
Abstract
Cadherin-13 (CDH13), a unique glycosylphosphatidylinositol-anchored member of the cadherin family of cell adhesion molecules, has been identified as a risk gene for attention-deficit/hyperactivity disorder (ADHD) and various comorbid neurodevelopmental and psychiatric conditions, including depression, substance abuse, autism spectrum disorder and violent behavior, while the mechanism whereby CDH13 dysfunction influences pathogenesis of neuropsychiatric disorders remains elusive. Here we explored the potential role of CDH13 in the inhibitory modulation of brain activity by investigating synaptic function of GABAergic interneurons. Cellular and subcellular distribution of CDH13 was analyzed in the murine hippocampus and a mouse model with a targeted inactivation of Cdh13 was generated to evaluate how CDH13 modulates synaptic activity of hippocampal interneurons and behavioral domains related to psychopathologic (endo)phenotypes. We show that CDH13 expression in the cornu ammonis (CA) region of the hippocampus is confined to distinct classes of interneurons. Specifically, CDH13 is expressed by numerous parvalbumin and somatostatin-expressing interneurons located in the stratum oriens, where it localizes to both the soma and the presynaptic compartment. Cdh13−/− mice show an increase in basal inhibitory, but not excitatory, synaptic transmission in CA1 pyramidal neurons. Associated with these alterations in hippocampal function, Cdh13−/− mice display deficits in learning and memory. Taken together, our results indicate that CDH13 is a negative regulator of inhibitory synapses in the hippocampus, and provide insights into how CDH13 dysfunction may contribute to the excitatory/inhibitory imbalance observed in neurodevelopmental disorders, such as ADHD and autism.
Similar content being viewed by others

Introduction
Brain development and plasticity are complex processes that require precise and specific interneuronal contacts to ensure the correct establishment of circuits at the cellular and systems level. To guarantee cellular and regional specification of synaptic contacts, the formation of neuronal networks is tightly controlled by a myriad of cell guidance and adhesion molecules, among which the cadherin superfamily of calcium-dependent cell adhesion molecules is of particular relevance.1 Furthermore, after development, cadherins regulate the function and plasticity of mature synapses by participating in synaptic vesicle dynamics or controlling the stability of synaptic receptors.1
Cadherin-13 (CDH13, also known as T-cadherin or H-cadherin) is an atypical member of the cadherin superfamily. It lacks both transmembrane and cytoplasmic domains and is linked to the plasma membrane via a glycosylphosphatidylinositol moiety.2, 3, 4 Genome-wide genetic approaches including linkage analyses, association studies and copy number variation studies have detected associations between the CDH13 gene and various neurodevelopmental disorders. Notably, in several independent studies, CDH13 has been associated with the risk for attention-deficit/hyperactivity disorder (ADHD) and frequent comorbid conditions including drug and alcohol abuse.4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17 In addition, CDH13 was implicated in depression,7, 18 autism spectrum disorders,19 schizophrenia20 and bipolar disorder.21 Variations in CDH13 have also been associated with the personality trait of extraversion22 and extremely violent behavior.23 The mechanism whereby CDH13 dysfunction contributes to the pathogenesis of neuropsychiatric conditions remains, however, unknown.
CDH13 is expressed in the developing and adult brain, as well as in the cardiovascular system, where it exerts its function via low-adhesive homophilic or heterophilic interactions to control cell migration, neurite outgrowth and axon guidance.24, 25, 26, 27, 28, 29 We therefore predicted that CDH13, as several other cell adhesion molecules, may have dual roles in the brain; specifically, as a guidance cue for migrating neurons and/or developing axons, and as a modulator of the establishment of specific synaptic contacts and function of mature synapses.4 The view that CDH13 may moderate synapse formation and plasticity is supported by a previous study,30 showing that Cdh13 knockdown via RNA interference in hippocampal cultures resulted in a decrease in the density of both excitatory and inhibitory synapses.
Here, we focused on the hippocampus to test in vivo the hypothesis that involvement of CDH13 in the susceptibility to neurodevelopmental/psychiatric disorders is associated with its potential role in the modulation of synaptic function. We investigated CDH13 expression and function in specific subtypes of hippocampal neurons in vitro and in vivo. Furthermore, we describe the electrophysiological and behavioral consequences of CDH13 deficiency in a mouse model of constitutive Cdh13 inactivation. Finally, by linking alterations in hippocampal GABAergic synaptic transmission with cognitive deficits, we provide evidence that supports a critical role for CDH13 in the synaptic activity of the hippocampal GABAergic circuitry.
Materials and methods
Generation of a constitutive Cdh13 knockout mouse line
A constitutive Cdh13 knockout mouse (Cdh13−/−) was generated through site-specific DNA recombination by crossing Cdh13loxP/loxP mice (C57Bl/6N background) with a constitutive Cre deleter line31 (Supplementary Figure S1 and Supplementary Table S1). The absence of CDH13 protein in Cdh13−/− mice was verified by western blot and immunohistochemistry. Animal protocols have been approved by the boards of the University of Würzburg and the government of Lower Franconia, and performed according to the European Community guidelines for animal care and use. Adult mice (10–16 weeks) were used, if not indicated otherwise.
Histological methods
In situ hybridization
Single chromogenic in situ hybridization to detect Cdh13 messenger RNA (mRNA) was carried out on 16 μm coronal brain sections. Double-fluorescent in situ hybridization was performed to detect Cdh13 (digoxigenin-labeled) and glutamic acid decarboxylase 1 (Gad1, fluorescein-labeled) mRNAs.
Immunofluorescence and colocalization assessment
Mice (n=3) were transcardially perfused, 40 μm free-floating serial coronal sections generated and co-stained for CDH13 plus one of the following interneuron markers: somatostatin (SOM), parvalbumin (PV), calbindin, neuropeptide Y, vasoactive intestinal peptide, neural nitric oxide synthase (nNOS) and calretinin. Some sections were triple-stained for CDH13, PV and SOM. Double immunofluorescence stainings were examined under an inverted epifluorescence microscope (Olympus, Hamburg, Germany). The numbers of single- and double-stained cells in single and merged images were counted using the ImageJ ‘Cell Counter’ plugin.
Immunohistochemistry
For single CDH13 staining, 20 μm sections of perfusion-fixed brains on slides were incubated with primary antibody. N-Histofine Simple Stain Mouse MAX PO (G) (Nichirei Biosciences, Tokyo, Japan) and 3,3′-diaminobenzidine (Roche Diagnostics Deutschland, Mannheim, Germany) were used for detection.
For single staining of interneuronal markers, immersion-fixed brains (n=7–9 per genotype) were serially sectioned into 50 μm free-floating coronal sections. Sections were treated with heat-induced epitope retrieval, incubated with primary antibodies against SOM, PV and nNOS and detected using biotinylated secondary antibodies (Vector Laboratories, Burlingame, CA, USA), AB complex (Vector Laboratories) and 3,3′-diaminobenzidine (Roche Diagnostics Deutschland) development. Stained sections were then analyzed using Stereo Investigator v.11 (MBF Bioscience, Williston, VT, USA). PV-, SOM- and nNOS-positive cells in the stratum oriens (SO) were counted with the optical fractionator method.
Immunocytochemistry
Primary hippocampal neuron cultures were prepared from embryonic day 18 (E18) old rats as previously described32 and maintained in neurobasal media supplemented with B27, penicillin, streptomycin and l-glutamine (GIBCO-Invitrogen, Darmstadt, Germany). Fixed (paraformaldehyde) and permeabilized (Triton X-100) cultures were co-stained for CDH13 together with one of the following synaptic markers: vesicular glutamate transporter, vesicular GABA transporter, postsynaptic density protein 95 (PSD95), gephyrin and GABA-A receptor α1 subunit (GABA-Aα1). Double immunofluorescence stainings were examined under a Fluoview FV1000 confocal microscope (Olympus).
RNA isolation and semiquantitative RT-PCR
Complementary deoxyribonucleic acid (cDNA) was synthesized using 1–2 μg total RNA from mouse brains (whole brain and hippocampus) of different ages. A 1:15 dilution of cDNA pool was used for semiquantitative RT-PCR analysis of Cdh13 expression. The semiquantitative RT-PCR data were analyzed using comparative quantification. Peptidyl prolyl isomerase (Ppia) and β-actin (Actb) served as reference genes for normalization of Cdh13 Q-values.
Electrophysiology
Electrophysiological experiments were conducted on horizontal slices (350-μm thick) from the ventral hippocampus of postnatal day (P) 20–22 mice of both genders. Miniature inhibitory postsynaptic currents (mIPSCs) were recorded in the presence of Tetrodotoxin (1 μm), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 5 μm) and D-(-)-2-amino-5-phosphonopentanoic acid (D-APV, 100 μm). Miniature excitatory postsynaptic currents were recorded in the presence of Tetrodotoxin and Picrotoxin (100 μm). Paired pulse ratio was recorded in the presence of 6-cyano-7-nitroquinoxaline-2,3-dione, D-(-)-2-amino-5-phosphonopentanoic acid, 4 mm CaCl2 and 4 mm MgCl2. Traces were recorded using a Multiclamp 700B amplifier (Molecular Devices, Wokingham, UK). Miniature recordings were analyzed using Mini Analysis Program (Synaptosoft, Decatur, GA, USA). Other traces were analyzed using Clampfit 10.2. Intrinsic cell parameters were recorded in current clamp using Signal (CED, Cambridge, UK) and traces were analyzed using Stimfit v0.13 following the parameter definitions used in Tricoire et al.33
Morphological reconstructions
Brain slices with biocytin-filled neurons were processed following a modified staining protocol based on Marx et al.34 Stained sections were imaged and somata, apical and basal dendrites of selected pyramidal neurons were reconstructed in Neurolucida v10 (MBF Bioscience). Reconstructions were analyzed in NeuroExplorer (MBF Bioscience) for intrinsic parameters and Sholl analysis.
Behavioral assessment
Four cohorts of adult male Cdh13+/+ (n=7–10), Cdh13+/− (n=8–10) and Cdh13−/− (n=7–10) mice were subjected to behavioral tests to assess anxiety-like behavior and conditioned fear responses, depression-like behavior, social interaction, locomotor activity, attention, impulsivity and cognitive flexibility. Stress hormone levels were measured from fecal samples and central blood.
Statistical analysis
Genotype effects were analyzed by univariate analysis of variance (two-sided) and, in cases of significance (P<0.05), post hoc comparisons using Bonferroni correction for multiple testing. In cases of non-normality and/or unequal variances, non-parametric statistical analysis using the Kruskal–Wallis test was performed instead of parametric analysis of variance. For those behavioral tests with repeated measures, a mixed analysis of variance was used, with genotype as between-subject factor and time or testing phase as repeated measure. Data analysis was performed using Prism 6.04 (GraphPad Software, La Jolla, CA, USA) or SPSS Statistics 21 (IBM, Armonk, NY, USA). A preliminary analysis revealed that the statistical power of our experimental setups to detect genotype-dependent differences with a medium effect size was of at least 0.8.
Detailed descriptions of all the methods are provided in the Supplementary Information.
Results
CDH13 is located in specific subtypes of hippocampal interneurons
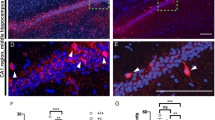
We detected Cdh13 mRNA in subregions and distinct cells of the hippocampus (Figure 1). In particular, the highest cellular expression was detected in the SO of the cornu ammonis (CA) region, whereas weaker labeling was observed in scattered cells of the CA3 pyramidal layer, as well as the hilus and the granule cell layer of the dentate gyrus. These in situ hybridization findings were complemented with an immunohistochemical analysis of CDH13 protein expression using an anti-CDH13 antibody which we validated for specificity using Cdh13 null mutant mice (Supplementary Figure S1). Of note, intense cellular immunoreactivity was observed in cell bodies located in the SO, in a pattern which closely resembled Cdh13 mRNA expression. In addition, CDH13 protein was also distributed in the hippocampus in a characteristic laminar pattern (Figure 1a), with the strongest staining in the stratum lucidum of CA3 and stratum lacunosum-moleculare, in the molecular layer and hilus of the dentate gyrus, as well as along the mossy fiber projections.

CDH13 expression in murine hippocampus. (a) CDH13 protein and mRNA expression in the hippocampus of the adult mouse, CDH13 protein and mRNA are present in cell somata of the stratum oriens (SO; a,a’). (ISH picture was shown as preliminary data in a review article by Rivero et al.;4 reproduced with permission from Elsevier). (b) Double FISH: Cdh13 is localized in Gad1 mRNA-positive cells of the SO (white arrows: colocalization; green arrows: only Gad1+). (c) IF images: colocalization study of CDH13 and parvalbumin/somatostatin/neural nitric oxide synthase (white arrows: colocalization, green arrowhead: single stained cell for PV/SOM/nNOS, red arrowhead: single stained cell for CDH13). (d) Colocalization of CDH13 with neurochemical markers of GABAergic interneurons in the SO of the dorsal (upper panel) and ventral (lower panel) hippocampus. A higher degree of colocalization is observed in the dorsal part of the hippocampus. n=3; 100%=CDH13+ cells; percentages calculated from median (see Supplementary Figure S2 for details). (e) Triple IF: CDH13 is colocalized with PV and SOM in some but not in all cells of the SO (white arrow: triple localization, empty arrowhead: double localization). Scale bars, 300 μm in a, 50 μm in a/a’, b, c and e. CB, calbindin; CR, calretinin; DG, dentate gyrus; FISH, fluorescent in situ hybridization; mRNA, messenger RNA; nNOS, neural nitric oxide synthase; NPY, neuropeptide Y; PV, parvalbumin; SOM, somatostatin; VIP, vasoactive intestinal peptide.
The unique and distinct cellular pattern of CDH13 labeling in the hippocampal SO prompted us to ascertain the nature of these CDH13-positive cells. Their sparse distribution suggested a GABAergic nature, as it is known that the bodies of a considerable number of GABAergic inhibitory interneurons are located in this deep layer of the hippocampus.35 As expected, a double-fluorescent in situ hybridization approach using probes for Cdh13 and the interneuron marker Gad1 showed that most of the Cdh13-positive somata of the SO were indeed interneurons (Figure 1b).
Hippocampal interneurons are a heterogeneous group of cells that differ in their morphological and electrophysiological properties.33 To better identify the nature of the CDH13-immunoreactive interneurons detected in the SO, we performed double-immunofluorescence experiments to label CDH13 with each of seven neurochemical markers (PV, calbindin, calretinin, SOM, neuropeptide Y, vasoactive intestinal peptide and nNOS) characteristically expressed in different subpopulations of hippocampal interneurons (Figures 1c and d). Initial qualitative assessment revealed that the extent of coexpression with CDH13 substantially varied depending on the analyzed interneuron marker (Figure 1c). Next, a stereological approach to precisely quantify the degree of coexpression (Figure 1d and Supplementary Figure S2a) was used. The highest levels of coexpression were found with SOM (63% of CDH13-immunoreactive cells were also positive for SOM in the dorsal hippocampus, 49% in the ventral hippocampus) and PV (with 50% of CDH13-immunoreactive cells being positive for PV in the dorsal hippocampus, 35% in the ventral part). In contrast, very low levels of coexpression were found with nNOS (2–6%) and calretinin (4%). In addition, the percentages of interneurons of each subpopulation that were positive for CDH13 were calculated (Supplementary Figure S2a). In this case, CDH13 was most frequently found in PV- (66% dorsal, 31% ventral), SOM- (55% dorsal, 39% ventral) and neuropeptide Y- (47% dorsal, 35% ventral) immunoreactive cells of the SO.
The above data indicate that, in the SO, CDH13 is most frequently found in PV- and SOM-positive cells. PV and SOM are thought to be representative of two interneuronal subpopulations that are distinct or have very little overlap. There is no coexpression of PV and SOM in the cerebral cortex,36 whereas there is a small degree of overlap in the hippocampus.33 Notably, we show via triple PV-SOM-CDH13 staining that some, but not all, of the CDH13-immunoreactive cells of the SO were positive for both SOM and PV (Figure 1e).
CDH13 is located at inhibitory synapses
To gain further insight into the function of CDH13 in the brain, we assessed its temporal expression pattern in whole brain as well as in hippocampus (Figure 2a). Cdh13 mRNA was detected from E13.5 onwards and increased to reach a peak at P7 to P21 for whole brain, as well as P14 for hippocampus, times of period that coincide with synapse formation.30, 37 Although previous studies showed that ectopically expressed CDH13 localized to synapses in hippocampal primary cultures,30 we explored endogenous labeling at the synapse in more detail. Hippocampal neurons at 3 days in vitro were transfected with a myc-tagged Cdh13.30 Co-staining for CDH13 and MYC at 14 days in vitro showed an exactly overlapping punctated pattern and confirmed antibody specificity for subsequent co-immunofluorescence analysis (Supplementary Figure S3). To determine the specific synaptic localization of endogenous CDH13, we co-stained hippocampal neurons at 14 days in vitro for CDH13 and the presynaptic markers vesicular glutamate transporter (VGLUT) or vesicular GABA transporter (VGAT) (excitatory and inhibitory, respectively) or the postsynaptic markers PSD95 or gephyrin (excitatory and inhibitory, respectively; Figure 2b). CDH13 colocalized exactly with the presynaptic marker VGAT, but not VGLUT. At the postsynaptic side, CDH13 showed juxtaposed labeling with gephyrin, but not with PSD95. As CDH13 is coexpressed in a high proportion of PV-positive cells (Supplementary Figure S2a), we investigated whether CDH13 colocalized with the GABA-A subunit primarily found in synapses formed by PV-positive cells, GABA-Aα1 (Figure 2c). Indeed, CDH13 puncta were found juxtaposed to GABA-Aα1 puncta, consistent with a presynaptic (CDH13) and postsynaptic (GABA-Aα1) localization. Conversely, not all GABA-Aα1-positive puncta were positive for CDH13, indicating that these puncta originate from another subpopulation of interneurons that is CDH13-negative. Together with our finding that CDH13 is expressed in inhibitory neurons in the SO, these data provide evidence that, in hippocampal neurons, CDH13 localizes at the presynaptic compartment of inhibitory synapses.

Temporal expression pattern and subcellular localization of CDH13. (a) Developmental timeline of Cdh13 mRNA expression for whole brain (black bars) and hippocampus (gray bars; mean±s.e.m.; n=2 for hippocampus E18.5, P7, P21 and adult, for all other time points n=3). (b) Immunofluorescence staining of cultured hippocampal neurons for CDH13 (green) and the synaptic markers VGLUT, VGAT, PSD95 or Gephyrin (red). CDH13-positive puncta are indicated by the triangles, colored yellow for colocalized foci (VGAT) and blue for juxtaposed foci (Gephyrin). CDH13 localizes at the presynaptic side of inhibitory synapses. Scale bar, 1 μm. (c) Immunofluorescence staining of cultured hippocampal neurons for CDH13 (green) and GABA-A receptor α1 subunit (GABA-Aα1, red). CDH13-positive puncta are indicated by the triangles, colored blue for juxtaposition with GABA-Aα1-positive puncta. White arrows indicate puncta that are CDH13-negative and GABA-Aα1-positive. Scale bar, 1 μm. mRNA, messenger RNA; VGAT, vesicular GABA transporter; VGLUT, vesicular glutamate transporter.
Cdh13 knockout mice display increased inhibitory synaptic transmission
To investigate the functional consequences of CDH13 deficiency for the hippocampal inhibitory network, we generated a constitutive Cdh13 knockout mouse line (Supplementary Figure S1). In agreement with a previously reported mouse model of constitutive CDH13 deficiency,38 Cdh13 null mutant (Cdh13−/−) mice exhibited normal life expectancy and breeding capacity; they did not present any gross morphological abnormalities of the brain (Supplementary Figure S4). Weight gain, daily food and water intake, as well as stress hormone levels were not affected by genotype (Supplementary Figure S5).
As CDH13 is localized at the presynaptic compartment of inhibitory synapses, we assessed basal inhibitory synaptic transmission by recording mIPSCs from CA1 pyramidal cells in acute slices from P20-22 Cdh13+/+, Cdh13+/− and Cdh13−/− mice (Figures 3a to c). We found an increased mIPSC frequency in the Cdh13−/− mice compared with Cdh13+/+ (Cdh13+/+: 3.29±0.47 Hz, Cdh13−/−: 4.93±0.29 Hz, P<0.001) and Cdh13+/− (3.73±0.29 Hz, P<0.05), but no change in mIPSC amplitude (Cdh13+/+: 18.25±0.42 pA, Cdh13−/−: 19.31±0.36 pA, P=0.07). Consistent with exclusive localization of CDH13 at inhibitory synapses, miniature excitatory postsynaptic currents were unchanged in both amplitude (Cdh13+/+: 16.43±0.67 pA, Cdh13+/−: 15.85±0.24 pA, Cdh13−/−: 16.00±0.32 pA, P>0.05) and frequency (Cdh13+/+: 0.34±0.03 Hz, Cdh13+/−: 0.40±0.03 Hz, Cdh13−/−: 0.40±0.04 Hz, P>0.05; Figures 3d to f). As our approach was constitutive rather than interneuron-specific Cdh13 inactivation, we aimed to exclude the possibility that knocking out Cdh13 in CA1 pyramidal cells has any effects on synaptic transmission. To do this, we prepared organotypic hippocampal slices from wild-type rats and injected a lentivirus carrying a shRNA against Cdh13 previously validated by Paradis et al.30 into the pyramidal cell layer of CA1 and then recorded simultaneously from an infected and a non-infected pyramidal neuron and compared the response to different stimulation locations (Supplementary Figures S6a, e and i). We found no difference in inhibitory input from SO (Supplementary Figures S6b to d) or stratum radiatum (Supplementary Figures S6f and g). Also, excitatory input from stimulation of the Schaffer collaterals was unaltered for both AMPA (Supplementary Figures S6j and k) and NMDA (Supplementary Figures S6l and m) receptor-mediated input. We therefore conclude that it is unlikely that the constitutive nature of our knockout model affects excitatory pyramidal cells directly, and the observed increase in mIPSC frequency is more likely caused by the inactivation of Cdh13 in presynaptic inhibitory neurons.

Hippocampal CA1 neurons from Cdh13 null mutant mice show increased inhibitory synaptic input and morphological changes. (a) Representative traces of miniature inhibitory postsynaptic currents (mIPSCs) recorded from CA1 pyramidal cells from acute brain slices from Cdh13+/+, Cdh13+/− and Cdh13−/− mice. (b and c) Cumulative distributions of mIPSC amplitudes (b) and inter-event intervals (IEI; c) from Cdh13+/+ (n=19/3), Cdh13+/− (n=18/3) and Cdh13−/− (n=23/4) hippocampal CA1 neurons. (d) Representative traces of miniature excitatory postsynaptic currents (mEPSCs) recorded from CA1 pyramidal cells from acute brain slices of Cdh13+/+, Cdh13+/− and Cdh13−/− mice. (e and f) Cumulative distributions of mIPSC amplitudes (e) and IEI (f) from Cdh13+/+ (n=12/3), Cdh13+/− (n=13/3) and Cdh13−/− (n=14/3) hippocampal CA1 neurons. (g) Representative overlaid traces of two IPSCs evoked by stimuli delivered at an inter-stimulus interval (ISI) of 50 ms. (h) Quantification of paired pulse ratio following stimulation in SO for Cdh13+/+ (n=15/4), Cdh13+/− (n=15/3) and Cdh13−/− (n=15/3). (i) Representative reconstructions of hippocampal CA1 neurons from Cdh13+/+ and Cdh13−/− mice. (j and k) Quantification of (j) basal dendritic and (k) apical dendritic nodes and endings for Cdh13+/+ (n=6) and Cdh13−/− (n=7). Bar graphs are presented as mean±s.e.m. *P<0.05; **P<0.01; ***P<0.001. n is shown as number of cells/number of animals.
Generally, changes in frequency are explained as either a change in presynaptic release probability, or as a change in synapse number. To enable distinction between both possibilities, we recorded the paired pulse ratio, a measure of the release probability, of inhibitory synapses following stimulation in SO at different inter-stimulus intervals. We found an increased paired pulse ratio in Cdh13−/− compared to Cdh13+/+ mice at the 50 ms inter-stimulus interval (ISI) only (ISI 50 ms: Cdh13+/+: 0.77±0.03, Cdh13−/−: 0.93±0.05, P<0.05, Figures 3g and h). An increased paired pulse ratio is associated with a decreased release probability. Therefore, the underlying cause for increased mIPSC frequency in Cdh13−/− mice is unlikely to be a change in release probability, but rather a change in the number of inhibitory synapses formed onto CA1 pyramidal cells.
To investigate whether the changes in inhibitory synapses could be due to altered development of interneuron subtypes we analyzed the density of PV-, SOM- and nNOS-positive interneurons of the SO in adult Cdh13−/−, Cdh13+/− and Cdh13+/+ mice. We found no effect of CDH13 deficiency on the densities of nNOS-positive (which rarely colocalize with CDH13), PV- or SOM-positive interneurons (P>0.05, Supplementary Figure S2b). These results exclude that the enhanced inhibitory synaptic input on CA1 neurons in Cdh13−/− mice is owing to an increased amount of interneurons in the SO. Furthermore, no differences in the hippocampal levels of transmitters (glutamate, GABA, dopamine, serotonin and norepinephrine) or their metabolites were found between genotypes (Supplementary Figure S7).
Next, we assessed the general dendritic morphology of CA1 pyramidal cells. To this end, CA1 pyramidal cells were loaded with biocytin for morphological reconstruction of apical and basal dendrites (Figure 3i). The basal dendrites were not different between Cdh13+/+ and Cdh13−/− animals (Figure 3j). The apical dendrites, on the other hand, were found to be significantly more complex in Cdh13−/− animals at the level of dendritic nodes and length (nodes: Cdh13+/+: 31.5±1.6, Cdh13−/−: 40.6±1.4, P<0.01; endings Cdh13+/+: 32.3±1.5, Cdh13−/−: 41.9±1.7, P<0.01; length Cdh13+/+: 3041.5±436.1 μm, Cdh13−/−: 4521.4±478.9 μm, P<0.05; Figure 3k, Supplementary Table S2). However, analysis of the space occupied by the dendrites (Convex Hull analysis, Supplementary Table S2) showed no difference between genotypes, indicating that the apical dendrite stays within the same envelope, but is more branched in Cdh13−/− animals. Intrinsic electrophysiological properties, such as membrane potential, input resistance and action potential properties, were identical between Cdh13+/+ and Cdh13−/− animals (Supplementary Table S3). Together, these data indicate that CDH13 is a negative regulator of inhibitory synapse function. Thus, loss of function of Cdh13 could lead to a distorted excitation/inhibition balance within the hippocampus.
Behavioral alterations of Cdh13 knockout mice correlate with hippocampal dysfunction and ADHD phenotypes
We assessed the effects of CDH13 deficiency on different behavioral domains. The analysis revealed no significant genotype effect on any variable measured to assess anxiety- and depression-like behavior as well as social interaction (Supplementary Figures S8–S10). We also tested locomotor activity in an open field for 30 min. In a cohort of mice with prior test experience, we detected a significant genotype effect for distance traveled (P=0.023, Figure 4a) and number of rears (P=0.039, Figure 4b), indicating that Cdh13−/− mice displayed higher locomotor activity than Cdh13+/+ mice. These findings were replicated in a cohort of naive mice (distance: P=0.069; rears: P=0.047). Center time, a measure of anxiety-like behavior, was not affected by genotype in either test-experienced or naive cohorts (Figure 4c).

Cdh13−/− mice display novelty-induced hyperlocomotion but unaltered anxiety-like behavior in the open field. (a) Total distance traveled and (b) number of rears as measures of locomotor activity. (c) Time spent in the center of the open field as measure of anxiety-like behavior. Experienced: n=7–8 per genotype, naive: n=10 per genotype. Data are presented as mean±s.e.m. #P<0.1, *P<0.05.
In the five-choice serial reaction time task (5-CSRTT), which evaluates attention and impulsivity, acquisition and performance of the task was similar in all genotypes, as indicated by gradually increasing accuracy, number of omission errors and pellets eaten, indicating unaltered attention in Cdh13−/− mice, as well as decreasing number of premature and timeout responses and faster response latencies, findings that support no genotype-dependent differences in impulsivity (Figure 5 and Supplementary Figure S11; session effects: all P<0.001). No significant genotype effects on any variable measured in the 5-CSRTT were detected. However, there was a session × genotype interaction for the number of timeout responses (Figures 5d, P=0.005), showing that during the first training session with longer stimulus duration of 20 s, Cdh13−/− mice made significantly more nose-pokes after an incorrect response, but only when compared with Cdh13+/− mice.

Cdh13−/− mice exhibit normal attentional performance in the 5-CSRTT but impaired spatial learning in the Barnes maze and deficits in cued fear memory following delay fear conditioning. (a) % number of correct responses, (b) % number of omission errors, (c) number of premature responses and (d) number of timeout responses at different stimulus durations in the 5-CSRTT (n=10 per genotype). (e) Total distance traveled, (f) escape latency and (g) number of primary errors during acquisition and reversal phase of the Barnes maze (n=7–10 per genotype). (h) Freezing (%) duration during presentation of the auditory cue and the original conditioning context following delay fear conditioning (n=9–10 per genotype). Data are presented as mean±s.e.m. #P<0.1, *P<0.05, **P<0.01, ***P<0.001. 5-CSRTT, five-choice serial reaction time task.
The assessment of hippocampus-dependent visuospatial memory in the Barnes maze showed that all mice learned to find the hidden escape box in both acquisition and reversal, as indicated by gradually decreasing distances, escape latencies and primary errors throughout the successive trials (significant session effects: all P<0.001; data not shown). There were no session × genotype interactions, thus data were pooled across sessions to assess genotype effects. Distance traveled, escape latency and primary errors (Figures 5e to g) did not differ between genotypes during acquisition. However, during reversal, where the escape box is located in a different position, Cdh13−/− mice made significantly more primary errors than Cdh13+/+ mice (P=0.027), indicating impaired cognitive flexibility.
During fear conditioning, all genotypes displayed similar responses to footshock as well as significantly increased post-shock freezing (time effect: P<0.001 compared with pre-shock), reflecting unaltered pain sensitivity and normal acquisition of conditioned fear in Cdh13−/− mice (Supplementary Figures S12a and b). Twenty-four hours cued fear memory was significantly impaired in Cdh13−/− mice compared with Cdh13+/+ mice, as indicated by decreased freezing (genotype main effect: P=0.047, Figure 5h) and increased activity (P=0.081, Supplementary Figure S12c) during tone presentation. Similarly, in the 48-h contextual fear memory test, Cdh13−/− mice tended to be more active than Cdh13+/+ mice (P=0.094), although freezing duration did not significantly differ between genotypes when exposed to the original conditioning chamber.
In summary, CDH13 deficiency impacts locomotor activity as well as learning and memory, two behavioral domains frequently associated with ADHD and related neurodevelopmental disorders.
Discussion
Variation in CDH13 has been associated with a wide range of neurodevelopmental and psychiatric disorders, ranging from ADHD to autism spectrum disorders, among others.4 Although functional single-nucleotide polymorphisms/variants or haplotypes in CDH13 have yet to be linked to neurodevelopmental/psychiatric disorders, several copy number variant studies revealed deletions in ADHD and autism spectrum disorder patients that may disrupt CDH13 expression and/or function.19, 39, 40 Our findings revealed that inhibitory interneurons are the primary locus of CDH13 in the murine hippocampus, and that CDH13 deficiency affects hippocampal inhibitory function and several behavioral domains that are fundamentally altered across various neuropsychiatric phenotypes, thus contributing to shed light on the role of CDH13 in the etiopathogenesis of neurodevelopmental and psychiatric disorders.
We confirmed that in the hippocampal CA region, CDH13 is predominantly expressed in Gad1 mRNA-positive cells, with preference for PV- and SOM-positive interneurons, thus allocating CDH13-related functions to several GABAergic interneuron subpopulations: axo-axonic cells, basket cells, bistratified cells, and also oriens lacunosum-moleculare or hippocampal-septal projection cells.33, 41 Alterations in PV interneurons have previously been linked to various neuropsychiatric and neurodevelopmental disorders as well as related animal models.42, 43 Furthermore, SOM-positive interneurons have been implicated in the acquisition of contextual fear memories and the regulation of hippocampal synaptic plasticity.44
Along with several other members of the cadherin superfamily that have been demonstrated to regulate interneuron development and hippocampal synaptic function,45, 46, 47, 48 CDH13 expression in the hippocampal CA region suggests a function in interneurons, particularly at the inhibitory synapse. Furthermore, we confirmed that Cdh13 mRNA expression reaches its peak in the developing brain of the mouse between P7 and P21, consistent with recent observations in lysates from rat cortex,29 thus supporting the view that CDH13 expression increments coincide with peaking synaptogenesis in the neocortex.30, 37 Our investigations also show that CA1 neurons from Cdh13−/− mice receive increased inhibitory input, whereas no changes were found in excitatory input, which is likely to result in an excitatory–inhibitory imbalance. This notion supports the view of CDH13 being a critical regulator of inhibitory function in the hippocampus consistent with its localization in the GABAergic presynaptic compartment. The fine-tuning of hippocampal circuitry depends greatly on the precise activity of the inhibitory system to control hyperexcitability in the hippocampus, which displays the lowest seizure threshold of the entire brain.49 However, alterations associated with an altered inhibitory function are pathophysiologically beyond epilepsy. Multiple lines of evidence support the role of the inhibitory GABAergic system in the pathogenesis of psychiatric disorders.42, 50 Accordingly, disturbances of the excitatory–inhibitory balance lead to deficits in information processing and signal gating, two key processes involved in attentional and executive control.42, 51, 52, 53, 54 Candidate risk genes studies further support the view that alterations in the inhibitory system likely account for a spectrum of neuropsychiatric manifestations, including schizophrenia, autism spectrum disorders and ADHD.42, 55, 56, 57, 58 As an example, ErbB4 receptor tyrosine kinase, which is almost exclusively expressed in interneurons, has strongly been implicated in schizophrenia.56, 59 In the hippocampus, ErbB4 is located at the postsynaptic compartment of glutamatergic synapses of PV-positive interneurons60 and has a critical role in the regulation of long-term potentiation at the SC-CA1 synapse via these interneurons.61, 62 In the thalamic reticular nucleus, ErbB4 is mainly expressed in SOM-positive interneurons and is important for sensory selection.51 Of note, we also observed strong expression of Cdh13 in thalamic reticular nucleus neurons.4 In autism, the different isoforms of the cell adhesion proteins neurexins and neuroligins have both been shown to have critical roles in inhibitory synapse function. Of particular relevance is the observation that neuroligins 2 (ref. 63) and 3 (ref. 64) have distinct functions depending on the interneurons with which they form a synapse. Analogous to the neuroligins, and as we show that CDH13 is expressed in both PV- and SOM-immunoreactive cells, CDH13 might have distinct functions depending on the interneurons in which it is expressed.
Although the concentrations of GABA in hippocampus appear to be unchanged in Cdh13−/− mice, CDH13 deficiency may have effects on the modulation of inhibitory synaptic activity and synapse turnover. Our electrophysiological findings support the view that CDH13 may act as a synapse destabilizer. Therefore, in the absence of CDH13, stability of GABAergic synapses might be augmented owing to a reduced synapse turnover, producing an increase in inhibitory synapse density, consistent with the observed increase in inhibitory synaptic transmission. The role of cadherins in synapse (de)stabilization and plasticity has been well documented.1, 65, 66 The mechanism whereby CDH13 would participate in synapse (de)stabilization might involve heterophilic interactions in trans with elements of the postsynaptic compartment and/or also interactions in cis with presynaptic transmembrane molecules in close proximity. Among the potential candidates to interact with CDH13 in trans is the GABA-Aα1 subunit, expressed in PV-positive hippocampal interneurons and whose association with CDH13 has already been described in vascular endothelial cells.67 Transmembrane molecules of the presynaptic (or postsynaptic) compartments that could potentially interact with CDH13 include integrin-linked kinase,68 integrins67 or receptor tyrosine kinases,4 among others. Such interactions may induce downstream changes in actin cytoskeleton organization,69 which destabilize the GABAergic synapse.
Cdh13 knockdown in dissociated primary hippocampal neurons was previously reported to lead to a reduced density of both excitatory and inhibitory synapses, as well as a decrease in miniature excitatory postsynaptic currents amplitude and frequency.30 In the present study, we show in organotypic slices that knockdown using the same shRNA exclusively in excitatory CA1 pyramidal cells did not affect inhibitory or excitatory input onto these cells, consistent with the expression pattern of CDH13 restricted to hippocampal CA inhibitory but not excitatory neurons. In addition, we show that in Cdh13−/− mice, miniature excitatory postsynaptic currents amplitude and frequency are unaffected. Together, our data confirm that excitatory synapses are not directly (via shRNA-mediated knockdown of Cdh13) or indirectly (secondary to changes in interneuronal activity and/or connectivity) affected. The difference between the two studies may thus be caused by the type of experimental model (postnatal brain sections and organotypic slices vs dissociated cells) rendering it difficult to accurately extrapolate conclusions from both studies.
The hippocampus is central to cognition, spatial navigation, as well as memory formation and specifically the regulation of emotional memories.35 Furthermore, altered verbal and visuospatial working memory is one of the neurocognitive alterations widely described in ADHD.70, 71, 72 Other mouse models for neurodevelopmental disorders, such as Down syndrome or neurofibromatosis type 1, also present increased inhibition, which correlates with spatial memory deficits.42, 73, 74, 75 Remarkably, the excitation/inhibition imbalance triggered by the loss of CDH13 function in the hippocampus was consistently reflected in the behavior of CDH13-deficient mice. We showed that Cdh13 null mutants display alterations in cognition and learning/memory (particularly, spatial memory deficits during the reversal phase of the Barnes maze when the position of the escape box is changed), suggesting either reduced cognitive flexibility in Cdh13−/− mice (that is, inability to change their search strategies during the reversal phase) or a learning deficit when the difficulty of the task increases. In addition, Cdh13−/− mice display other minor alterations in conditioned fear responses as well as in the initial phase of the 5-CSRTT, which support the notion of learning deficits as a consequence of CDH13 deficiency. For example, the increased number of timeout responses during the training session may also support the notion of cognitive inflexibility in these mice.76 By contrast, during the 5-CSRTT, no modifications in attention or impulsivity were detected in Cdh13−/− mice. In the context of neurodevelopmental/psychiatric disorders, this suggests that the contribution of CDH13 variation to these disorders would be restricted to certain behavioral dimensions, that is, learning ability and cognitive flexibility, whereas other aspects (for example, attentional processes) would be less affected by CDH13 variation, at least under baseline conditions.
The alteration in cognitive functions observed in Cdh13-deficient mice are in general mild but not unexpected, given the observation that single genetic risk variants for neuropsychiatric disorders tend to display very modest effect sizes.77 Moreover, considering that deficits and impairments associated with neurodevelopmental disorders, such as ADHD, are more poorly compensated during childhood, the effects of CDH13 deficiency on mouse behavior might even be more pronounced in adolescent mice compared with adult animals investigated in our study. Finally, it is also possible that CDH13 deficiency may induce more extensive inhibitory dysfunction under conditions of environmental adversity, similar to its role as a cardioprotector under acute or chronic cardiac stress or its function in angiogenesis and tumor growth in a mouse model of mammary tumor.38, 78 Further studies will be necessary to disentangle the contribution of CDH13 under different environmental conditions in more detail. Finally, we provided evidence that Cdh13−/− mice display increased locomotor activity, one of the core phenotypes in ADHD,79 which is even more pronounced in mice following adaptation to the environment, suggesting that CDH13 deficiency may impair the hippocampal-dependent learning process that underlies the habituation to consecutive behavioral tests.80
We are aware that our study presents some limitations. Although the current work focused on CDH13 in the hippocampus, we and others have previously reported that CDH13 is also expressed in other regions of the mouse brain.4, 29 Therefore, the use of conditional Cdh13-deficient mice will clarify whether inactivation of CDH13 in other brain regions and neuronal cells impacts at the synaptic and behavioral levels. A cell-specific, such as PV- or SOM-specific, Cdh13 knockout will facilitate dissection of the specific contribution of CDH13 to each interneuron type where it is expressed. Furthermore, following this line of reasoning, Cdh13 is also expressed in serotonin (5-HT)-specific neurons of the raphe complex.4 Genetic variants of CDH13 have been associated with criminal behavior with extreme violence,23 a domain that is profoundly influenced by the brain 5-HT system.81 However, we did not find any indication for alterations in phenotypes related to emotion regulation in Cdh13-deficient mice including baseline levels of the stress hormone corticosterone. This also excludes the possibility that observed alterations in the cognitive domain are masked by emotional dysregulation. Finally, no functional CDH13 variants associated with violent behavior have been identified yet and it is still unknown whether the nature of these variants results in a loss or a gain of CDH13 function.
In conclusion, our findings demonstrate that CDH13 localizes to the presynaptic compartment of hippocampal GABAergic synapses and safeguards the excitatory–inhibitory balance in the CA1 region. CDH13 is thus essential for cognitive flexibility function and correct memory formation. CDH13 deficiency results in behavioral alterations associated with symptoms observed in neurodevelopmental disorders such as ADHD, including learning and memory deficits as well as locomotor hyperactivity. The present findings therefore contribute to the understanding of how dysregulation of CDH13 function may predispose individuals to ADHD and related comorbidities.
References
Hirano S, Takeichi M . Cadherins in brain morphogenesis and wiring. Physiol Rev 2012; 92: 597–634.
Philippova M, Joshi MB, Kyriakakis E, Pfaff D, Erne P, Resink TJ . A guide and guard: the many faces of T-cadherin. Cell Signal 2009; 21: 1035–1044.
Ranscht B, Dours-Zimmermann MT . T-cadherin, a novel cadherin cell adhesion molecule in the nervous system lacks the conserved cytoplasmic region. Neuron 1991; 7: 391–402.
Rivero O, Sich S, Popp S, Schmitt A, Franke B, Lesch KP . Impact of the ADHD-susceptibility gene CDH13 on development and function of brain networks. Eur Neuropsychopharmacol 2013; 23: 492–507.
Arias-Vasquez A, Altink ME, Rommelse NN, Slaats-Willemse DI, Buschgens CJ, Fliers EA et al. CDH13 is associated with working memory performance in attention deficit/hyperactivity disorder. Genes Brain Behav 2011; 10: 844–851.
Doyle A, Faraone SV, MgGrath L, Thermenos H, Juelich R, Chaponis J et al. Multivariate association of CDH13 variants and cortical thickness in ADHD. Biol Psychiatry 2014; 75: 347S.
Edwards AC, Aliev F, Bierut LJ, Bucholz KK, Edenberg H, Hesselbrock V et al. Genome-wide association study of comorbid depressive syndrome and alcohol dependence. Psychiatr Genet 2012; 22: 31–41.
Hart AB, Engelhardt BE, Wardle MC, Sokoloff G, Stephens M, de Wit H et al. Genome-wide association study of d-amphetamine response in healthy volunteers identifies putative associations, including cadherin 13 (CDH13). PLoS One 2012; 7: e42646.
Lesch KP, Timmesfeld N, Renner TJ, Halperin R, Roser C, Nguyen TT et al. Molecular genetics of adult ADHD: converging evidence from genome-wide association and extended pedigree linkage studies. J Neural Transm 2008; 115: 1573–1585.
Mavroconstanti T, Halmoy A, Haavik J . Decreased serum levels of adiponectin in adult attention deficit hyperactivity disorder. Psychiatry Res 2014; 216: 123–130.
Neale BM, Lasky-Su J, Anney R, Franke B, Zhou K, Maller JB et al. Genome-wide association scan of attention deficit hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet 2008; 147B: 1337–1344.
Neale BM, Medland SE, Ripke S, Asherson P, Franke B, Lesch KP et al. Meta-analysis of genome-wide association studies of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry 2010; 49: 884–897.
Treutlein J, Cichon S, Ridinger M, Wodarz N, Soyka M, Zill P et al. Genome-wide association study of alcohol dependence. Arch Gen Psychiatry 2009; 66: 773–784.
Uhl GR, Drgon T, Johnson C, Li CY, Contoreggi C, Hess J et al. Molecular genetics of addiction and related heritable phenotypes: genome-wide association approaches identify ‘connectivity constellation’ and drug target genes with pleiotropic effects. Ann N Y Acad Sci 2008; 1141: 318–381.
Uhl GR, Drgon T, Liu QR, Johnson C, Walther D, Komiyama T et al. Genome-wide association for methamphetamine dependence: convergent results from 2 samples. Arch Gen Psychiatry 2008; 65: 345–355.
Yang J, Wang S, Yang Z, Hodgkinson CA, Iarikova P, Ma JZ et al. The contribution of rare and common variants in 30 genes to risk nicotine dependence. Mol Psychiatry; e-pub ahead of print 2 December 2014; doi: 10.1038/mp.2014.156.
Zhou K, Dempfle A, Arcos-Burgos M, Bakker SC, Banaschewski T, Biederman J et al. Meta-analysis of genome-wide linkage scans of attention deficit hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet 2008; 147B: 1392–1398.
Sibille E, Wang Y, Joeyen-Waldorf J, Gaiteri C, Surget A, Oh S et al. A molecular signature of depression in the amygdala. Am J Psychiatry 2009; 166: 1011–1024.
Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 2011; 70: 863–885.
Borglum AD, Demontis D, Grove J, Pallesen J, Hollegaard MV, Pedersen CB et al. Genome-wide study of association and interaction with maternal cytomegalovirus infection suggests new schizophrenia loci. Mol Psychiatry 2014; 19: 325–333.
Xu W, Cohen-Woods S, Chen Q, Noor A, Knight J, Hosang G et al. Genome-wide association study of bipolar disorder in Canadian and UK populations corroborates disease loci including SYNE1 and CSMD1. BMC Med Genet 2014; 15: 2.
Terracciano A, Sanna S, Uda M, Deiana B, Usala G, Busonero F et al. Genome-wide association scan for five major dimensions of personality. Mol Psychiatry 2010; 15: 647–656.
Tiihonen J, Rautiainen MR, Ollila HM, Repo-Tiihonen E, Virkkunen M, Palotie A et al. Genetic background of extreme violent behavior. Mol Psychiatry 2014; 20: 786–792.
Bai S, Ghoshal K, Jacob ST . Identification of T-cadherin as a novel target of DNA methyltransferase 3B and its role in the suppression of nerve growth factor-mediated neurite outgrowth in PC12 cells. J Biol Chem 2006; 281: 13604–13611.
Ciatto C, Bahna F, Zampieri N, VanSteenhouse HC, Katsamba PS, Ahlsen G et al. T-cadherin structures reveal a novel adhesive binding mechanism. Nat Struct Mol Biol 2010; 17: 339–347.
Fredette BJ, Miller J, Ranscht B . Inhibition of motor axon growth by T-cadherin substrata. Development 1996; 122: 3163–3171.
Fredette BJ, Ranscht B . T-cadherin expression delineates specific regions of the developing motor axon-hindlimb projection pathway. J Neurosci 1994; 14: 7331–7346.
Takeuchi T, Misaki A, Liang SB, Tachibana A, Hayashi N, Sonobe H et al. Expression of T-cadherin (CDH13, H-Cadherin) in human brain and its characteristics as a negative growth regulator of epidermal growth factor in neuroblastoma cells. J Neurochem 2000; 74: 1489–1497.
Hayano Y, Zhao H, Kobayashi H, Takeuchi K, Norioka S, Yamamoto N . The role of T-cadherin in axonal pathway formation in neocortical circuits. Development 2014; 141: 4784–4793.
Paradis S, Harrar DB, Lin Y, Koon AC, Hauser JL, Griffith EC et al. An RNAi-based approach identifies molecules required for glutamatergic and GABAergic synapse development. Neuron 2007; 53: 217–232.
Schwenk F, Baron U, Rajewsky K . A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res 1995; 23: 5080–5081.
Nadif Kasri N, Nakano-Kobayashi A, Van Aelst L . Rapid synthesis of the X-linked mental retardation protein OPHN1 mediates mGluR-dependent LTD through interaction with the endocytic machinery. Neuron 2011; 72: 300–315.
Tricoire L, Pelkey KA, Erkkila BE, Jeffries BW, Yuan X, McBain CJ . A blueprint for the spatiotemporal origins of mouse hippocampal interneuron diversity. J Neurosci 2011; 31: 10948–10970.
Marx M, Gunter RH, Hucko W, Radnikow G, Feldmeyer D . Improved biocytin labeling and neuronal 3D reconstruction. Nat Protoc 2012; 7: 394–407.
Andersen P, Morris R, Amaral D, Bliss T, O'Keefe J . The Hippocampus Book. Oxford University Press: New York, NY, USA, 2006.
Miyoshi G, Butt SJ, Takebayashi H, Fishell G . Physiologically distinct temporal cohorts of cortical interneurons arise from telencephalic Olig2-expressing precursors. J Neurosci 2007; 27: 7786–7798.
Steward O, Falk PM . Selective localization of polyribosomes beneath developing synapses: a quantitative analysis of the relationships between polyribosomes and developing synapses in the hippocampus and dentate gyrus. J Comp Neurol 1991; 314: 545–557.
Hebbard LW, Garlatti M, Young LJ, Cardiff RD, Oshima RG, Ranscht B . T-cadherin supports angiogenesis and adiponectin association with the vasculature in a mouse mammary tumor model. Cancer Res 2008; 68: 1407–1416.
Williams NM, Zaharieva I, Martin A, Langley K, Mantripragada K, Fossdal R et al. Rare chromosomal deletions and duplications in attention-deficit hyperactivity disorder: a genome-wide analysis. Lancet 2010; 376: 1401–1408.
Lionel AC, Crosbie J, Barbosa N, Goodale T, Thiruvahindrapuram B, Rickaby J et al. Rare copy number variation discovery and cross-disorder comparisons identify risk genes for ADHD. Sci Transl Med 2011; 3: 95ra75.
Jinno S, Kosaka T . Cellular architecture of the mouse hippocampus: a quantitative aspect of chemically defined GABAergic neurons with stereology. Neurosci Res 2006; 56: 229–245.
Marin O . Interneuron dysfunction in psychiatric disorders. Nat Rev Neurosci 2012; 13: 107–120.
Ramamoorthi K, Lin Y . The contribution of GABAergic dysfunction to neurodevelopmental disorders. Trends Mol Med 2011; 17: 452–462.
Kluge C, Stoppel C, Szinyei C, Stork O, Pape HC . Role of the somatostatin system in contextual fear memory and hippocampal synaptic plasticity. Learn Mem 2008; 15: 252–260.
Fiederling A, Ewert R, Andreyeva A, Jungling K, Gottmann K . E-cadherin is required at GABAergic synapses in cultured cortical neurons. Neurosci Lett 2011; 501: 167–172.
Li Y, Xiao H, Chiou TT, Jin H, Bonhomme B, Miralles CP et al. Molecular and functional interaction between protocadherin-gammaC5 and GABAA receptors. J Neurosci 2012; 32: 11780–11797.
Najarro EH, Wong L, Zhen M, Carpio EP, Goncharov A, Garriga G et al. Caenorhabditis elegans flamingo cadherin fmi-1 regulates GABAergic neuronal development. J Neurosci 2012; 32: 4196–4211.
Nikitczuk JS, Patil SB, Matikainen-Ankney BA, Scarpa J, Shapiro ML, Benson DL et al. N-cadherin regulates molecular organization of excitatory and inhibitory synaptic circuits in adult hippocampus in vivo. Hippocampus 2014; 24: 943–962.
Johnston D, Amaral DG . Hippocampus. In: Sheperd GM (ed). The Synaptic Organization of the Brain. Oxford University Press: New York, NY, USA, 2003, pp 455–498.
Edden RA, Crocetti D, Zhu H, Gilbert DL, Mostofsky SH . Reduced GABA concentration in attention-deficit/hyperactivity disorder. Arch Gen Psychiatry 2012; 69: 750–753.
Ahrens S, Jaramillo S, Yu K, Ghosh S, Hwang GR, Paik R et al. ErbB4 regulation of a thalamic reticular nucleus circuit for sensory selection. Nat Neurosci 2015; 18: 104–111.
Kremkow J, Aertsen A, Kumar A . Gating of signal propagation in spiking neural networks by balanced and correlated excitation and inhibition. J Neurosci 2010; 30: 15760–15768.
Olshausen BA, Anderson CH, Van Essen DC . A neurobiological model of visual attention and invariant pattern recognition based on dynamic routing of information. J Neurosci 1993; 13: 4700–4719.
Vogels TP, Abbott LF . Signal propagation and logic gating in networks of integrate-and-fire neurons. J Neurosci 2005; 25: 10786–10795.
Farzan F, Barr MS, Levinson AJ, Chen R, Wong W, Fitzgerald PB et al. Evidence for gamma inhibition deficits in the dorsolateral prefrontal cortex of patients with schizophrenia. Brain 2010; 133 (Pt 5): 1505–1514.
Fazzari P, Paternain AV, Valiente M, Pla R, Lujan R, Lloyd K et al. Control of cortical GABA circuitry development by Nrg1 and ErbB4 signalling. Nature 2010; 464: 1376–1380.
Lewis DA, Hashimoto T, Volk DW . Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci 2005; 6: 312–324.
Ongur D, Prescot AP, McCarthy J, Cohen BM, Renshaw PF . Elevated gamma-aminobutyric acid levels in chronic schizophrenia. Biol Psychiatry 2010; 68: 667–670.
Neddens J, Buonanno A . Selective populations of hippocampal interneurons express ErbB4 and their number and distribution is altered in ErbB4 knockout mice. Hippocampus 2010; 20: 724–744.
Vullhorst D, Neddens J, Karavanova I, Tricoire L, Petralia RS, McBain CJ et al. Selective expression of ErbB4 in interneurons, but not pyramidal cells, of the rodent hippocampus. J Neurosci 2009; 29: 12255–12264.
Chen YJ, Zhang M, Yin DM, Wen L, Ting A, Wang P et al. ErbB4 in parvalbumin-positive interneurons is critical for neuregulin 1 regulation of long-term potentiation. Proc Natl Acad Sci USA 2010; 107: 21818–21823.
Shamir A, Kwon OB, Karavanova I, Vullhorst D, Leiva-Salcedo E, Janssen MJ et al. The importance of the NRG-1/ErbB4 pathway for synaptic plasticity and behaviors associated with psychiatric disorders. J Neurosci 2012; 32: 2988–2997.
Gibson JR, Huber KM, Sudhof TC . Neuroligin-2 deletion selectively decreases inhibitory synaptic transmission originating from fast-spiking but not from somatostatin-positive interneurons. J Neurosci 2009; 29: 13883–13897.
Foldy C, Malenka RC, Sudhof TC . Autism-associated neuroligin-3 mutations commonly disrupt tonic endocannabinoid signaling. Neuron 2013; 78: 498–509.
Aloy EM, Weinmann O, Pot C, Kasper H, Dodd DA, Rulicke T et al. Synaptic destabilization by neuronal Nogo-A. Brain Cell Biol 2006; 35: 137–156.
Andreyeva A, Nieweg K, Horstmann K, Klapper S, Muller-Schiffmann A, Korth C et al. C-terminal fragment of N-cadherin accelerates synapse destabilization by amyloid-beta. Brain 2012; 135 (Pt 7): 2140–2154.
Philippova M, Ivanov D, Joshi MB, Kyriakakis E, Rupp K, Afonyushkin T et al. Identification of proteins associating with glycosylphosphatidylinositol- anchored T-cadherin on the surface of vascular endothelial cells: role for Grp78/BiP in T-cadherin-dependent cell survival. Mol Cell Biol 2008; 28: 4004–4017.
Joshi MB, Ivanov D, Philippova M, Erne P, Resink TJ . Integrin-linked kinase is an essential mediator for T-cadherin-dependent signaling via Akt and GSK3beta in endothelial cells. FASEB J 2007; 21: 3083–3095.
Philippova M, Ivanov D, Allenspach R, Takuwa Y, Erne P, Resink T . RhoA and Rac mediate endothelial cell polarization and detachment induced by T-cadherin. FASEB J 2005; 19: 588–590.
Oosterlaan J, Logan GD, Sergeant JA . Response inhibition in AD/HD, CD, comorbid AD/HD+CD, anxious, and control children: a meta-analysis of studies with the stop task. J Child Psychol Psychiatry 1998; 39: 411–425.
Oosterlaan J, Sergeant JA . Response inhibition and response re-engagement in attention-deficit/hyperactivity disorder, disruptive, anxious and normal children. Behav Brain Res 1998; 94: 33–43.
Willcutt EG, Doyle AE, Nigg JT, Faraone SV, Pennington BF . Validity of the executive function theory of attention-deficit/hyperactivity disorder: a meta-analytic review. Biol Psychiatry 2005; 57: 1336–1346.
Kleschevnikov AM, Belichenko PV, Villar AJ, Epstein CJ, Malenka RC, Mobley WC . Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of Down syndrome. J Neurosci 2004; 24: 8153–8160.
Reeves RH, Irving NG, Moran TH, Wohn A, Kitt C, Sisodia SS et al. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat Genet 1995; 11: 177–184.
Costa RM, Federov NB, Kogan JH, Murphy GG, Stern J, Ohno M et al. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature 2002; 415: 526–530.
Amitai N, Markou A . Comparative effects of different test day challenges on performance in the 5-choice serial reaction time task. Behav Neurosci 2011; 125: 764–774.
Gratten J, Wray NR, Keller MC, Visscher PM . Large-scale genomics unveils the genetic architecture of psychiatric disorders. Nat Neurosci 2014; 17: 782–790.
Denzel MS, Scimia MC, Zumstein PM, Walsh K, Ruiz-Lozano P, Ranscht B . T-cadherin is critical for adiponectin-mediated cardioprotection in mice. J Clin Invest 2010; 120: 4342–4352.
Geissler J, Lesch KP . A lifetime of attention-deficit/hyperactivity disorder: diagnostic challenges, treatment and neurobiological mechanisms. Expert Rev Neurother 2011; 11: 1467–1484.
Bolivar VJ . Intrasession and intersession habituation in mice: from inbred strain variability to linkage analysis. Neurobiol Learn Mem 2009; 92: 206–214.
Lesch KP, Araragi N, Waider J, van den Hove D, Gutknecht L . Targeting brain serotonin synthesis: insights into neurodevelopmental disorders with long-term outcomes related to negative emotionality, aggression and antisocial behaviour. Philos Trans R Soc Lond B Biol Sci 2012; 367: 2426–2443.
Acknowledgements
We are grateful to Nicole Steigerwald, Astrid Oudakker, Andrea Dietzel, Dmitrij Nagel and Nick van Bakel for their valuable assistance in animal handling, genotyping and other experiments. This work was supported by Deutsche Forschungsgemeinschaft (SFB TRR 58/A5 to KPL; RTG 1253/2 to KPL, fellowship to SP), the European Community (EC: AGGRESSOTYPE FP7/No. 602805 to KPL, OR, DvdH and TS), the Fritz Thyssen Foundation (Az.10.13.1185 to KPL), the Jacobs Foundation (to OR), the ‘Donders Center for Neuroscience fellowship award of the Radboudumc’ (to NNK), the ‘FP7-Marie Curie International Reintegration Grant’ (No.277091 to NNK) and the Jerome Lejeune Foundation (to NNK). OR was the recipient of a postdoctoral fellowship from Generalitat Valenciana, Spain. LB and DK were supported by grants of the German Excellence Initiative to the Graduate School of Life Sciences, University of Würzburg. OR and KPL are members of the international multicentre persistent ADHD collaboration (IMpACT). IMpACT unites major research centers working on the genetics of ADHD persistence across the lifespan and has participants in the Netherlands, Germany, Spain, Norway, the United Kingdom, the United States, Brazil and Sweden. The funding entities had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Rivero, O., Selten, M., Sich, S. et al. Cadherin-13, a risk gene for ADHD and comorbid disorders, impacts GABAergic function in hippocampus and cognition. Transl Psychiatry 5, e655 (2015). https://doi.org/10.1038/tp.2015.152
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2015.152
This article is cited by
-
Aging and age-related diseases with a focus on therapeutic potentials of young blood/plasma
Naunyn-Schmiedeberg's Archives of Pharmacology (2024)
-
Functional convergence of genomic and transcriptomic architecture underlies schooling behaviour in a live-bearing fish
Nature Ecology & Evolution (2023)
-
In the developing cerebral cortex: axonogenesis, synapse formation, and synaptic plasticity are regulated by SATB2 target genes
Pediatric Research (2023)
-
Cadherin-13 is a critical regulator of GABAergic modulation in human stem-cell-derived neuronal networks
Molecular Psychiatry (2022)
-
Genetic variations influence brain changes in patients with attention-deficit hyperactivity disorder
Translational Psychiatry (2021)
