
Abstract
Working memory (WM) is an important endophenotype in neuropsychiatric research and its use in genetic association studies is thought to be a promising approach to increase our understanding of psychiatric disease. As for any genetically complex trait, demonstration of sufficient heritability within the specific study context is a prerequisite for conducting genetic studies of that trait. Recently developed methods allow estimating trait heritability using sets of common genetic markers from genome-wide association study (GWAS) data in samples of unrelated individuals. Here we present single-nucleotide polymorphism (SNP)-based heritability estimates (h2SNP) for a WM phenotype. A Caucasian sample comprising a total of N=2298 healthy and young individuals was subjected to an N-back WM task. We calculated the genetic relationship between all individuals on the basis of genome-wide SNP data and performed restricted maximum likelihood analyses for variance component estimation to derive the h2SNP estimates. Heritability estimates for three 2-back derived WM performance measures based on all autosomal chromosomes ranged between 31 and 41%, indicating a substantial SNP-based heritability for WM traits. These results indicate that common genetic factors account for a prominent part of the phenotypic variation in WM performance. Hence, the application of GWAS on WM phenotypes is a valid method to identify the molecular underpinnings of WM.
Similar content being viewed by others
Introduction
Working memory (WM) has a pivotal role in human cognition and cognitive performance, allowing the integration of information from instantly perceived stimuli, long-term-memory and thought-processes. A wide variety of psychological testing procedures have been developed to render WM performance quantifiable.1, 2, 3, 4 Assessment of WM performance along with brain imaging techniques have been used to elucidate and delimit its different components, aiming at a more circumscribed understanding of the neuronal processes at the basis of WM.5 The integration of extensive research on WM yields a common consensus that it constitutes a complex trait and that the buffer size for the transiently stored content varies interindividually, which is partly due to genetic factors. WM is an important endophenotype in neuropsychiatric research and its use in genetic association studies is thought to be a promising approach to increase our understanding of psychiatric disease.6, 7, 8, 9, 10, 11 Namely, two recent studies have corroborated the genetic link between schizophrenia and WM, demonstrating the validity of this endophenotype for schizophrenia research: Stefansson et al.12 report that control subjects carrying Copy Number Variants, which predispose to schizophrenia and autism, perform at a level that is in between patients and population controls in cognitive tasks including a spatial WM test. A genome-wide gene set enrichment study conducted by our group, identified a set of voltage-gated cation channel activity genes that were robustly linked to performance in WM tasks in healthy individuals and also to risk for schizophrenia in a large case–control sample.13 Both these studies suggest that the findings of cognitive deficits are translatable between healthy subjects and cohorts of psychiatric patients. Improving our understanding of the molecular basis of this endophenotype might be key for future drug discovery and treatment options in psychiatry.14,15 As for any genetically complex trait, demonstration of sufficient heritability within the specific study context is a prerequisite for conducting genetic studies of that trait. Heritability is a concept that summarizes how much of the phenotypic variation in a given trait is attributable to heritable factors, the majority of them being genetic. Since inter-individual trait differences attributable to genetic variability are a prerequisite for quantitative trait loci mapping, estimation of trait heritability is important to demonstrate the validity of a quantitative trait loci study of a given trait. Conventionally, heritability estimates in humans are derived from phenotypic data by comparing correlations between relatives, where the extent of genetic relatedness is derived from the degree of relationship. Results from previous twin and family studies that used a variety of tasks to measure WM performance have shown heritability estimates ranging between 15 and 72%.16, 17, 18, 19 Recently developed methods propose inferring genetic identity from high-throughput SNP data and correlating these estimates with phenotypic resemblance among unrelated individuals.20,21 This allows the estimation of an SNP-based heritability measure (h2SNP) for any specific genome-wide association study data set. Importantly, the h2SNP value quantifies the amount of phenotypic variation that can be explained by the common SNPs represented in genome-wide association study data sets. Of note, h2SNP hereby represents a lower bound heritability estimate, because causal variants that are neither genotyped nor tagged by the used set of markers are completely disregarded. In the present study we show that a substantial part of phenotypic variance of WM performance can be explained by using the common marker set represented on the Affymetrix 6.0 Human SNP array to estimate h2SNP values for N-back-derived WM phenotypes.
Materials and Methods
Ethics statement
The experiments were approved by the ethics committee of the Cantons of Basel-City and Basel-Country. Written informed consent was obtained from all subjects before participation.
Participants and genotyping procedure
A sample of 2703 young healthy Swiss individuals (1721 females, 982 males; mean age: 22.5 years; median age: 22 years; range: 18–38 years) was assessed for WM performance using an N-back task. Saliva samples were obtained from each study participant, using an Oragene DNA Self-Collection Kit (DNA Genotek, Ottawa, Ontario, Canada). DNA was extracted from saliva using standard protocols. All subjects were individually genotyped using the Affymetrix Human SNP assay 6.0 according to the manufacturer’s recommendation.
N-back task
All subjects completed the 0-back and 2-back version of the N-back task, after they were instructed and trained on the task. The 0-back condition served as a non-memory-guided control condition, measuring general attention, concentration and reaction time. The N-back task was presented on a computer screen and consisted of 12 blocks (six 0-back and six 2-back blocks). Per block, 14 stimuli consisting of 3 targets and 11 non-targets, were presented in a randomized order. Each stimulus was presented for 500 ms, followed by a black screen for 1500 ms. For each stimulus participants had to indicate as fast as possible whether it was a target or a non-target by pressing the corresponding button. At the beginning of each block the instruction was shown for 5 s. There was a 20 s break after every second block. The two blocks between the breaks consisted of a 0-back and a 2-back block, which were presented in a randomized order. Each block lasted for 33 s. The N-back task had a total duration of roughly 10 min, including instructions presented at the beginning of the task. Performance in the 0-back and the 2-back tasks was assessed by the mean correct response accuracy. The difference in mean accuracy between 2-back and the 0-back condition served as main phenotype (2-back mean accuracy attention corrected: 2-back−0-back). We also tested the uncorrected 2-back mean accuracy and the d-prime for the 2-back condition. The d-prime was calculated according to the following formula:

The maximal number of hits (that is, correct responses to targets) was N=36 and the maximal number of false alarms (that is, incorrect responses to non-targets) was N=132. To allow for a probit-transformation, the hit and false-alarm-rates of 0 or 1 had to be avoided by replacing these extreme values, which was done as follows: 100% of hits were set to 1−1/36 and 0% were set to 1/36. Accordingly, 100% of false alarms were set to 1−1/132 and 0% of false alarms were set to 1/132.22 In addition, we also estimated h2SNP for the mean reaction time of the 2-back task (2-back mean reaction time). See Table 1 for descriptive statistics of uncorrected task performance measures.
Genotypic outlier analysis
We identified and subsequently excluded subjects with technical biases or non-Caucasian ancestry. A Bayesian clustering algorithm was applied to genome-wide summary statistics to identify and exclude samples affected by a potential technical bias. Briefly, considering a combination of genome-wide call rate and heterozygosity rate, the algorithm infers each sample's posterior probability to belong to the outliers' class, as extreme values for these summary statistics may be indicative of a genotyping bias. Similarly, the Bayesian clustering algorithm was applied on the coordinates of the subjects on the first two PCA components inferred from HapMap data using the YRI, CEU and CHB-JPT populations. Thus, samples of non-Caucasian ancestry were identified and removed, excluding N=272 individuals.23
Phenotypic and genetic quality controls
To adjust for sex and age, all N-back derived performance measurements (accuracy and reaction time) entered a linear model and residuals were extracted, which were used for all further calculations. As the presence of outliers in the phenotypic data can affect heritability estimation for complex traits,24 all subjects whose performance measurements in any of the four N-back performance measurements differed more than 3.5 standard deviations from the sample mean were excluded (Nexcluded=40).
Due to this procedure, the exact same number of subjects was included in all subsequent analyses for all investigated phenotypes.
The genotype data set was filtered (MAF⩾0.01; HWE pFisher⩾0.001; per SNP call rate⩾95%; per individual call rate⩾95%) to obtain a set of 702’744 autosomal and 27’265 X-chromosomal SNPs for further analysis.
Estimation of the pairwise genetic similarity using all autosomal markers and subsequently removing one of each pairs showing genetic relatedness >0.025 (~second-degree cousins) led to the exclusion of N=93 subjects, yielding a sample size of N=2298 for h2SNP estimation.
h2SNP Estimation
To obtain the genome-wide heritability estimates h2SNP, we calculated the genetic relationship between all individuals on the basis of the autosomal SNP data and performed restricted maximum likelihood analyses for variance component estimation using the GCTA software package.20 In a second step, h2SNP estimates were calculated for all chromosomes separately including the X-chromosome. The genetic relationship matrix for the X-chromosome was obtained using the designated option in the GCTA toolset.
Results
Heritability estimates for the three 2-back derived WM performance measures on the basis of autosomal chromosomes ranged between 31 and 41%, indicating substantial SNP-based heritabilities (Figure 1). The proportion of phenotypic variance explained by all autosomal SNPs for difference in response accuracy between the 2-back and the 0-back condition was 41% (s.e.=0.139; P-value=0.0008). h2SNP for the mean response accuracy in the 2-back alone (that is, without correction for 0-back performance) and the false-response corrected d-prime condition were 31% with s.e.=0.138; P-value=0.006 and s.e.=0.140; P-value=0.01, respectively. Due to several reports linking processing speed to WM-related cognitive abilities,25,26 we also investigated heritability of the mean reaction time in the 2-back condition (h2SNP: 24%; s.e.=0.142; P-value=0.04). A descriptive analysis of correlations between phenotypes shows strong intercorrelations between the three accuracy-related 2-back WM phenotypes (r2>0.65), whereas correlation coefficients between the mean reaction time in the 2-back condition and the WM phenotypes are negligible (r2<0.01) by means of shared phenotypic variance (Table 2).

h2SNP estimates for 2-back derived WM measures in percent for a total of N=2298. The error bars represent standard errors. 2-Back mean accuracy attention corrected (corr.) h2SNP 41%, s.e. 14%, P-value=0.0008; 2-back mean accuracy h2SNP 31%, s.e. 14%, P-value=0.006; 2-back d-prime h2SNP 31%, s.e. 14%, P-value=0.01; 2-back mean reaction time h2SNP 24%, s.e. 14%, P-value=0.04. SNP, single-nucleotide polymorphism; WM, working memory.
Chromosomewise h2SNP estimates
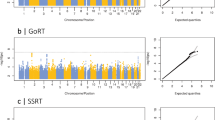
In a further step, we partitioned heritability into contributions from single chromosomes, including the X-chromosome. Figure 2 shows the h2SNP values per chromosome, presenting an approximation to the amount of phenotypic variance explained by the single chromosomes. For each phenotype, we derived a vector containing h2SNP estimates per chromosome. To investigate whether the correlational pattern of the phenotypes is reflected also in a similar chromosomal heritability profile, we calculated the Pearson’s correlation coefficients for these vectors. The highly correlated WM phenotypes show a similar profile for the chromosomewise h2SNP estimates. Regarding the single chromosome h2SNP values for the mean reaction time in the 2-back condition, we observe a different distribution of h2SNP estimates per chromosome. Table 3 depicts the descriptive Pearson’s correlation coefficients between the chromosomewise h2SNP estimates for all phenotypes. We also investigated whether the chromosomal length and the number of SNPs are correlated with the phenotypic variance that each chromosome explains. We observed significant correlations (Pearson’s r in the range between 0.53 and 0.68) for the WM phenotypes with the chromosomal length and number of SNPs, whereas no significant correlation was observed for the mean reaction time of the 2-back task (See Table 4).

Chromosomewise h2SNP estimates for N-back phenotypes and mean reaction time h2SNP estimates per chromosome are plotted on the y axis. The x axis depicts the chromosomal length in Mb. Circle size indicates the number of SNPs used in the analysis after quality control. Color scheme from red to blue reflects the chromosomal ordering from chromosome 1 to 22, with the X-chromosome colored in black. The black line depicts the regression line for chromosomal length and explained variance (regression coefficients (df=21): (a) r=0.53, P-value=0.009, (b) r=0.56, P-value=0.006, (c) r=0.65, P-value=0.0009, (d) r=0.25, P-value=0.3). SNP, single-nucleotide polymorphism.
Discussion
The present data demonstrate that a substantial proportion of variance in WM performance is captured by common genome-wide association study SNPs in a sample of healthy young unrelated individuals. The h2SNP estimates for the N-back-derived WM phenotypes range from 31 to 41% with a mean standard error of 0.14. Thus, the h2SNP estimates are consistent with the previously reported heritability for WM on the basis of twin-studies.27 Taken together, these findings add further support for the hypothesis that WM is a highly heritable trait. The chromosomal partitioning analysis (Figure 2), which depicts h2SNP estimates for all chromosomes analyzed individually, provides a clear hint for the pronounced polygenicity of the investigated WM phenotype. This finding is in line with the previously reported ubiquitious polygenicity of human complex traits.28 The amount of genetic variation that explains the variance of WM performance is proportional to the chromosomal length and the number of investigated SNPs per chromosome (see Table 4). The correlational pattern for the investigated phenotypes (see Table 2) indicates that the WM performance measures and the mean reaction time are independent behavioral metrics. The h2SNP estimate based on genome-wide data (see Figure 1) are lower for the mean reaction time and the h2SNP estimates per chromosome also show a different distribution compared with the WM performance derived estimates (see Figure 2). Furthermore, although there is an observable trend for the proportionality of h2SNP estimates with chromosomal length, the correlation fails to reach statistical significance (see Table 2), which may be due to a lower overall heritability of this trait. Hence, we conclude that the WM performance measured with the 2-back task is independent of the mean response time under the cognitive load of performing the 2-back test.
Of note, the heritability estimation for complex traits using genome-wide data sets is rather a complement than a substitution to studies on twin- and family-based heritability. Marker-based heritability estimation represents a lower bound for the true trait heritability as it relies only on the effects of common variants assuming an additive variance model. On one hand, the investigation of large pools of unrelated individuals allows to assess heritability without the undermining effects due to shared environment or familiality. Yet, on the other hand, it will not take the potential effects of rare variants into account. The SNP-based heritability estimates for WM suggest that a large share of phenotypic variance can be explained by common SNPs rendering well-powered genome-wide association study data sets a promising tool to discover molecular players that act in concert to form this complex trait. It has been repeatedly shown that familial risk for psychiatric diseases like schizophrenia is often accompanied by reduced cognitive abilities.29, 30, 31 Results from a long-term study suggest that people who reported psychotic-like experiences in late adulthood performed poorer in cognitive tasks during childhood and adolescence.32 In a recent twin study investigating healthy twins, Goldberg et al.6 report that the phenotypic correlation between intelligence quotient and WM can be almost entirely attributed to shared genetic variance. In an effort to investigate dimensions of observable behavior and neurobiological measures that can be used to classify psychopathology, the National Institute of Mental Health established the Research Domain Criteria project. The N-back task used in the present study is deemed appropriate to measure the sub-constructs of WM of active maintenance, limited capacity and with some, albeit not definitive evidence, for flexible updating according to the Research Domain Criteria project. Given the implication of cognitive and especially WM deficits in schizophrenia and other psychiatric disorders, the genetically guided decomposition of WM-related molecular pathways might pave the way for a better understanding of the genetic architecture implicated in these mental disorders.
References
Barch DM, Moore H, Nee DE, Manoach DS, Luck SJ . CNTRICS imaging biomarkers selection: working memory. Schizophr Bull 2012; 38: 43–52.
Daneman M, Carpenter P . Individual differences in working memory and reading. J Verbal Learn Verbal Behav 1980; 19: 450–466.
Gur RC, Richard J, Hughett P, Calkins ME, Macy L, Bilker WB et al. A cognitive neuroscience-based computerized battery for efficient measurement of individual differences: standardization and initial construct validation. J Neurosci Methods 2010; 187: 254–262.
Kirchner WK . Age differences in short-term retention of rapidly changing information. J Exp Psychol 1958; 55: 352–358.
Rissman J, Wagner AD . Distributed representations in memory: insights from functional brain imaging. Annu Rev Psychol 2012; 63: 101–128.
Goldberg X, Alemany S, Rosa A, Picchioni M, Nenadic I, Owens SF et al. Substantial genetic link between iq and working memory: implications for molecular genetic studies on schizophrenia. the european twin study of schizophrenia (EUTwinsS). Am J Med Genet B Neuropsychiatr Genet 2013; 162: 413–418.
Snitz BE, Macdonald AW, Carter CS . Cognitive deficits in unaffected first-degree relatives of schizophrenia patients: a meta-analytic review of putative endophenotypes. Schizophr Bull 2006; 32: 179–194.
Gottesman II, Gould TD . The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry 2003; 160: 636–645.
Gould TD . Gottesman II. Psychiatric endophenotypes and the development of valid animal models. Genes Brain Behav 2006; 5: 113–119.
Dick DM, Jones K, Saccone N, Hinrichs A, Wang JC, Goate A et al. Endophenotypes successfully lead to gene identification: results from the collaborative study on the genetics of alcoholism. Behav Genet 2006; 36: 112–126.
Murrough JW, Iacoviello B, Neumeister A, Charney DS, Iosifescu DV . Cognitive dysfunction in depression: neurocircuitry and new therapeutic strategies. Neurobiol Learn Mem 2011; 96: 553–563.
Stefansson H, Meyer-Lindenberg A, Steinberg S, Magnusdottir B, Morgen K, Arnarsdottir S et al. CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature 2014; 505: 361–366.
Heck A, Fastenrath M, Ackermann S, Auschra B, Bickel H, Coynel D et al. Converging genetic and functional brain imaging evidence links neuronal excitability to working memory, psychiatric disease, and brain activity. Neuron 2014; 81: 1203–1213.
Papassotiropoulos A, de Quervain DJ-F . Genetics of human episodic memory: dealing with complexity. Trends Cogn Sci 2011; 15: 381–387.
Hyman SE . Psychiatric drug development: diagnosing a crisis. Cerebrum 2013; 2013: 5.
Vinkhuyzen AAE, van der Sluis S, Boomsma DI, de Geus EJC, Posthuma D . Individual differences in processing speed and working memory speed as assessed with the Sternberg memory scanning task. Behav Genet 2010; 40: 315–326.
Lee T, Mosing MA, Henry JD, Trollor JN, Ames D, Martin NG et al. Genetic influences on four measures of executive functions and their covariation with general cognitive ability: the Older Australian Twins Study. Behav Genet 2012; 42: 528–538.
Karlsgodt KH, Kochunov P, Winkler AM, Laird AR, Almasy L, Duggirala R et al. A multimodal assessment of the genetic control over working memory. J Neurosci 2010; 30: 8197–8202.
Wilson RS, Barral S, Lee JH, Leurgans SE, Foroud TM, Sweet RA et al. Heritability of different forms of memory in the Late Onset Alzheimer’s Disease Family Study. J Alzheimers Dis 2011; 23: 249–255.
Yang J, Lee SH, Goddard ME, Visscher PM . GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 2011; 88: 76–82.
Yang J, Lee SH, Goddard ME, Visscher PM . Genome-wide complex trait analysis (GCTA): methods, data analyses, and interpretations. Methods Mol Biol 2013; 1019: 215–236.
Stanislaw H, Todorov N . Calculation of signal detection theory measures. Behav Res Methods Instrum Comput 1999; 31: 137–149.
Bellenguez C, Strange A, Freeman C, Donnelly P, Spencer CC, Wellcome Trust Case Control Consortium. A robust clustering algorithm for identifying problematic samples in genome-wide association studies. Bioinformatics 2012; 28: 134–135.
Hayat Y, Yang J, Xu H, Zhu J . Influence of outliers on QTL mapping for complex traits. J Zhejiang Univ Sci B 2008; 9: 931–937.
Baker LA, Vernon PA, Ho HZ . The genetic correlation between intelligence and speed of information processing. Behav Genet 1991; 21: 351–367.
Ogata S, Kato K, Honda C, Hayakawa K . Common genetic factors influence hand strength, processing speed, and working memory. J Epidemiol 2013; 24: 31–38.
Ando J, Ono Y, Wright MJ . Genetic structure of spatial and verbal working memory. Behav Genet 2001; 31: 615–624.
Yang J, Lee T, Kim J, Cho M-C, Han B-G, Lee J- et al. Ubiquitous polygenicity of human complex traits: genome-wide analysis of 49 traits in Koreans. PLoS Genet 2013; 9: e1003355.
Wiener H, Klei L, Calkins M, Wood J, Nimgaonkar V, Gur R et al. Principal components of heritability from neurocognitive domains differ between families with schizophrenia and control subjects. Schizophr Bull 2013; 39: 464–471.
Toulopoulou T, Goldberg TE, Mesa IR, Picchioni M, Rijsdijk F, Stahl D et al. Impaired intellect and memory: a missing link between genetic risk and schizophrenia? Arch Gen Psychiatry 2010; 67: 905–913.
Conklin HM, Curtis CE, Calkins ME, Iacono WG . Working memory functioning in schizophrenia patients and their first-degree relatives: cognitive functioning shedding light on etiology. Neuropsychologia 2005; 43: 930–942.
Barnett JH, McDougall F, Xu MK, Croudace TJ, Richards M, Jones PB . Childhood cognitive function and adult psychopathology: associations with psychotic and non-psychotic symptoms in the general population. Br J Psychiatry 2012; 201: 124–130.
Acknowledgements
This work was supported by the Swiss National Science Foundation (Sinergia Grants CRSI33_130080 and CRSII1_136227 to AP and DJ-FdeQ.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Vogler, C., Gschwind, L., Coynel, D. et al. Substantial SNP-based heritability estimates for working memory performance. Transl Psychiatry 4, e438 (2014). https://doi.org/10.1038/tp.2014.81
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2014.81
This article is cited by
-
Working memory and reaction time variability mediate the relationship between polygenic risk and ADHD traits in a general population sample
Molecular Psychiatry (2022)
-
Genetic influence on cognitive development between childhood and adulthood
Molecular Psychiatry (2021)
-
Heritability of human visual contour integration—an integrated genomic study
European Journal of Human Genetics (2019)
-
Multi-level genomic analyses suggest new genetic variants involved in human memory
European Journal of Human Genetics (2018)
-
Genetics and Functional Imaging: Effects of APOE, BDNF, COMT, and KIBRA in Aging
Neuropsychology Review (2015)
