pressing a designated key on the keyboard. With this information and the video-tracked xy-coordinates (EthoVision 2.6), the following measures were computed: total duration of trial, time spent moving, duration of arm visits, time spent in pellet area, decision time between visits, average velocity, velocity in/out of arms, number of omitted arms, number of bait neglect errors (failure to consume pellet in a baited arm), number of aborted visits (not reaching pellet area), number of working-memory errors (visits to already emptied arms), number of correct choices before first error, correct choices out of first eight, relative frequency of serial choices, relative frequency of repeated angles, relative frequency of a favorite choice angle and number of visits to a favorite arm.
For all experiments on learning and memory, mice were habituated for 7 days to a reversed light/dark cycle, and experiments were performed during their normal active phase.
Rotarod and grip test
Rotarod
Mice were tested on a RotaRod device (TSE Systems, Bad Homburg, Germany) using an accelerating 10–40-r.p.m. protocol. Each mouse performed 12 trials over 2 days, with 6 trials per day separated into two batches of three trials. The mice were allowed to rest for at least 10 min between trials and the two daily batches of trials were separated by at least 2 h. Data were analyzed using the Prism software (GraphPad Software, San Diego, CA, USA) and expressed as average latency to fall, mean±s.e.m.
Grip test
Forepaw grip strength was measured using a newtonmeter that was attached horizontally and to a metallic ring of 5.5 cm diameter and 3 mm thickness. Mice were held by the tail and allowed to grasp the ring with both forepaws. They were then gently pulled away until they released the ring. Five measurements were obtained each on two consecutive sessions and averaged.41
Open field
Sixteen PV+/+ and 16 PV−/− mice were tested, with approximately balanced representation of genders in both groups. Age at the begining of testing was 10–11 weeks. Prior to testing, mice were habituated for 7 days to reversed 12-h light/dark cycle and experiments performed during the normal active (dark) phase. The large open-field arena had a diameter of 150 and 35-cm-high walls. Mice were released near the wall and observed for 10 min. This was repeated the following day for a total duration of 20 min, divided into four periods of 5 min for time course analysis. The square-shaped small open-field arena measured 50 × 50 cm. Twenty-four hours prior to testing, the mice were familiarized with a small home box in their homecages. They were then transferred to the open-field arena together with this home box and observed for 30 min.42 In both experiments, animals were video-tracked using EthoVision 2.3 and data were transferred to Wintrack 2.4 for analysis. To obtain an index of linearity of locomotion, the distance from start to end point was measured for every path segment of 50 cm length and averaged along the path. As a measure of small-scale accelerations and decelerations, a ‘jerkiness’ index was calculated as the sum of unsigned length differences between successive path segments, divided by the distance moved. Because the tracking system also monitored apparent subject area, vertical activity could be estimated by counting reductions of subject area deeper than 250 mm2 while the animal was not engaged in locomotion.
Anxiety—light/dark box, O-maze
Light/dark box
The light/dark box consisted of a 20 × 30 × 20-cm brightly illuminated chamber with transparent walls connected via a 7.5 × 7.5-cm opening to a 20 × 15 × 20-cm enclosed, dark polyvinyl chloride box. Each subject was released in the middle of the lit compartment and observed for 5 min.
O-maze
The O-maze was a 5.5-cm-wide annular runway with an outer diameter of 46 cm, placed at 40 cm above the floor. Two opposing 90° sectors were protected by 16-cm-high inner and outer walls of gray polyvinyl chloride. Animals were released in one of the closed sectors and observed for 10 min. In both experiments, animals were video-tracked using EthoVision 2.3 and data were transferred to Wintrack 2.4 for analysis.
For experiments on anxiety-related behavior, mice were habituated for 7 days to a reversed light/dark cycle and experiments were performed during their normal active phase.
Startle response and prepulse inhibition
Testing was conducted using a Hamilton-Kinder SM100 startle monitor system (http://www.hamiltonkinder.com, Poway, CA, USA). The rectangular animal restrainer (3.8 × 8.8 cm) was made of clear Perspex and had an adjustable ceiling that was set to prevent the animal from rearing. The restrainer rested on a sensing plate that carried a piezoelectric accelerometer at its bottom. The unit was mounted on a heavy metallic base plate by four mounting pins and enclosed in a sound-attenuated ventilated cabinet (internal dimensions 29 × 29 × 18 cm). The loudspeaker was located 22 cm above the animal and produced white noise pulses. A microcomputer interface controlled the loudspeaker and performed A/D conversion of the signals from the accelerometer. Signal calibration was done using a Newton impulse calibrator. Sound levels were verified using a digital sound level meter (RadioShack). The background noise level inside the closed cabinet was maintained at 70 dB. Subjects were placed in the thoroughly cleaned and dried restrainer and left undisturbed for 5 min before the session began. To record a startle response profile, 66 trials were presented in total after a habituation time of 300 s. Nine different sound levels (dB) were used: 64, 68, 72, 76, 80, 90, 100, 110 and 120. Each stimulus was 40 ms and presented six times in pseudorandom order, such that each sound level occurred once within a block of nine trials. The series began and ended with six presentations each of the 120-dB stimulus. The average inter-trial interval was 15 s (ranging from 10 to 20 s). Background noise is set at 64 dB during the entire session. In order to assess prepulse inhibition, after a habituation period of 300 s, 60 trials were presented in total, which consisted of six trial types. One was a 40-ms, 120-dB startle stimulus. There were four different prepulse plus startle stimulus trials presented, so that the onset of a prepulse stimulus was 100 ms before the onset of the startle stimulus. The 20-ms prepulse stimuli were sounds of 68, 72, 76 or 80 B. Finally, trials where no stimulus was presented served to measure baseline movements. After six presentations of the plain 120-dB startle stimulus, the six trial types were presented nine times in pseudorandom order such that each type occurred once within a block of nine trials. The average inter-trial interval was 15 s (ranging from 10 to 20 s). Mice were habituated for 7 days to a reversed light/dark cycle and experiments were performed during their nor mal active phase.
Nociception
Hot-plate test
The hot-plate test was used to evaluate sensitivity to a painful hind paw stimulus. Nine PV+/+ mice (4 males, 5 females) and 10 PV−/− (5 males, 5 females) were individually placed in a transparent cylinder (20 cm diameter) on a 55.0±0.5 °C hot plate, and the latency to the first hind paw response—shaking or licking the paw—was recorded. The latency of the response was measured with a stopwatch by an experienced observer. If no such response was observed within 30 s, the animal was removed from the plate to avoid skin damage.
Tail-flick test
To test the heat sensitivity of the tail, each mouse was gently wrapped in a soft towel and placed on a transparent platform with its tail extending into a shallow grove. An infrared beam was applied from below (7370 plantar test; Ugo Basile, Gemonio, Italy, infrared intensity of 50), and tail-flick latencies were automatically recorded. Three trials were run on different areas of the tail with about 1-min inter-trial intervals. If no tail flick was observed within 15 s, the animal was removed from the plate to avoid skin damage.
MRI sequence
A total of 42 male mice were used for the structural magnetic resonance imaging (MRI) analysis. The first groups of mice were imaged in vivo (C57BL/6J (PV+/+); juvenile (PND20): n=6, adult: n=12, age=14.1±2.8 months; PV−/−: juvenile: n=6 adult: n=6, age=12.9±3.6 months). The second groups consisted of fixed brains within the intact cranium (C57BL/6J (PV+/+); juvenile (PND20): n=6 and PV+/−; juvenile: n=6). All mice were imaged with a Bruker AVANCE 400WB (Bruker, NMR, Billerica, MA) spectrometer (nuclear magnetic resonance) outfitted with an 89-mm-bore 9.4 tesla vertical Bruker magnet (Oxford Instruments, Oxford, UK), a birdcage RF probe with 30 mm inner diameter, and a shielded gradient system (100 G cm−1). For the in vivo group, anesthesia was used to immobilize the animals, and to minimize the fear and anxiety induced by the loud environment of the scanner. We used isoflurane for anesthesia; induction phase: 3 volume percent (vol%) and maintenance: 1.1–1.5 vol% at 1 l min−1 air flow, via a nose cone, under conditions that do not significantly affect heart rate, respiratory rate or oxygen saturation (SaO2) in C57BL/6J mice.43
Three scout scans were first acquired that enabled to position the subsequent T2-weighted images along the standard anatomical orientations in a reproducible manner. Optimal horizontal images were determined empirically by repositioning the animal to ensure that the animal’s brain was located at the center of the MRI coils. Shimming was performed to optimize B0 field homogeneity. By running the auto-shim procedure, the three linear shim channels (x, y and z) were automatically adjusted by means of an iterative software routine that maximized the area of the free inductive decay. T2-weighted images were obtained with a fast spin echo sequence with TR/effective TE=2000 ms/70 ms, rapid acquisition with relaxation enhancement factor=16, FOV=20 mm, acquisition matrix=256 × 256, 17 slices, with a slice thickness=0.5 mm and number of excitations=28. Each set of images required 15 min, and five sets of images were acquired sequentially to improve SNR.
Fixed brains were imaged using the same MRI sequence; only two sets of images were required to obtain an optimal SNR.
MRI processing
Labeling of the neocortex, cerebellum and hippocampus was conducted in a semi-automated, registration-dependent framework. Such a framework relies on the coregistration of a source image (that is, PV−/− mouse) to a labeled template image. We coregistered T2-weighted MRI volumes of PND20 PV+/+, PV−/− and PV+/− mice independently to a PND0 T2-weighted atlas developed at the Laboratory of NeuroImaging (www.loni.usc.edu). A PND0 C57BL/6J atlas, as opposed to an adult mouse atlas, was used here in an effort to best match the myelination and gross structural/morphological profiles of the PND20 mice. Labels in atlas space were modified to include the hippocampus as a region distinct from its original (cortical) label, as defined by Laboratory of NeuroImaging.
As the z-plane slice thickness (500 μ) of the T2-weighted source acquisitions introduced a sampling and coregistration confound, the coregistration procedure was first carried out as a series of two-dimensional (2D) operations. First, the P0 atlas (50 × 50 × 50 μ) and the source T2 volume (86 × 86 × 500 μ) were each split into a series of 2D slices along the dorsal–ventral axis, producing 200 2D slices of the P0 atlas, and 24 slices of the source T2-weighted volumes.
Subsequently, an iterative three-degree of freedom 2D coregistration was carried out using the fMRIB analysis group’s linear image registration tool (FLIRT—http://fsl.fmrib.ox.ac.uk/fsl/flirt/). Each slice24 of the source image served as the source slice in the coregistration, whereas each slice of the atlas (200) served as the target slice in the coregistration. Subsequent computation of the correlation ratio of the image pair (transformed source slice and target atlas slice) was computed, and the atlas slice with the best (lowest) similarity score to a transformed source slice was chosen to represent an equivalent section of the source brain. Such a calculation was carried out for each of the coregistrations (200 × 24) to determine the 24 atlas slices that best represented the source brain (Supplementary Figure S7A).
Subsequently, 24 atlas slices were then reassembled into a three-dimensional volume and served as the target of a three-dimensional coregistration of the three-dimensional source volume to the reassembled, 24-slice atlas. In addition to a 12-degree of freedom linear registration conducted by FLIRT, the FMRIB’s nonlinear registration tool (fNIRT—http://fsl.fmrib.ox.ac.uk/fsl/fnirt/index.html) was used to register local deformations necessary to regularize the transformation. The inverse of the linear and nonlinear transformations were then applied both to the template image and its corresponding labels with nearest-neighbor interpolation, to produce a segmented (neocortex, cerebellum and hippocampus) source brain (Supplementary Figure S7B).
Electrophysiology
Slice preparation and FSI visualization
To facilitate FSI identification in control and PV-reduced/deficient conditions, most experiments were performed on mice aged 18–24 days (PND18–24) from the PV-enhanced green fluorescent protein (EGFP) BAC transgenic mouse line,44 PV-EGFP PV+/− and PV-EGFP PV−/− mice, respectively (all lines on a C57Bl/6J background). The reduced/deficient lines were obtained by crossing the PV-EGFP mice with PV−/− mice.27 Some recordings were obtained from PV+/+, PV+/− and PV−/− mice at PND18–24, mice as used for all other experiments. Recordings were obtained at least from 6–8 different animals per genotype, which belonged to 5–6 different litters. A set of experiments with n⩾8 embraced at least five different animals per genotype. Mice were decapitated after deep halothane anesthesia. After dissection, brains were rapidly immerged in an ice-cold bicarbonate-buffered saline solution (BBS) at 4 °C with the following composition: 125 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 26 mM NaHCO3, 2 mM CaCl2, 1 mM MgCl2, 10 mM glucose equilibrated with a 95% O2–5% CO2 mixture (pH=7.3). Cortico-striatal coronal slices were cut in 250-μm-thick sections using a vibratome (Leica VT1000S; Leica Microsystemes SAS, Nanterre, France) and transferred to an incubation chamber containing the same BBS at 32 °C for at least 1 h before recordings and protected from light. Slices were transferred to a submerged recording chamber perfused with BBS at 22–24 °C with a 1.5 ml min−1 flux rate. FSIs were visualized in the striatum with a × 63 water immersion objective of a Zeiss upright microscope (Axioskop 2FS Plus, Zeiss, Oberkochen, Germany) and the intrinsic EGFP fluorescence detected using short pulses of blue light from a source composed of an LED array (OptoLED, Cairn Research, Faversham, UK) emitting at 488 nm and coupled to the microscope that was equipped with a dichroic mirror and a high-pass emission filter centered at 505 and 507 nm, respectively. FSIs were visualized with a back-illuminated charge-coupled device camera (iXon+; Andor Technology, Belfast, UK).
Perforated-patch recordings
All recordings were made with a Dual EPC-10 operational amplifier and data were acquired using the software Patchmaster (HEKA, Lambrecht-Pfalz, Germany). Using the built-in filter of EPC10, voltage-clamp recordings were filtered at 2.9 kHz and digitized at 20 kHz. Patch pipettes were made from borosilicate glass capillaries (Hilgenberg Malsfeld, Germany) with a two-stage vertical puller (PIP 5, Heka). Pipette input resistances were in the 5–7-MΩ range. Perforated-patch recordings on FSI were made using a pipette solution containing the following (in mM): 80 K2SO4, 10 NaCl, 15 glucose, 5 HEPES, pH=7.2 with KOH, 225–230 mOsm l−1 and 400 μg ml−1 amphotericin B. Fresh stocks of the ionophore were prepared every 2 h (4 mg amphotericine B in 50 μl dimethyl sulfoxide). Series resistance was monitored to ensure that voltage attenuation in current-clamp mode was always <10%. Electrical stimulations were performed with a bipolar electrode (Phymep, Paris, France) in the presence of the GABAA blocker, gabazine (10 μM). Pulses of 500 μs duration were delivered at 0.25 Hz by increasing the stimulation intensity from 0 to 9 V. These pulses were generated by an Iso-flex stimulus isolation unit (AMPI, Jerusalem, Israel) connected to an output channel of the amplifier and triggered by the Patchmaster software. When the stimulation amplitude evoked stable excitatory postsynaptic currents (EPSCs) in time, protocols to study unitary properties of EPSC, paired-pulse ratio (PPR) and trains were started. NBQX (2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione) and gabazine were dissolved in the bath solution and applied to the preparation by superfusion. The drugs solution reached a steady-state concentration in the experimental chamber within 2 min. After reaching this steady-state period, the response to the drug was measured after a prolonged application (up to 5 min). NBXQ and gabazine were purchased from Sigma-Aldrich (St Louis, MO, USA). Stocks were prepared in water and stored at −20 °C. They were diluted in BBS before bath application.
Dendritic branching counts and measurements
For morphological reconstructions, FSIs were subjected to whole-cell recordings with a pipette solution containing the following (in mM): 150 K gluconate, 4.6 MgCl2, 10 Hepes-K, 1 EGTA-K, 0.4 Na-GTP, 4 Na-ATP, 0.1 CaCl2, pH=7.2 and complemented with 0.4% biocytin. A period of 20 min after whole-cell break-in was respected and a high-resistance outside-out patch was obtained when the pipette was withdrawn. Slices were fixed by overnight immersion in 4% paraformaldehyde at 4 °C, rinsed in phosphate-buffered saline at 0.1 mM and, later, immersed for 2-h periods in a mixture of phosphate-buffered saline/Triton X-100 (0.1 mM and 0.1%, respectively). Biocytin was revealed in red with streptavidin-conjugated NL557 (R&D systems, Minneapolis, MN, USA), diluted 1:5000, always in phosphate-buffered saline/Triton X-100. After a final rinse in Tris-buffered saline (TBS), slices were mounted on coverslips with FlourSave reagent (EMD Millipore, Billerica, MA, USA) and secured with nail polish. Experiments were obtained from 8 to 18 different animals per genotype, which belonged to 5–9 different litters. A set of experiments with n=8 embraced at least five different animals per genotype. Confocal images were acquired using an Axiovert 200M-LSM 510 META microscope (Zeiss) equipped with a C-Apochromat × 40/1.2 and a 543-nm helium–neon laser. Band-pass emission filters were used for selective detection of the endogenous EGFP (500–550 nm) and red-biocytin labeling (565–615 nm). Single FSI images were acquired as 50–70-μm-thick Z-stacks composed of 2048 × 2048 pixel images (pixel size: 0.22 μm) with a Z-step of 0.62 μm. Reconstructions of neuronal projections were performed using the LSM Image Browser (Zeiss) and processed with a median filter (2 pixel radius) from Image J. Dendritic branching was quantified by Sholl analysis45 and their elongation measured with ‘moment calculator’, an ImageJ plug-in (Francois Richard, University of Ottawa; http://imagej.nih.gov/ij/plugins/moments.html). Dendritic distribution was evaluated by tracing vectors from the soma (as the center) and the crossing dendrites within a 100-μm-radius circle. The angular variance of those vectors was calculated for each FSI according to Batschelet.46
Statistical analysis
For analysis of isolation-induced USV, body weight, body temperature and somatosensory reflexes in mouse pups, two-way analyses of variance (ANOVAs) with the between-subjects factors genotype' (PV+/+, PV+/− and PV−/−) and sex (male or female) were calculated. In order to test whether differences in pup USV emitted in isolation emerged over time during testing, ANOVAs for repeated measurements with the same between-subject factor and the within-subject factor test duration were performed. For analysis of interaction-induced USV and reciprocal social interaction behavior in juvenile mice, two-way ANOVAs with the between-subject factors' genotype (PV+/+, PV+/− and PV−/−) and sex (male or female) plus the covariate age of subject mice in PND were calculated. In order to test whether differences in juvenile USV and social interaction behavior displayed during reciprocal social interaction emerged over time during testing, ANOVAs for repeated measurements with the same between-subject factors plus the covariate age of subject mice in PND and the within-subject factor test duration were performed. For analysis of female-induced USV in adult male mice, one-way ANOVAs with the between-subject factors' genotype (PV+/+, PV+/− and PV−/−) plus the covariate age of subject mice in PND were calculated. ANOVAs were followed by least significant difference post hoc analysis when appropriate. A paired t-test was used to compare the likelihood of the occurrence of a social behavior in response to a social behavior.
For analysis of repetitive and stereotyped patterns of behavior, motor functions and locomotor activity, anxiety-related behavior and sensory information processing, data were analyzed using mixed ANOVA models with genotype (mutant, control) as between-subject factors and within-subject factors to explore the dependence of genotype effects on place, time or stimulus. Significant interactions and where necessary significant main effects were further explored by Tukey–Kramer post hoc tests or by splitting the ANOVA model, as appropriate. One-sample t-tests were used for follow-up comparisons against chance levels. Variables known to produce strongly skewed distributions and/or frequent outliers were subjected to a log transformation before ANOVA analysis (as indicated, for example, latency measures). Sex and, where applicable, cohorts were added to the ANOVA model as additional between-subject factors to check for sex or cohort dependence of any genotype effect. Main effects of sex and cohort were not studied; interactions are only reported when significant. Student’s t-test was performed to compare rotarod performance between genotypes, and pain sensitivity was analyzed by means of an ANOVA, including the factors sex and genotype.
For statistical analysis of the MRI data, ANOVAs with a post hoc Bonferroni test were used to compare groups (that is, genotypes, using their specific controls). For electrophysiological recordings and Sholl analysis, statistical analyses were performed using the Neuromatic software package (http://www.neuromatic.thinkrandom.com) and custom routines within the IgorPro environment (Wavemetrics, Lake Oswego, OR, USA). Statistical tests were performed using the Student's t-test within Excel software package (Microsoft, Redmond, WA, USA) and repeated-measures ANOVA were performed using Prism 4.0. (GraphPad Software). Gaussian statistics were used for comparisons of angular variances. All values are expressed as mean±s.e.m. A P<0.050 was considered statistically significant.
Results
ASD core symptoms are manifest in mice with reduced/absent PV expression
With a focus on behavioral phenotypes of PV−/− mice with relevance to ASD core symptoms, we performed a detailed analysis of their social behavioral repertoire during reciprocal social interactions, measured the production of USV in pups isolated from mother and littermates, juvenile mice during reciprocal social interactions and adult male mice exposed to females, along with the assessment of repetitive and stereotyped patterns of behavior by means of a T-maze working-memory task and the MWM place navigation task, with all paradigms known to be sensitive for detecting behavioral deficits relevant for ASD.14, 15, 16, 17
Social behavior deficits
Reduced levels (PV+/−) or absence of PV (PV−/−) caused a clear reduction in the time spent in reciprocal social interaction behavior in juvenile mice at PND25±1 as compared with PV+/+ littermate controls (Figure 1a). Although reciprocal social behavior was overall reduced in PV+/− and PV−/− mice, its temporal pattern was similar in all genotypes, with most intensive social interaction behavior during the first 2 min of reciprocal social interaction (Figure 1b). Importantly, similar genotype effects were obtained for the total numbers of social interactions (Figure 1c). When analyzing the social behavioral repertoire in detail, its richness and reciprocal character were found to be strongly affected by genotype. PV+/+ mice displayed a significant preference for engaging in another social behavior following a previous one in ~61% of the cases (~39% for nonsocial behavior), whereas no such preference was seen in PV+/− and PV−/− mice, with social behaviors following in ~53% and ~49% of cases, respectively (Figure 1d; typical ethograms are depicted in Figure 1e). Genotypes did not differ in nonsocial behaviors, including rearing, grooming and digging behavior during reciprocal social interactions (Supplementary Figures S1A–C). Moreover, the type and frequency of nonsocial behavior following a social one was not different between genotypes (not shown).

Social behavior deficits: PV−/− null mutant and PV+/− heterozygous mice display social behavior deficits during reciprocal social interactions as juveniles. (a) Total social interaction time displayed during the 5-min social interaction period (genotype: F2,23=5.405, P=0.012; sex: NS; genotype × sex: NS). (b) Time course for the social interaction time displayed during each 1-min time bin across the 5-min social interaction period, plus 1 min habitation (dashed line indicates introduction of partner mouse). (c) Total number of social interactions displayed during the 5-min social interaction period (genotype: F2,23=3.620, P=0.043; sex: NS; genotype × sex: NS). Black bar: PV+/+ wild-type littermate control mice; striped bar: PV+/− heterozygous mice; white bar: PV−/− null mutant mice. Data are presented as means±s.e.m., bars denoting the s.e.m. *P<0.050 vs PV+/+. (d) Percentage of nonsocial vs social behavior following social behavior (dashed line indicates 50% chance level). Black bar: social; striped bar: nonsocial. *P<0.050 vs nonsocial. #P<0.050 vs 50% chance level (PV+/+: t8=2.408, P=0.043; PV+/−: NS; PV−/−: NS). (e) Representative ethograms of social and nonsocial behavior displayed during juvenile reciprocal social interactions by a PV+/+ wild-type littermate control mouse, a PV+/− heterozygous mouse and a PV−/− null mutant mouse. NS, not significant; PV, parvalbumin.
Impairments in communication
USV emission appears to be a major mode of communication in rodents47 and ASD mouse models are typically characterized by altered USV production.48 We therefore tested USV production at various developmental time points. USV emission during juvenile reciprocal social interaction (PND25±1) was reduced approximately fourfold in PV−/− mice as compared with PV+/+ littermate controls (Figure 2a), with a similar, yet overall reduced temporal USV emission pattern, characterized by the highest USV rates during the first 2 min of reciprocal social interaction (Figure 2b). Remarkably, USV emission in PV+/− animals was diminished to the same extent as in PV−/− mice. Also duration of USV was found to be shorter in PV+/− and PV−/− mice than in PV+/+ littermate controls (Figure 2c). Peak frequency and frequency modulation were not affected (Figures 2d and e; typical spectrograms are depicted in Figures 2f–h). Thus, a reduction of PV to ~30–40% of PV+/+ (ref. 24) is sufficient for inducing communication deficits with relevance to ASD. USV production in adult mice was also affected by genotype. Unlike at PND25±1, however, PV+/− vocalized like PV+/+ mice in these experiments. When adult male mice (PND~120) were exposed to females for 5 min, PV+/+ and PV+/− mice emitted ~320 USV and ~390 USV, respectively, whereas PV−/− mice emitted ~10 USV only (Supplementary Figures S2A and B). Furthermore, USV emitted by PV−/− mice were shorter in duration and less frequency-modulated than those emitted by PV+/+ littermate controls (Supplementary Figures S2C and E). Peak frequency was not affected (Supplementary Figure S2D; typical spectrograms are depicted in Supplementary Figures S2F–H). Importantly, deficits in USV emission were found to be specific for developmental stages in which PV supports important physiological functions. When testing for pup isolation-induced USV (Supplementary Figures S3A–H), somatosensory reflexes and body development (Supplementary Figures S4A–D) at PND8, no genotype differences were observed, as expected. At PND8, PV protein expression levels in PV+/+ mice are very low, often below the detection limits of immunohistochemistry and/or western blot analysis, for example, in neocortex49 and cerebellum.25

Impairments in communication: PV−/− null mutant and PV+/− heterozygous mice display ultrasonic vocalization deficits during reciprocal social interactions as juveniles. (a) Total number of ultrasonic vocalizations emitted during the 5-min social interaction period (genotype: F2,21=9.240, P=0.001; sex: NS; genotype × sex: NS). (b) Time course for the number of ultrasonic vocalizations emitted for each 1-min time bin across the 5-min social interaction period, plus 1 min habitation (dashed line indicates introduction of partner mouse). (c) Duration of calls (genotype: F2,21=3.870, P=0.037; sex: NS; genotype × sex: NS), (d) peak frequency (genotype: NS; sex: NS; genotype × sex: NS) and (e) frequency modulation of calls (genotype: NS; sex: NS; genotype × sex: NS) emitted during the 5-min social interaction period. Black bar: PV+/+ wild-type littermate control mice; striped bar: PV+/− heterozygous mice; white bar: PV−/− null mutant mice. Data are presented as means±s.e.m., bars denoting the s.e.m. *P<0.050 vs PV+/+. (f–h) Representative spectrograms of ultrasonic vocalizations emitted during juvenile reciprocal social interactions by (f) a PV+/+ wild-type littermate control mouse, (g) a PV+/− heterozygous mouse and (h) a PV−/− null mutant mouse. NS, not significant; PV, parvalbumin.
Repetitive and stereotyped patterns of behavior
To model ASD-associated perseverative thinking and behavioral inflexibility, we tested PV+/+ and PV−/− mice in reversal learning paradigms using a T-maze working-memory task and the MWM place navigation task.37 No genotype differences were detected during the acquisition phase of the T-maze working-memory task. Both genotypes demonstrated equal ability to learn the test as revealed by the number of correct daily trials, a plateau of ~8 correct trials was reached within 4 days. Also the time required to reach criterion performance (⩾8/10 correct trials during three consecutive days in 70% of the mouse cohorts) was similar, with PV+/+ and PV−/− mice achieving criterion at 14 and 12 days, respectively (Figure 3a). Similarly, PV−/− mice performed normally in the acquisition phase of the MWM place navigation task (Figure 3c). Consistently, no evidence for learning and memory deficits were observed in the radial-maze working-memory task (Supplementary Figure S5A); the number of correct choices and the learning rate across trials over the 10-day procedure, as well as the errors per collected bait were not different between groups. This indicates that both spatial reference and spatial working memory are intact in PV−/− mice. Repetitive and stereotyped patterns of behavior could hence be tested using these tasks. In the T-maze working-memory task, seven PV+/+ and six PV−/− mice were able to proceed to the reversal phase. During reversal learning, PV−/− mice exhibited a clear deficit in their ability to reverse their behavior to obtain the reward from the opposite arm of the maze. Analysis of trials to reach criterion for acquisition and reversal showed a significant difference in reversal learning (Figure 3b). Similar findings were obtained in the MWM place navigation task. Despite intact acquisition learning of a submerged escape-platform location, PV−/− mice were more strongly disturbed by the relocation of the platform during the reversal phase than PV+/+ mice and performed worse during the first trial block (Figure 3d).

Repetitive and stereotyped patterns of behavior: PV−/− null mutant mice display reversal deficits in adulthood. (a) During acquisition of the T-maze task, no effect of genotype on learning was observed (% mice not reaching criterion by day, log-rank Mantel–Cox; genotype: χ12=0.4; NS). (b) During T-maze reversal, the rate of improvement was significantly lower in PV−/− mice (log-rank Mantel–Cox; genotype: χ12=3.9; P=0.048), with only 2/6 animals reaching criterion by 20 days. In contrast, 6/7 PV+/+ mice reached criterion with a learning rate similar to the acquisition phase. (c) During place navigation acquisition in the MWM, neither performance level nor learning rate were affected by genotype (escape latency by blocks of three trials; ANOVA, genotype: F1,27=0.5, NS; time: F5,135=20.8, P<0.0001; time × genotype: F5,135=0.4, NS). (d) In the reversal stage, PV−/− were more strongly disturbed by the relocation of the platform and performed worse during the first trial block (ANOVA, genotype: F1,27=1.9, NS; time: F3,81=17.5, P<0.0001; time × genotype: F3,81=3.5, P=0.020; *PV−/− vs PV+/+ P=0.041). Data are presented as means±s.e.m., bars denoting the s.e.m. ANOVA, analysis of variance; MWM, Morris water maze; NS, not significant; PV, parvalbumin.
ASD-linked comorbidities are prevalent in PV−/− mice
Besides behavioral paradigms with relevance to ASD core symptoms, mice were subjected to a battery of assays to detect potentially confounding factors, such as motor deficits and reduced exploratory activity, and to study relevant ASD comorbidities, such as anxiety and sensorimotor impairments.1 In line with human studies indicating that seizures are present in ~25% of individuals with ASD,50 increased susceptibility to seizures induced by pentylenetetrazole in PV−/− mice has previously been reported, yet these mice do not show spontaneous seizures.24
Motor function and locomotor activity
Motor performance in adult (>3 months) PV−/− mice was normal on the acceleration rotarod (10–40 r.p.m.), in the force grip test, and normal swim parameters were found in the MWM place navigation task (not shown). General locomotor parameters were determined by measuring exploratory activity in a large open-field arena over two consecutive days. PV+/+ and PV−/− mice were indistinguishable in terms of within- and between-session habituation (Supplementary Figures S5B and C). Essentially all parameters including moving time and average speed were similar between genotypes, with two exceptions: bouts of locomotion in PV−/− mice were less structured than in PV+/+ mice, being more linear (linearity index: F1,52=9.503, P=0.003) and showing less small-scale accelerations and decelerations (jerkiness index: F1,52=9.745, P=0.003), and, in line with previous observations,51 rearing activity was reduced in PV−/− (estimated vertical movements: F1,28=4.743, P=0.038), indicating mildly changed patterns of spontaneous locomotion. Level and habituation rate of activity in the small open field with home box were unchanged (Supplementary Figure S5D).
Anxiety-related and depression-like behavior
In the large open field, PV−/− mice showed a larger speed difference between centrifugal and centripetal locomotion (genotype: F1,52=8.599, P=0.005), a finding sometimes associated with increased anxiety. Similarly, PV−/− mice displayed reduced rearing activity in the center field of the small open field (estimated vertical movements: genotype: F1,28=7.02, P=0.013), whereas center time and the time spent inside the home box were unchanged (genotype: not significant; Supplementary Figure S5E). Also, no genotype differences were observed in the light/dark transition and the O-maze test (Supplementary Figures S5F and G). In the sucrose preference test used to assess anhedonia, a core symptom of depression, no evidence for genotype effects were observed (not shown).
Sensory information processing
Altered responses to sensory stimuli are commonly observed in ASD patients, particularly in the auditory domain and in response to pain. Thus, we determined sensitivity to acoustic and heat stimuli in PV+/+ and PV−/− mice. In the prepulse inhibition paradigm, no evidence for significantly altered prepulse inhibition was obtained (Supplementary Figure S5H), yet PV−/− mice showed a reduced startle response to strong stimuli (110 and 120 dB; Supplementary Figure S6A). In response to heat stimuli, PV−/− mice showed reduced nociception, as reflected in an increased latency to withdrawal from a painful heat stimulus in both the hot-plate and tail-flick assays as compared with PV+/+ controls (Supplementary Figures S6B and C).
Age-dependent brain morphology alterations in PV−/− mice show similarities to structural changes reported in human ASD patients
Evidence has accumulated that the time course of brain development may be altered in ASD, with cortical lobes and cerebellum most consistently affected.31, 32, 52 Using MRI cerebral blood volume imaging, an in vivo correlate of brain metabolism in the basal state, an age-independent impairment of metabolic status is observed in the dentate gyrus and CA1 region of the hippocampal formation in PV−/− mice.33 Here we performed MRI volumetric analysis of neocortex, cerebellum, hippocampus and whole intracranial content (Figure 4a). PV+/+ and PV−/− mice were imaged in vivo at PND20 (juvenile) and between 9 and 16 months (adult). Segmentation and volumetry of the aforementioned regions of interest was conducted using a custom-built semi-automated approach (see Materials and methods and Supplementary Figures S7A and B). Region of interest volumes were normalized to total intracranial content.

Absence of parvalbumin in PV−/− mice induces transient neocortical hypertrophy and cerebellar hypoplasia in male juvenile (PND20) mice. (a) Representative axial sections acquired with T2-weighted imaging. Semi-automatically labeled cerebellar (purple), neocortical (green) and hippocampal (red) ROIs are highlighted. (b) (From left to right) neocortical volume, which was normalized to total intracranial content, from control and PV−/− mice at the specified age groups; at PND20, the neocortical volume in PV−/− mice was increased (genotype: F1,29=4.65, P=0.021). Normalized cerebellar volume at the specified age groups: at PND20, the cerebellar volume in PV−/− mice was significantly smaller (genotype: F1,29=13.10, P=0.0029). Normalized hippocampal volume and total intracranial content at the specified age groups. No significant differences were observed in the normalized hippocampal or total intracranial content of juveniles (PND20) or in any of the ROIs analyzed in the adult group. Data are presented as means±s.e.m., bars denoting the s.e.m. *P<0.05, **P<0.01 two-way ANOVA followed by Bonferroni’s t-test. ANOVA, analysis of variance; ICC, intracranial content; PND, postnatal day; PV, parvalbumin; ROI, region of interest.
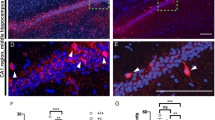
At PND20, neocortical volume was 21±4.4% larger and cerebellum 14.9±2.1% smaller in PV−/− than in PV+/+ mice (Figure 4b). In the adult groups, neocortical volumes were similar in PV−/− and PV+/+ mice, indicative of a transient hypertrophy at PND20; also no differences persisted in cerebellar volumes between PV+/+ and PV−/− mice at adult age. No genotypic differences in hippocampal volumes were observed in both age groups (Figure 4b). Moreover, there was no genotype difference in the whole-brain volume at the two time points investigated (Figure 4b). Thus, PV deficiency results in transient neocortical hypertrophy and cerebellar atrophy in PND20 mice. As a clear behavioral phenotype was observed in PV+/− mice (Figures 1 and 2), morphological changes were also investigated in PND20 PV+/− mice (Supplementary Figure 8). The PV+/− neocortical volume was increased by 11.3±2.9%, whereas volumes of cerebellum and hippocampus were unaltered, as was the total intracranial volume. To assess whether the neocortical hypertrophy was linked to putative morphological changes of PV interneurons in PV−/− and PV+/− mice, we selected the subpopulation of PV+ neurons in the striatum termed FSI, where the lower density, compared with the high density of PV+ neurons in the cortex,21 allowed to better visualize the dendritic arbor of individual FSIs. In cortico-striatal coronal slices (250 μm thickness; PND18–24), the number of branches was clearly increased in PV−/− FSI (Supplementary Figure S9A). Sholl analysis revealed a higher number of dendrites in a region between 40 and 150 μm from the soma (Supplementary Figure S9B) and also more branches from the 3rd to 5th order (Supplementary Figure S9C). However, the elongation or eccentricity of the dendritic arbors was similar in all three genotypes, that is, the increase in dendrites was not accompanied with a polarization of the main dendrites (PV+/+ 5.79±1.88 vs PV+/− 4.85±0.89 vs PV−/− 6.29±1.65; genotype: not significant), indicating that the additional dendrites in PV−/− and PV+/− FSIs were homogeneously distributed in space. The FSI morphology of PV+/− mice revealed differences, when compared with either PV+/+ or PV−/− neurons. In PV+/− FSI, the number of intersections was increased 40–70 μm from the somata (as in PV−/−), whereas at distances >70 μm, intersection numbers were nearly identical as seen in PV+/+ controls (Supplementary Figure S9B). This indicates that a decrease in PV levels most strongly affects proximal branching, whereas its complete absence also affects distal branching. With respect to branching, in PV+/− FSIs more branches from the 2nd to 5th order were observed, reminiscent of the situation in PV−/− FSIs (Supplementary Figure S9C).
Absence of PV affects E/I balance involving pre- and postsynaptic mechanisms
The effect of presynaptic PV in preventing short-term facilitation of synaptic transmission by acting as a Ca2+ buffer has been demonstrated in cerebellar interneurons,25, 53 PV+ interneurons in the hippocampus23, 26 and striatal FSIs.27 Results from all these studies in PV−/− mice are in line with a shift toward activity-dependent increased inhibition. Here, the effect of PV deficiency at the postsynaptic level was investigated by stimulating cortical excitatory inputs onto striatal PV+ FSIs and recording EPSCs a in perforated-patch configuration (Figure 5a1). Excitatory currents in FSIs were mostly mediated by AMPA receptors that were reversibly blocked by NBQX (10 μM) (Figure 5a2). No differences in single evoked EPSCs were evident in all three genotypes, suggesting that PV neither exerts an influence on the kinetics of individual EPSCs nor on the variance to mean ratio, the latter representing a valuable estimate of the quantal size (Supplementary Table 1). In a paired-pulse protocol, the cortical neuron–FSI short-term plasticity was investigated at different inter-spike intervals (ISIs) ranging from 10 to 200 ms in PV+/+, PV+/− and PV−/− mice. Significantly reduced EPSC facilitation was observed in PV−/− and PV+/− FSIs at 10, 20 and 50 ms ISI; (Figure 5b2) when compared with PV+/+ cells, for example, a PPR of 1.03±0.03 (PV+/+) vs 1.36±0.06 (PV−/−) (P<0.001 at 50 ms; Figure 5b1). Nearly identical results were obtained in PV+/− cells, demonstrating that a reduction in PV levels is sufficient to induce an adaptive (likely homeostatic) mechanism at this synapse (Figure 5b2). Moreover, analysis of cortically evoked EPSC trains (8 stimuli at 50 Hz) revealed depression of FSI EPSCs in all three genotypes (Figures 5c1 and c2). However, stronger depression starting from the 2nd EPSC was evident in PV−/− and PV+/− FSIs; point per point comparisons of pooled data demonstrated that only the second EPSC in PV−/− and PV+/− FSIs was significantly smaller, compared with the one in PV+/+ neurons (Figure 5c2). Comparison of all averaged EPSCs resulted in a statistically significant difference between PV+/+ and either PV+/− or PV−/− FSI (P<0.001 for both comparisons). Similar results were also obtained for EPSCs elicited by 100-Hz stimulation (Figures 5d1 and d2). A comparison of all recorded EPSC demonstrated a statistically significant difference between FSI from the three genotypes (two-way ANOVA; P<0.001) and the depression of the second EPSC was stronger in PV+/− and PV−/− FSIs (P<0.01 and P<0.001, respectively, for both comparisons). The CV−2 evaluation test was applied to identify pre- versus postsynaptic influences on EPSC changes, as previously used for analysis of synaptic events evoked by extracellular stimulation.54 The PPR series at 50 ms ISI was used to calculate ratios between CV−2 amplitudes for the 2nd EPSC (CV−2 2-EPSC) and the 1st EPSC (CV−2 1-EPSC). In PV+/+, this ratio was 1.68±0.15, indicative of an increase in the probability of release for the 2nd EPSC, which is coherent with the PPR facilitation observed at this ISI (Figure 5b2). In PV−/− FSI, this ratio was 0.98±0.13 (P<0.01 vs PV+/+), indicating a reduction in the probability of release for the 2nd EPSC. This is in accordance with the absence of PPR facilitation in PV−/− FSI at 50 ms ISI; however, at this synapse with no changes in the 2nd EPSC mean amplitude. These results indicate that short-term plasticity at the cortical neuron–FSI synapse might have a presynaptic adaptation, that is, homeostatic plasticity55, 56 in the cortical neuron caused by the absence/reduction of PV in the postsynaptic FSI, besides PV’s more direct role at the presynaptic side in FSI and other PV+ interneurons.

Short-term plasticity of excitatory cortical inputs to FSI is modulated during fast frequencies by postsynaptic PV. (a1) Experimental configuration to evoke EPSC in FSI (striatum, CPu; corpus callosum, cc). (a2) An averaged EPSC and its blockage by NBQX (10 μM) are shown. (b1) Averaged EPSC from a PPR protocol at a 50-ms interval. The gray arrow indicates a decrease of the second EPSC in PV−/− FSI compared with PV+/+. Amplitudes were normalized to the first EPSC. (b2) Pooled data for all PPR protocols. PV+/+: n=10; PV+/−: n=8; PV−/−: n=12. (c1) Averaged EPSC from a 50-Hz train. In the PV−/− FSI, the progressive reduction of EPSC amplitudes is more accentuated than in the PV+/+ FSI. Amplitudes were normalized to the first EPSC. (c2) Pooled data for 50-Hz trains. The second EPSC is significantly reduced in PV−/− and PV+/− FSIs compared with PV+/+. A two-way ANOVA test demonstrated a significant difference between genotypes (PV+/+: n=10; PV+/−: n=8; PV−/−: n=12; P<0.001). (d1) Averaged EPSCs from a 100-Hz train. At higher frequency, a marked reduction of the second EPSC is observed in PV−/− and PV+/− FSIs compared with PV+/+. Amplitudes were normalized to the first EPSC. (d2) Pooled data for 100-Hz trains. The second EPSC is also significantly reduced in PV−/− and PV+/− FSIs compared with PV+/+. The two-way ANOVA test confirmed a significant difference between genotypes (PV+/+: n=10; PV+/−: n=8; PV−/−: n=11; P<0.001). The same color code (black and red) applies for b1–d2. Averages are from 20 tests recorded at 4-s intervals in a FSI (b1, c1 and d1). Dotted line in b2 represents unity. All values are presented as means±s.e.m., bars denoting the s.e.m. *P<0.05, **P<0.01, ***P<0.001 Student’s t-test. ANOVA, analysis of variance; EPSC, excitatory postsynaptic current; FSI, fast-spiking interneuron; ISI, inter-spike interval; NBQX, 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione; PPR, paired-pulse ratio; PV, parvalbumin.
Discussion
The etiology of ASD remains unclear, likely resulting from the extremely heterogeneous nature with contributions from various genetic and environmental factors. Several unifying defects have been suggested: synaptic dysfunction,57 E/I imbalance58 and more generally, impairment in activity-dependent neuronal signaling3, 4 are among the most prominent ones. A computational systems biology approach led to an integrative network model for gene/environment interactions, and the most relevant node for ASD was found to be the Ca2+ node;59 a link between altered Ca2+ signaling and ASD was suggested previously.60 PV is a key component of the Ca2+ homeostatic mechanisms required for the correct functioning of PV+ interneurons, and other studies (genome-wide association studies and transcriptomic co-expression network analyses)5, 6, 7 concluded that many ASD-linked gene products are expressed in PV+ neurons, making this neuron population a prime target in ASD research.61
Our study revealed behavioral deficits in PV−/− mice with relevance to all human ASD core symptoms. First, social interaction behavior was clearly lower among both, PV−/− and PV+/− juveniles. Moreover, a social interaction between PV+/+ animals was likely followed by another one, in contrast to mice without or reduced PV levels. Overall, the richness and heterogeneity of the social behavioral repertoire was reduced in PV−/− and PV+/− mice. These results comprise a quantitative and qualitative impairment in social behavior. Social dysfunction is also a key phenotypic trait in other ASD mouse models, where ‘a reduction in PV+ neurons’ was reported.18, 19, 20, 21, 22, 62
Second, USV emission was considerably reduced in PV+/− and PV−/− mice, indicating communication deficits.47, 48 Interestingly, the impairment in social interaction and USV was of similar magnitude in PV+/− and PV−/− mice, suggesting that even a reduction in PV expression is sufficient to elicit social and communication deficits with relevance to ASD in juveniles. Although the USV phenotype in PV−/− mice persisted in 3-month-old males, it was close to normal in PV+/− mice exposed to females, possibly because of adaptive mechanisms induced to cope with the decrease in PV expression resulting in the ‘normalization’ of USV or due to the fact that spontaneous USV emitted during female exposure, a situation of high biological relevance, are less vulnerable to PV depletion than USV emitted during reciprocal social interactions in juveniles.
Third, PV−/− mice were found to display deficits in reversal learning, modeling resistance to change in routine, insistence on sameness or rigid habits observed in ASD patients.1 PV−/− mice showed a distinct deficit in the T-maze reversal assay. This cannot be interpreted as deficits in olfaction, learning or working memory, as both PV+/+ and PV−/− mice performed equally well in the acquisition phase, as well as in the radial-maze working-memory task and during learning the MWM place navigation task. In the latter, the resistance to revert was also observed during the initial reversal learning.
Hypoalgesia, that is, increased pain threshold, is associated with a subset of ASD patients and is also present in the rat valproic acid ASD model, as well as in PV−/− mice. PV−/− mice also showing a reduced startle response at high decibel levels (>100 dB) would also point to a diminished response to aversive sensory stimuli. The reticular nucleus, a densely packed region of largely PV+ neurons, is implicated in relaying sensory information between the somatosensory cortex and the thalamus and thus, absence of PV and associated altered firing behavior29 could be related to the hypoalgesia phenotype of PV−/− mice. Noteworthy, no evidence for behavioral phenotypes with relevance to anxiety, depression and schizophrenia was obtained in PV−/− mice.
Brain abnormalities have been documented in ASD, with increased cortical volume in young children and cerebellar hypoplasia (for example, Purkinje cell loss) being among the most consistent ones.31, 32 In line with the observed accelerated brain development that results in cerebral overgrowth in juvenile (PND20) PV−/− mice, macrocephaly occurs in ~20% of ASD patients and enlargements were found practically in all cortical lobes.31, 32 Also the reduced size of PV−/− cerebella fits well with the human literature. Disturbances to the cerebellum are commonly observed in ASD, with postmortem studies consistently showing decreased numbers of Purkinje cells.31, 63, 64, 65, 66, 67 In addition, damage to the cerebellum is followed by reduced sociability66 and communication impairments64 in humans. In mouse studies, altered cerebellar development had been linked to social deficits,65, 67 impaired communication, as well as repetitive self-grooming and impaired reversal learning.67, 68 Recently, in a comprehensive neuroanatomical study comprising 26 ASD mouse models, morphological changes were reported in various brain regions and the most affected ones were found to include the parieto-temporal lobe, cerebellar cortex, frontal lobe, hypothalamus and striatum.69 Clustering of the 26 models resulted in three large groups, thus significantly reducing the heterogeneity; group 1 was characterized by an increase in the frontal and parieto-temporal lobes and decreases in cerebellar cortex, strongly reminiscent of the morphological phenotype of PV−/− mice. Moreover, in ASD models of this group including En2, Fmr1 and Shank3, decreases in PV staining/PV+ neurons/PV+ puncta were reported, respectively (Supplementary Table 1). It remains to be investigated, whether such a decrease is also present in other group 1 members including Nrxn1a. If so, the decrease in PV expression might represent a common mechanistic link in ASD, possibly for group 1 ASD models.
Is the decrease in PV expression a possible common end point in some forms of ASD? A decrease in the number or density of PV+ neurons was reported in genetic18, 19, 20, 22 and environmental21, 62 mouse ASD models (see Supplementary Table 1), as well as in human patients.10, 11 Yet it is currently unknown whether such a reduction in PV+ neurons is the result of neuron loss or PV downregulation. Functionally, a loss of PV+ neurons is expected to globally decrease inhibition, thus increasing the E/I ratio, whereas downregulation of PV leads to increased inhibition. We consider a clarification of this point of high relevance, not primarily for the interpretation of the findings in PV−/− mice, but more general, for the field of ASD research. Whether in ASD the E/I imbalance results from altered inhibition or excitation remains a matter of debate and cases for both have been reported, even in the same ASD mouse model. In NL3R451C mice, inhibition is increased in the somatosensory cortex, whereas AMPA receptor-mediated excitation is increased in hippocampal CA1 region, indicating that in ASD excitatory and inhibitory synapse properties are altered in a region- and circuit-specific manner by a mutation of a single synaptic protein.30 In PV−/− mice, the E/I ratio is even altered within the same neuronal (cortico-striatal) circuit. Although the lack of PV augments inhibition, that is, frequency-specific short-term plasticity at inhibitory FSI to MSN synapses,27 at the same time the excitatory drive onto FSI by neocortical neurons is decreased. Hypothesizing that also in other ASD mouse models the reported ‘decrease in PV+ neurons’ is the result of PV downregulation in order to increase the inhibitory drive by the PV-expressing (mostly fast-spiking) neurons, one may conjecture that decreased PV levels might represent a point of convergence, at least in some forms of ASD (for example, group 1 ASD-associated genes69). If correct, treatments of ‘low-PV’ ASD patients aimed to restoring/increasing PV expression may then be considered as a novel and possibly unifying therapeutic avenue in ASD research, at least in cases, where the genetic defect is linked to altered PV expression.
References
APA DSM-5 - Diagnostic And Statistical Manual Of Mental Disorders. American Psychiatric Publishing: Arlington, VA, USA, 2013.
Abrahams BS, Geschwind DH . Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet 2008; 9: 341–355.
Ebert DH, Greenberg ME . Activity-dependent neuronal signalling and autism spectrum disorder. Nature 2013; 493: 327–337.
Toro R, Konyukh M, Delorme R, Leblond C, Chaste P, Fauchereau F et al. Key role for gene dosage and synaptic homeostasis in autism spectrum disorders. Trends Genet 2010; 26: 363–372.
Weiss LA, Arking DE, Daly MJ, Chakravarti A . A genome-wide linkage and association scan reveals novel loci for autism. Nature 2009; 461: 802–808.
Ben-David E, Shifman S . Networks of neuronal genes affected by common and rare variants in autism spectrum disorders. PLoS Genet 2012; 8: e1002556.
Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011; 474: 380–384.
Kawaguchi Y, Kubota Y . Neurochemical features and synaptic connections of large physiologically-identified GABAergic cells in the rat frontal cortex. Neuroscience 1998; 85: 677–701.
Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S et al. Functional organization of the transcriptome in human brain. Nat Neurosci 2008; 11: 1271–1282.
Zikopoulos B, Barbas H . Altered neural connectivity in excitatory and inhibitory cortical circuits in autism. Front Hum Neurosci 2013; 7: 609.
Stoner R, Chow ML, Boyle MP, Sunkin SM, Mouton PR, Roy S et al. Patches of disorganization in the neocortex of children with autism. N Engl J Med 2014; 370: 1209–1219.
Berridge MJ . Dysregulation of neural calcium signaling in Alzheimer disease, bipolar disorder and schizophrenia. Prion 2013; 7: 2–13.
Lewis DA, Hashimoto T, Volk DW . Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci 2005; 6: 312–324.
Jamain S, Radyushkin K, Hammerschmidt K, Granon S, Boretius S, Varoqueaux F et al. Reduced social interaction and ultrasonic communication in a mouse model of monogenic heritable autism. Proc Natl Acad Sci USA 2008; 105: 1710–1715.
Ey E, Leblond CS, Bourgeron T . Behavioral profiles of mouse models for autism spectrum disorders. Aut Res 2011; 4: 5–16.
Silverman JL, Yang M, Lord C, Crawley JN . Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci 2010; 11: 490–502.
Yang M, Bozdagi O, Scattoni ML, Wohr M, Roullet FI, Katz AM et al. Reduced excitatory neurotransmission and mild autism-relevant phenotypes in adolescent Shank3 null mutant mice. J Neurosci 2012; 32: 6525–6541.
Penagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 2011; 147: 235–246.
Martins GJ, Shahrokh M, Powell EM . Genetic disruption of Met signaling impairs GABAergic striatal development and cognition. Neuroscience 2011; 176: 199–209.
Selby L, Zhang C, Sun QQ . Major defects in neocortical GABAergic inhibitory circuits in mice lacking the fragile X mental retardation protein. Neurosci Lett 2007; 412: 227–232.
Gogolla N, Leblanc JJ, Quast KB, Sudhof TC, Fagiolini M, Hensch TK . Common circuit defect of excitatory-inhibitory balance in mouse models of autism. J Neurodev Disord 2009; 1: 172–181.
Gant JC, Thibault O, Blalock EM, Yang J, Bachstetter A, Kotick J et al. Decreased number of interneurons and increased seizures in neuropilin 2 deficient mice: implications for autism and epilepsy. Epilepsia 2009; 50: 629–645.
Vreugdenhil M, Jefferys JG, Celio MR, Schwaller B . Parvalbumin-deficiency facilitates repetitive IPSCs and gamma oscillations in the hippocampus. J Neurophysiol 2003; 89: 1414–1422.
Schwaller B, Tetko IV, Tandon P, Silveira DC, Vreugdenhil M, Henzi T et al. Parvalbumin deficiency affects network properties resulting in increased susceptibility to epileptic seizures. Mol Cell Neurosci 2004; 25: 650–663.
Collin T, Chat M, Lucas MG, Moreno H, Racay P, Schwaller B et al. Developmental changes in parvalbumin regulate presynaptic Ca2+ signaling. J Neurosci 2005; 25: 96–107.
Eggermann E, Jonas P . How the 'slow' Ca(2+) buffer parvalbumin affects transmitter release in nanodomain-coupling regimes. Nat Neurosci 2012; 15: 20–22.
Orduz D, Bischop DP, Schwaller B, Schiffmann SN, Gall D . Parvalbumin tunes spike-timing and efferent short-term plasticity in striatal fast spiking interneurons. J Physiol 2013; 591: 3215–3232.
Manseau F, Marinelli S, Mendez P, Schwaller B, Prince DA, Huguenard JR et al. Desynchronization of neocortical networks by asynchronous release of GABA at autaptic and synaptic contacts from fast-spiking interneurons. PLoS Biol 2010; 8: e1000492.
Alberi L, Lintas A, Kretz R, Schwaller B, Villa AE . The calcium-binding protein parvalbumin modulates the firing 1 properties of the reticular thalamic nucleus bursting neurons. J Neurophysiol 2013; 109: 2827–2841.
Etherton M, Foldy C, Sharma M, Tabuchi K, Liu X, Shamloo M et al. Autism-linked neuroligin-3 R451C mutation differentially alters hippocampal and cortical synaptic function. Proc Natl Acad Sci USA 2011; 108: 13764–13769.
Amaral DG, Schumann CM, Nordahl CW . Neuroanatomy of autism. Trends Neurosci 2008; 31: 137–145.
Palmen SJ, van Engeland H, Hof PR, Schmitz C . Neuropathological findings in autism. Brain 2004; 127: 2572–2583.
Moreno H, Burghardt NS, Vela-Duarte D, Masciotti J, Hua F, Fenton AA et al. The absence of the calcium-buffering protein calbindin is associated with faster age-related decline in hippocampal metabolism. Hippocampus 2012; 22: 1107–1120.
Terranova ML, Laviola G . Scoring of social interactions and play in mice during adolescence. Curr Protoc Toxicol 2005; Chapter 13: Unit13 0.
Wohr M, Roullet FI, Hung AY, Sheng M, Crawley JN . Communication impairments in mice lacking Shank1: reduced levels of ultrasonic vocalizations and scent marking behavior. PloS One 2011; 6: e20631.
Wohr M, Silverman JL, Scattoni ML, Turner SM, Harris MJ, Saxena R et al. Developmental delays and reduced pup ultrasonic vocalizations but normal sociability in mice lacking the postsynaptic cell adhesion protein neuroligin2. Behav Brain Res 2013; 251: 50–64.
Moy SS, Nadler JJ, Young NB, Perez A, Holloway LP, Barbaro RP et al. Mouse behavioral tasks relevant to autism: phenotypes of 10 inbred strains. Behav Brain Res 2007; 176: 4–20.
Mohajeri MH, Madani R, Saini K, Lipp HP, Nitsch RM, Wolfer DP . The impact of genetic background on neurodegeneration and behavior in seizured mice. Genes Brain Behav 2004; 3: 228–239.
Olton DS, Samuelson RI . Remembrance of places passed: spatial memory in rats. J Exp Psychol Anim Behav Process 1976; 2: 97–116.
Lang UE, Wolfer DP, Grahammer F, Strutz-Seebohm N, Seebohm G, Lipp HP et al. Reduced locomotion in the serum and glucocorticoid inducible kinase 3 knock out mouse. Behav Brain Res 2006; 167: 75–86.
Tremml P, Lipp HP, Muller U, Ricceri L, Wolfer DP . Neurobehavioral development, adult openfield exploration and swimming navigation learning in mice with a modified beta-amyloid precursor protein gene. Behav Brain Res 1998; 95: 65–76.
Madani R, Kozlov S, Akhmedov A, Cinelli P, Kinter J, Lipp HP et al. Impaired explorative behavior and neophobia in genetically modified mice lacking or overexpressing the extracellular serine protease inhibitor neuroserpin. Mol Cell Neurosci 2003; 23: 473–494.
Moreno H, Hua F, Brown T, Small S . Longitudinal mapping of mouse cerebral blood volume with MRI. NMR Biomed 2006; 19: 535–543.
Meyer AH, Katona I, Blatow M, Rozov A, Monyer H . In vivo labeling of parvalbumin-positive interneurons and analysis of electrical coupling in identified neurons. J Neurosci 2002; 22: 7055–7064.
Sholl DA . Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat 1953; 87: 387–406.
Batschelet E . Circular Statistics in Biology. Academic Press: London, UK, 1981.
Wohr M, Schwarting RK . Affective communication in rodents: ultrasonic vocalizations as a tool for research on emotion and motivation. Cell Tissue Res 2013; 354: 81–97.
Scattoni ML, Crawley J, Ricceri L . Ultrasonic vocalizations: a tool for behavioural phenotyping of mouse models of neurodevelopmental disorders. Neurosci Biobehav Rev 2009; 33: 508–515.
del Rio JA, de Lecea L, Ferrer I, Soriano E . The development of parvalbumin-immunoreactivity in the neocortex of the mouse. Brain Res Dev Brain Res 1994; 81: 247–259.
Tuchman R, Cuccaro M, Alessandri M . Autism and epilepsy: historical perspective. Brain Dev 2010; 32: 709–718.
Farre-Castany MA, Schwaller B, Gregory P, Barski J, Mariethoz C, Eriksson JL et al. Differences in locomotor behavior revealed in mice deficient for the calcium-binding proteins parvalbumin, calbindin D-28k or both. Behav Brain Res 2007; 178: 250–261.
Wang SS, Kloth AD, Badura A . The cerebellum, sensitive periods, and autism. Neuron 2014; 83: 518–532.
Caillard O, Moreno H, Schwaller B, Llano I, Celio MR, Marty A . Role of the calcium-binding protein parvalbumin in short-term synaptic plasticity. Proc Natl Acad Sci USA 2000; 97: 13372–13377.
Pedroarena CM, Schwarz C . Efficacy and short-term plasticity at GABAergic synapses between Purkinje and cerebellar nuclei neurons. J Neurophysiol 2003; 89: 704–715.
Vitureira N, Letellier M, Goda Y . Homeostatic synaptic plasticity: from single synapses to neural circuits. Curr Opin Neurobiol 2012; 22: 516–521.
Turrigiano G . Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb Persp Biol 2012; 4: a005736.
Baudouin SJ, Gaudias J, Gerharz S, Hatstatt L, Zhou K, Punnakkal P et al. Shared synaptic pathophysiology in syndromic and nonsyndromic rodent models of autism. Science 2012; 338: 128–132.
Rubenstein JL, Merzenich MM . Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav 2003; 2: 255–267.
Zeidan-Chulia F, Rybarczyk-Filho JL, Salmina AB, de Oliveira BH, Noda M, Moreira JC . Exploring the multifactorial nature of autism through computational systems biology: calcium and the Rho GTPase RAC1 under the spotlight. Neuromol Med 2013; 15: 364–383.
Krey JF, Dolmetsch RE . Molecular mechanisms of autism: a possible role for Ca2+ signaling. Curr Opin Neurobiol 2007; 17: 112–119.
Marin O . Interneuron dysfunction in psychiatric disorders. Nat Rev Neurosci 2012; 13: 107–120.
Meyer U, Nyffeler M, Schwendener S, Knuesel I, Yee BK, Feldon J . Relative prenatal and postnatal maternal contributions to schizophrenia-related neurochemical dysfunction after in utero immune challenge. Neuropsychopharmacology 2008; 33: 441–456.
Fatemi SH, Aldinger KA, Ashwood P, Bauman ML, Blaha CD, Blatt GJ et al. Consensus paper: pathological role of the cerebellum in autism. Cerebellum 2012; 11: 777–807.
Gudrunardottir T, Sehested A, Juhler M, Schmiegelow K . Cerebellar mutism: review of the literature. Childs Nerv Syst 2011; 27: 355–363.
Kim YS, Harry GJ, Kang HS, Goulding D, Wine RN, Kissling GE et al. Altered cerebellar development in nuclear receptor TAK1/ TR4 null mice is associated with deficits in GLAST(+) glia, alterations in social behavior, motor learning, startle reactivity, and microglia. Cerebellum 2010; 9: 310–323.
Riva D, Giorgi C . The cerebellum contributes to higher functions during development: evidence from a series of children surgically treated for posterior fossa tumours. Brain 2000; 123: 1051–1061.
Tsai PT, Hull C, Chu Y, Greene-Colozzi E, Sadowski AR, Leech JM et al. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 2012; 488: 647–651.
Martin LA, Goldowitz D, Mittleman G . Repetitive behavior and increased activity in mice with Purkinje cell loss: a model for understanding the role of cerebellar pathology in autism. Eur J Neurosci 2010; 31: 544–555.
Ellegood J, Anagnostou E, Babineau BA, Crawley JN, Lin L, Genestine M et al. Clustering autism: using neuroanatomical differences in 26 mouse models to gain insight into the heterogeneity. Mol Psychiatry 2014; 20: 118–125.
Acknowledgements
This study was supported by grants from the Swiss National Science Foundation (SNF grant: 130680 to BS; SNF NCCR Neural Plasticity and Repair to DPW), the Deutsche Forschungsgemeinschaft (DFG WO 1732/1-1 to MW), the National Institutes of Health (NIH grant number: AG027476 to HM), Fondation Médicale Reine Elisabeth (FMRE-Belgium), Van Buuren Foundation, FRS–FNRS (Belgium) and IUAP from Belspo (P7/10) to SNS and DG and the FP7 Collaborative Project EUROSPIN (HEALTH-F2-2009-241498 to DPW). DPW is a member of the Neuroscience Center Zurich (ZNZ) and the Zurich Center for Integrative Human Physiology (ZIHP). We would also like to thank Scott A Small, MD and Elisa Konofagou, PhD (Columbia University) for allowing the use of the mouse MRI facility, and Luna Buitrago (SUNY Downstate) and Maria Eleni Karakatsani (Department of Biomedical Engineering, Columbia University) for help with MR image processing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Wöhr, M., Orduz, D., Gregory, P. et al. Lack of parvalbumin in mice leads to behavioral deficits relevant to all human autism core symptoms and related neural morphofunctional abnormalities. Transl Psychiatry 5, e525 (2015). https://doi.org/10.1038/tp.2015.19
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2015.19
