
Abstract
Current pharmacological treatments of depression and related disorders suffer from major problems, such as a low rate of response, slow onset of therapeutic effects, loss of efficacy over time and serious side effects. Therefore, there is an urgent need to explore new therapeutic approaches that address these issues. Interestingly, the atypical antidepressant tianeptine already meets in part these clinical goals. However, in spite of three decades of basic and clinical investigations, the molecular target of tianeptine, as well as its mechanism of action, remains elusive. Herein, we report the characterization of tianeptine as a μ-opioid receptor (MOR) agonist. Using radioligand binding and cell-based functional assays, including bioluminescence resonance energy transfer-based assays for G-protein activation and cAMP accumulation, we identified tianeptine as an efficacious MOR agonist (Ki Human of 383±183 nM and EC50 Human of 194±70 nM and EC50 Mouse of 641±120 nM for G-protein activation). Tianeptine was also a full δ-opioid receptor (DOR) agonist, although with much lower potency (EC50 Human of 37.4±11.2 μM and EC50 Mouse of 14.5±6.6 μM for G-protein activation). In contrast, tianeptine was inactive at the κ-opioid receptor (KOR, both human and rat). On the basis of these pharmacological data, we propose that activation of MOR (or dual activation of MOR and DOR) could be the initial molecular event responsible for triggering many of the known acute and chronic effects of this agent, including its antidepressant and anxiolytic actions.
Similar content being viewed by others

Introduction
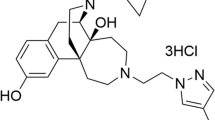
On the basis of shortcomings of existing therapies, the broad field of depression research has formulated several key goals for the development of novel therapeutics, including faster onset of antidepressant effects, efficacy in treatment-resistant subjects and minimization of side effects.1 The atypical antidepressant tianeptine (Figure 1a), an agent with established clinical efficacy, already addresses several limitations of standard antidepressants.2 Namely, it shows fast effects against some depressive symptoms (cognitive and anxiety symptoms), is effective in patients resistant to selective serotonin reuptake inhibitor therapy3 and shows a better side effect profile compared with selective serotonin reuptake inhibitors and tricyclic antidepressants.2 Interestingly, unlike other tricyclic antidepressants, tianeptine does not inhibit biogenic amine transporters, and despite longstanding interest in this agent, its direct target, and thus its molecular mechanism of action, have remained elusive.2

Summary of tianeptine’s activity at the opioid receptors. (a) Chemical structure of tianeptine. (b) Radioligand displacement binding assay of tianeptine at μ-opioid receptor (MOR; n=3), δ-opioid receptor (DOR; n=4) and κ-opioid receptor (KOR; n=4). Tianeptine’s functional activity at MOR, DOR and KOR are also summarized. Data represent mean±s.e.m.
In preclinical studies, tianeptine has beneficial effects in a number of behavioral models related to depression, anxiety and other stress-related disorders.2,4 Tianeptine has been shown to modulate glutamatergic transmission, and its effects on neuroplasticity have been extensively studied in the hippocampus and amygdala.2 For example, tianeptine restores stress-induced reduction of dendritic arborization in hippocampal neurons. Moreover, tianeptine reverses stress-induced inhibition of long-term potentiation at excitatory synapses in the hippocampus and prefrontal cortex.2 Although tianeptine has been shown to modulate N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor function,5 several reports have shown that tianeptine has no measurable affinity for these receptors2 and also does not bind appreciably to monoamine neurotransmitter receptors, kainate receptors or monoamine transporters.6
Driven by the impressive and wide-reaching biological effects of tianeptine, we set out to elucidate its primary molecular target. We report here that tianeptine is an efficacious μ-opioid receptor (MOR) and δ-opioid receptor (DOR) agonist, and we propose that MOR-agonism (or combined MOR/DOR-agonism) underlies the clinical, preclinical and in vitro effects of tianeptine.
Materials and methods
Materials
HEK-293T cells were obtained from the American Type Culture Collection (Rockville, MD, USA) and were cultured in a 5% CO2 atmosphere at 37 °C in Dulbecco’s Modified Eagle Medium (high glucose no. 11965; Life Technologies; Grand Island, NY, USA) supplemented with 10% Fetal Bovine Serum (Premium Select, Atlanta Biologicals; Atlanta, GA, USA) and 100 U ml−1 penicillin and 100 μg ml−1 streptomycin (no. 15140, Life Technologies). Tianeptine sodium salt was purchased from Selleck Chemicals (Houston, TX, USA); [D-Ala2, N-Me-Phe4, Gly5-ol]-Enkephalin (DAMGO) acetate salt, naltrexone hydrochloride and forskolin were purchased from Sigma-Aldrich (Saint Louis, MO, USA); [D-Pen(2,5)]Enkephalin (DPDPE) and nor-binaltorphimine dihydrochloride were purchased from Tocris Bioscience (Minneapolis, MN, USA); U-50,488 and TIPP[psi] were obtained from the National Institute on Drug Abuse Drug Supply Program; coelenterazine H was purchased from Dalton Pharma Services (Toronto, ON, Canada); polyethylenimine was purchased from Polysciences (Warrington, PA, USA).
Receptor screening and Ki determination
Receptor screening and Ki determination was generously performed by the National Institute of Mental Health's Psychoactive Drug Screening Program, Contract no. HHSN-271-2008-00025-C (NIMH PDSP). The NIMH PDSP is directed by Bryan L Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscoll at NIMH, Bethesda, MD, USA. For experimental details, please refer to the PDSP website: http://pdsp.med.unc.edu/. Tianeptine was evaluated in a primary radioligand binding screen against a panel of 51 human central nervous system receptors and transporters, including serotonin receptors (5-HT1a,b,d,e; 5-HT2a,b,c; 5-HT3,5a,6,7); nicotinic acetylcholine receptors (α2β2, α3β2, α3β4, α4β2, α4β4, α7); adrenergic receptors (α1A, 1B, 1D, 2A, 2B, 2C; β1,2,3); benzodiazepine receptor (BZP); cannabinoid receptors (CB1,2); dopamine receptors (D1–5); GABAA; dopamine transporter (DAT); norepinephrine transporter (NET); serotonin transporter (SERT); DOR, κ-opioid receptor (KOR), MOR; histamine receptors (H1,3,4); muscarinic acetylcholine receptors (M1–5); peripheral benzodiazepine receptor (PBR); sigma 1,2. The primary screen was used to select ligands that showed >50% inhibition of binding of a radiolabeled reference compound at a single 10 μM test concentration. This primary screen was followed by Ki determination when appropriate (>50% inhibition). Tianeptine was negative at all examined receptors (<50% inhibition by 10 μM tianeptine) except for MOR. Kis were determined at MOR, as well as at DOR and KOR. The reported Ki values are the average of 4 (DOR), 3 (MOR) or 4 (KOR) independent experiments±s.e.m. Tianeptine also showed no agonist or antagonist activity at mGluR1a,2,4,5,6,8 receptors in primary functional assays at a 10 μM test concentration (determined by PDSP).
DNA constructs
The mouse MOR (mMOR), the mouse DOR (mDOR) and the rat KOR (rKOR) were provided by Dr Lakshmi Devi at Mount Sinai Hospital. The human MOR (hMOR), human DOR (hDOR) and human KOR (hKOR) were obtained from the Missouri S&T Resource Center. The human G protein constructs used here have been previously described7 and were obtained from the Missouri S&T Resource Center unless otherwise noted. The G proteins used were: untagged GαoB (GαoB); GαoB with Renilla luciferase 8 (RLuc8) inserted at position 91 (GαoB-RLuc8, generously provided by C Galés7,8); Gβ1 (β1); untagged Gγ2 (γ2); Gγ2, which we fused to full-length mVenus at its N terminus via the amino acid linker GSAGT (mVenus-γ2). YFP-Epac-RLuc (CAMYEL) was obtained from ATCC (no. MBA-277).9 All constructs were sequence confirmed.
Transfection
A total of 15 μg of cDNA was transiently transfected into HEK-293T cells (5 × 106 cells per plate) in 10 cm dishes for G-protein activation (2.5 μg MOR/DOR/KOR, 0.125 μg GαoB-RLuc8, 6.25 μg β1, 6.25 μg mVenus-γ2) or cAMP inhibition assays (1.25 μg MOR/DOR/KOR, 10 μg CAMYEL, 1.25 μg GαoB, 1.25 μg β1, 1.25 μg γ2) using polyethylenimine in a 1:1 ratio (diluted in Opti-MEM, Life Technologies). Cells were maintained in the HEK-293T media described above. After 24 h, the media was changed, and the experiment was performed 24 h later (48 h after transfection). Saturation curves using 3H-diprenorphine were performed to determine the expression of KOR, MOR and DOR. We estimated KOR density to be approximately five hundred thousand receptors per cell and the MOR and DOR cellular concentration to be approximately one million receptors per cell (data not shown).
Bioluminescence resonance energy transfer
Bioluminescence resonance energy transfer (BRET) experiments were performed as described previously.7,8,10 Briefly, transfected cells were dissociated and resuspended in phosphate-buffered saline. Approximately 200,000 cells per well were added to a black-framed, white well 96-well plate (no. 60050; Perkin Elmer; Waltham, MA, USA). The microplate was centrifuged and the cells were resuspended in either phosphate-buffered saline (G-protein activation) or phosphate-buffered saline containing 1 μM forskolin (cAMP accumulation). After 5 min, 5 μM solution of the luciferase substrate coelenterazine H was added to each well. After 5 min, ligands were added and the BRET signal was measured at 5 min on a PHERAstar FS plate reader (BMG Labtech, Cary, NC, USA). The BRET signal was calculated as the ratio of the light emitted by the energy acceptor, mVenus (510–540 nm), over the light emitted by the energy donor, RLuc8 (485 nm). After agonist stimulation, a decrease of the BRET signal was measured, which reflected a change of conformation or dissociation between the different subunits of the G protein. This drug-induced BRET signal was transformed (multiplied by −1) and normalized using the Emax of the full agonists DAMGO (MOR), DPDPE (DOR) or U-50,488 (KOR), which was defined as the 100% maximal response for G-protein activation or as 0% for the maximal inhibition of forskolin-stimulated cAMP accumulation in the cAMP inhibition assay. Dose–response curves were fit using a three-parameter logistic equation in GraphPad Prism 6 (Graphpad Software, La Jolla, CA, USA). All experiments were performed three to six times, and data represent mean±s.e.m. of those independent trials.
Results
On the basis of in vivo pharmacological experiments, tianeptine has been proposed to act as an adenosine A1 receptor (A1R) agonist.11 However, we found that tianeptine had no direct activity at A1R (Supplementary Information). Therefore, in an attempt to elucidate its primary molecular target, we screened tianeptine at a concentration of 10 μM against a broad panel of human brain receptors (>50 receptors, Psychoactive Drug Screening Program, University of North Carolina). Tianeptine bound to hMOR, the only hit identified in the entire panel, with a Ki of 383±183 nM (Figure 1b). We therefore further investigated its activity at this receptor (both human and mouse isoforms), as well as at the related DOR and KOR, which also couple to Gi/o proteins. Using previously reported BRET-based assays, tianeptine showed full agonism at mMOR for G-protein activation (Figure 2a, EC50 (mMOR, G Protein)=641±120 nM), as well as downstream inhibition of cAMP accumulation (Figure 2b, EC50 (mMOR, cAMP inhibition)=1.03±0.10 μM).7, 8, 9, 10 In addition, the tianeptine-induced activation of MOR was blocked by the opioid antagonist naltrexone in a dose-dependent manner (Figure 2c). The measured potency of tianeptine at hMOR was higher than that at mMOR in both G protein and cAMP inhibition (Figure 1b, EC50 (hMOR, G Protein)=194±70 nM; EC50 (hMOR, cAMP Inhibition)=151±45 nM, Supplementary Information).

Tianeptine is a full agonist at μ-opioid receptors (MORs). (a) The mouse MOR was co-expressed with GαoB-RLuc8, β1, and mVenus-γ2 to assay G-protein activation. MOR agonist [D-Ala2, N-Me-Phe4, Gly5-ol]-Enkephalin (DAMGO) was used as a positive control. (b) The mouse MOR was co-expressed with GαoB, β1, γ2 and the bioluminescence resonance energy transfer (BRET) sensor CAMYEL to assay inhibition of forskolin-stimulated cAMP accumulation. (c) The mouse MOR was co-expressed with GαoB, β1, γ2 and the BRET sensor CAMYEL to measure the inhibition of cAMP inhibition by tianeptine and the MOR agonist DAMGO. Naltrexone dose-dependently inhibits cAMP inhibition by both DAMGO and tianeptine with an IC50 of 35.3±2.3 and 23.3±3.9 nM, respectively. Data represent mean±s.e.m. of six independent experiments.
Next, we studied the activity of tianeptine at the DOR. We found that tianeptine binds to hDOR (Figure 1b, Ki>10 μM) and is also a full agonist at mouse DOR, although with an order of magnitude lower potency compared with MOR for both G-protein activation (Figure 3a, EC50 (mDOR, G Protein)=14.5±6.6 μM) and inhibition of cAMP accumulation (Figure 3b, EC50 (mDOR, cAMP inhibition)=9.46±1.34 μM). The DOR antagonist TIPP[psi]12 dose-dependently blocked the tianeptine-induced activation of mDOR (Figure 3c). Interestingly, we also found a difference in the potency of tianeptine between the human and mouse DOR; in this case, tianeptine was less potent at the hDOR (EC50 (hDOR, G protein)=37.4±11.2 μM). Thus, while the selectivity of MOR over DOR is approximately 20-fold in mouse, it is nearly 200-fold in human.

Tianeptine is a full agoinst at δ-opioid receptors (DORs). (a) The mouse DOR was co-expressed with GαoB-RLuc8, β1 and mVenus-γ2 to assay G-protein activation. DOR agonist [D-Pen(2,5)]Enkephalin (DPDPE) was used as a positive control. (b) The mouse DOR was co-expressed with GαoB, β1, γ2 and the bioluminescence resonance energy transfer (BRET) sensor CAMYEL to assay inhibition of forskolin-stimulated cAMP accumulation. (c) The mouse DOR was co-expressed with GαoB, β1, γ2 and the BRET sensor CAMYEL to measure the inhibition of cAMP inhibition by tianeptine and the DOR agonist DPDPE. TIPP-psi dose-dependently inhibits the cAMP inhibition by both DPDPE and tianeptine with an IC50 of 232±159 and 95.4±76.6 nM, respectively. Data represent mean±s.e.m. of three independent experiments.
In order to complete our characterization of tianeptine’s functional activity at the opioid receptors, we assessed its activation of KOR. In both G-protein activation and cAMP inhibition assays, we found tianeptine to be inactive at rat KOR (Figure 4). Tianeptine was inactive at human KOR as well (Supplementary Information). Previous reports have highlighted the antidepressant effects of KOR antagonists.13, 14, 15 In order to eliminate the possibility that tianeptine’s antidepressant effects may in fact result from activity as a KOR antagonist, we tested tianeptine’s ability to dose-dependently block the activation of KOR by control agonist U-50,488. In contrast to the known KOR antagonist, nor-binaltorphimine, tianeptine failed to inhibit the activation of KOR by U-50,488 (Figure 4c). Therefore, it is clear through both binding studies (Figure 1b) and functional assays that tianeptine has no activity at KOR.

Tianeptine has no apparent activity at κ-opioid receptors (KORs). (a) The rat KOR was co-expressed with GαoB-RLuc8, β1 and mVenus-γ2 to assay G-protein activation. KOR agonist U-50,488 was used as a positive control. Tianeptine has no significant agonistic activity at KOR. (b) The rat KOR was co-expressed with GαoB, β1, γ2 and the bioluminescence resonance energy transfer (BRET) sensor CAMYEL to assay inhibition of forskolin-stimulated cAMP accumulation. In this assay, tianeptine also has no significant agonistic activity at KOR. (c) The rat KOR was co-expressed with GαoB, β1, γ2 and the BRET sensor CAMYEL to measure the inhibition of cAMP inhibition by the KOR agonist U-50,488. Tianeptine does not show any antagonistic activity at KOR. Nor-binaltorphimine (nor-BNI) showed an IC50 of 28.1±27.2 nM. Data represent mean±s.e.m. of three independent experiments.
Discussion
We have described the first identification of specific molecular targets for the atypical antidepressant tianeptine, namely MOR and DOR. These are unexpected results as previous mechanistic studies have focused on tianeptine’s modulation of aminergic and glutamatergic neurotransmission.2 With regard to modulation of the glutamatergic system, previous reports have shown that tianeptine does not have appreciable affinity (Ki>10 μM) for NMDA, AMPA and kainate receptors.6 In the present work, we have demonstrated that tianeptine also has no agonist or antagonist activity at metabotropic glutamate receptors (including mGluR1a,2,4,5,6,8 receptors). It is therefore unlikely that tianeptine modulates glutamatergic neurons and synapses via a direct interaction with glutamate receptors, at least in the concentration range examined in the present study (<10 μM). In contrast, our evidence suggests that tianeptine’s modulation of the glutamatergic system may occur indirectly, via activation of opioid receptor signaling.
In humans, a single dose of tianeptine (12.5 mg) results in ~1 μM maximal concentration of the drug in the plasma, whereas in rodents, standard acute and chronic dosing (10 mg kg−1 per day) leads to plasma concentrations of ~10 μM (in vivo brain concentrations have not been determined; an estimate in ex vivo tissue is low micromolar).16,17 Therefore, the in vivo concentration range appears sufficient for the activation of MOR (EC50~0.2–1 μM, Figures 1 and 2), whereas activation of DOR (EC50~12–34 μM) may only become relevant with higher dosing. Although it has been previously shown that DOR agonists have antidepressant-like effects in vivo,18 the evidence that MOR activation may have antidepressant/anxiety effects has only been suggestive.19
Taken together, these results lead us to hypothesize that activation of MOR (or possibly dual activation of MOR and DOR) is the initial molecular event responsible for tianeptine’s modulation of the glutamatergic system and for triggering many of the known acute and chronic effects of this agent, including its antidepressant/anxiety actions. MORs are widely expressed in the hippocampus and are known to modulate glutamatergic neurons and synapses through a number of mechanisms. For example, MOR activation in dentate granule cells is known to decrease protein kinase A activity, which results in a decrease of NMDA receptor phosphorylation and activity.20 This may underlie the corrective effect of tianeptine on increased NMDA receptor signaling in stressed animals.2 In addition, activation of MORs (as well as DORs) in hippocampal inhibitory interneurons decreases their activity, thereby disinhibiting CA1 glutamatergic neurons,21 consistent with reports of tianeptine’s enhancing effects on excitability and synaptic plasticity in CA1.2 We note a striking similarity between these cellular and circuit-level effects of tianeptine and those exerted by direct NMDA receptor antagonists, which also show rapid onset of antidepressant effects.1,22
Other circuit-level effects likely contribute to tianeptine’s modulation of the glutamatergic system and antidepressant action. For example, tianeptine, like known MOR and DOR agonists, increases dopamine release in the nucleus accumbens,23 and dopamine can in turn modulate glutamate release through action at presynaptic dopamine receptors on glutamatergic presynaptic terminals.
Taken together, the pharmacological results and mechanistic hypothesis presented here provide a new perspective for interpreting tianeptine’s extensive clinical and preclinical data. The proposed hypothesis suggests specific experiments to test its validity in different experimental paradigms (for example, MOR knockout animals and MOR pharmacological inhibition). Our data are also in agreement with growing evidence regarding the importance of the opioid system in depression, anxiety and stress-related disorders.18, 19, 24, 25, 26
References
Martinowich K, Jimenez DV, Zarate CA Jr, Manji HK . Rapid antidepressant effects: moving right along. Mol Psychiatry 2013; 18: 856–863.
McEwen BS, Chattarji S, Diamond DM, Jay TM, Reagan LP, Svenningsson P et al. The neurobiological properties of tianeptine (Stablon): from monoamine hypothesis to glutamatergic modulation. Mol Psychiatry 2010; 15: 237–249.
Woo YS, Bahk WM, Jeong JH, Lee SH, Sung HM, Pae CU et al. Tianeptine combination for partial or non-response to selective serotonin re-uptake inhibitor monotherapy. Psychiatry Clin Neurosci 2013; 67: 219–227.
Zoladz PR, Fleshner M, Diamond DM . Differential effectiveness of tianeptine, clonidine and amitriptyline in blocking traumatic memory expression, anxiety and hypertension in an animal model of PTSD. Prog Neuropsychopharmacol Biol Psychiatry 2013; 44: 1–16.
Kole MHP, Swan L, Fuchs E . The antidepressant tianeptine persistently modulates glutamate receptor currents of the hippocampal CA3 commissural associational synapse in chronically stressed rats. Eur J Neurosci 2002; 16: 807–816.
Svenningsson P, Bateup H, Qi H, Takamiya K, Huganir RL, Spedding M et al. Involvement of AMPA receptor phosphorylation in antidepressant actions with special reference to tianeptine. Eur J Neurosci 2007; 26: 3509–3517.
Rives M-L, Rossillo M, Liu-Chen L-Y, Javitch JA . 6'-Guanidinonaltrindole (6'-GNTI) is a G protein-biased κ-opioid receptor agonist that inhibits arrestin recruitment. J Biol Chem 2012; 287: 27050–27054.
Negri A, Rives M-L, Caspers MJ, Prisinzano TE, Javitch JA, Filizola M . Discovery of a novel selective kappa-opioid receptor agonist using crystal structure-based virtual screening. J Chem Inf Model 2013; 53: 521–526.
Jiang LI, Collins J, Davis R, Lin K-M, DeCamp D, Roach T et al. Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. J Biol Chem 2007; 282: 10576–10584.
Vezzi V, Onaran HO, Molinari P, Guerrini R, Balboni G, Calò G et al. Ligands raise the constraint that limits constitutive activation in G protein-coupled opioid receptors. J Biol Chem 2013; 288: 23964–23978.
Uzbay TI, Kayir H, Ceyhan M . Effects of tianeptine on onset time of pentylenetetrazole-induced seizures in mice: possible role of adenosine A1 receptors. Neuropsychopharmacology 2007; 32: 412–416.
Schiller PW, Weltrowska G, Nguyen TM, Wilkes BC, Chung NN, Lemieux C . TIPP[psi]: a highly potent and stable pseudopeptide delta opioid receptor antagonist with extraordinary delta selectivity. J Med Chem 1993; 36: 3182–3187.
Beardsley PM, Howard JL, Shelton KL, Carroll FI . Differential effects of the novel kappa opioid receptor antagonist, JDTic, on reinstatement of cocaine-seeking induced by footshock stressors vs cocaine primes and its antidepressant-like effects in rats. Psychopharmacology (Berl) 2005; 183: 118–126.
Mague SD, Pliakas AM, Todtenkopf MS, Tomasiewicz HC, Zhang Y, Stevens WC Jr et al. Antidepressant-like effects of kappa-opioid receptor antagonists in the forced swim test in rats. J Pharmacol Exp Ther 2003; 305: 323–330.
Carr GV, Bangasser DA, Bethea T, Young M, Valentino RJ, Lucki I . Antidepressant-like effects of kappa-opioid receptor antagonists in Wistar Kyoto rats. Neuropsychopharmacology 2009; 35: 752–763.
Grislain L, Gele P, Bertrand M, Luijten W, Bromet N, Salvadori C et al. The metabolic pathways of tianeptine, a new antidepressant, in healthy volunteers. Drug Metabol Dispos 1990; 18: 804–808.
Couet W, Girault J, Latrille F, Salvadori C, Fourtillan JB . Kinetic profiles of tianeptine and its MC5 metabolite in plasma, blood and brain after single and chronic intraperitoneal administration in the rat. Eur J Drug Metab Pharmacokinet 1990; 15: 69–74.
Jutkiewicz EM . The antidepressant-like effects of delta-opioid receptor agonists. Mol Interv 2006; 6: 162–169.
Besson A, Privat AM, Eschalier A, Fialip J . Effects of morphine, naloxone and their interaction in the learned-helplessness paradigm in rats. Psychopharmacology (Berl) 1996; 123: 71–78.
Xie CW, Lewis DV . Involvement of cAMP-dependent protein kinase in mu-opioid modulation of NMDA-mediated synaptic currents. J Neurophysiol 1997; 78: 759–766.
Svoboda KR, Adams CE, Lupica CR . Opioid receptor subtype expression defines morphologically distinct classes of hippocampal interneurons. J Neurosci 1999; 19: 85–95.
Duman RS, Li N, Liu R-J, Duric V, Aghajanian G . Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology 2012; 62: 35–41.
Invernizzi R, Pozzi L, Garattini S, Samanin R . Tianeptine increases the extracellular concentrations of dopamine in the nucleus accumbens by a serotonin-independent mechanism. Neuropharmacology 1992; 31: 221–227.
Colasanti A, Rabiner EA, Lingford-Hughes A, Nutt DJ . Opioids and anxiety. J Psychopharmacol 2011; 25: 1415–1433.
Devoize JL, Rigal F, Eschalier A, Trolese J-F, Renoux M . Influence of naloxone on antidepressant drug effects in the forced swimming test in mice. Psychopharmacology (Berl) 1984; 84: 71–75.
Onali P, Dedoni S, Olianas MC . Direct agonist activity of tricyclic antidepressants at distinct opioid receptor subtypes. J Pharmacol Exp Ther 2010; 332: 255–265.
Acknowledgements
We would like to thank Professor Bryan L Roth and the National Institute of Mental Health’s Psychoactive Drug Screening Program for their assistance in determining the binding affinities of tianeptine at the opioid receptors and for screening tianeptine at 51 central nervous system receptors. We would also like to thank Prashant Donthamsetti for making the mVenus-γ2 construct and Professor Nevin Lambert for helpful discussion.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Gassaway, M., Rives, ML., Kruegel, A. et al. The atypical antidepressant and neurorestorative agent tianeptine is a μ-opioid receptor agonist. Transl Psychiatry 4, e411 (2014). https://doi.org/10.1038/tp.2014.30
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2014.30
This article is cited by
-
Tianeptine promotes lasting antiallodynic effects in a mouse model of neuropathic pain
Neuropsychopharmacology (2023)
-
The atypical antidepressant tianeptine confers neuroprotection against oxygen–glucose deprivation
European Archives of Psychiatry and Clinical Neuroscience (2023)
-
Mu opioid receptors on hippocampal GABAergic interneurons are critical for the antidepressant effects of tianeptine
Neuropsychopharmacology (2022)
-
Opioid-like adverse effects of tianeptine in male rats and mice
Psychopharmacology (2022)
-
Tianeptine modulates synaptic vesicle dynamics and favors synaptic mitochondria processes in socially isolated rats
Scientific Reports (2021)
