
Abstract
Dysfunctional glutamatergic neurotransmission has been implicated in autism spectrum disorder (ASD). However, relatively few studies have directly measured brain glutamate in ASD adults, or related variation in glutamate to clinical phenotype. We therefore set out to investigate brain glutamate levels in adults with an ASD, comparing these to healthy controls and also comparing results between individuals at different points on the spectrum of symptom severity. We recruited 28 adults with ASD and 14 matched healthy controls. Of those with ASD, 15 fulfilled the ‘narrowly’ defined criteria for typical autism, whereas 13 met the ‘broader phenotype’. We measured the concentration of the combined glutamate and glutamine signal (Glx), and other important metabolites, using proton magnetic resonance spectroscopy in two brain regions implicated in ASD—the basal ganglia (including the head of caudate and the anterior putamen) and the dorsolateral prefrontal cortex—as well as in a parietal cortex ‘control’ region. Individuals with ASD had a significant decrease (P<0.001) in concentration of Glx in the basal ganglia, and this was true in both the ‘narrow’ and ‘broader’ phenotype. Also, within the ASD sample, reduced basal ganglia Glx was significantly correlated with increased impairment in social communication (P=0.013). In addition, there was a significant reduction in the concentration of other metabolites such as choline, creatine (Cr) and N-acetylaspartate (NAA) in the basal ganglia. In the dorsolateral prefrontal cortex, Cr and NAA were reduced (P<0.05), although Glx was not. There were no detectable differences in Glx, or any other metabolite, in the parietal lobe control region. There were no significant between-group differences in age, gender, IQ, voxel composition or data quality. In conclusion, individuals across the spectrum of ASD have regionally specific abnormalities in subcortical glutamatergic neurotransmission that are associated with variation in social development.
Similar content being viewed by others


Introduction
Autism spectrum disorder (ASD) is characterized by deficits in social reciprocity, communication impairments, and restricted, repetitive interests and behaviours.1 Recent research suggests an approximate prevalence of 0.6–1.5% in the general population.2
At present, however, therapeutic options for ASD are limited because the pathophysiology of ASD is unclear, leading to a paucity of treatment targets for the core symptoms. Numerous studies have reported abnormalities in brain anatomy and function of ASD individuals (e.g. see Hallahan et al.3 and Barttfeld et al.4), but the underlying molecular basis of these differences is unknown.
There is, however, emerging evidence suggesting that ASD may be associated with abnormalities in excitatory glutamate and inhibitory γ-amino-butyric acid (GABA) neurotransmission.5 The balanced interaction between glutamate and GABA transmission is essential for regulating cognition, learning, memory and emotional behaviours. An imbalance between glutamate excitation and GABA inhibition, leading to hyperexcitation, has been linked to ASD.6, 7, 8
There is also evidence for an association between ASD and genetic variation in the glutamatergic and GABAergic systems. For example, there are reports of associations between ASD and variants in genes coding for glutamate receptors9, 10 and glutamate transporter proteins,11 although not in all studies.12 Also, recent work on fragile X syndrome, the most common monogenetic syndrome associated with ASD, points to the potential importance of metabotropic glutamate receptors (mGluR1) as possible treatment targets in ASD.13
Unfortunately, it is not possible to quantify glutamate and GABA concentrations in post-mortem studies, because they degrade rapidly after death. Progress can be made, however, as in vivo proton magnetic resonance spectroscopy ([1H]MRS) can be used to quantify a range of neural metabolites, including glutamate and its metabolic product glutamine (Glu+Gln—henceforth abbreviated Glx).
There are six published [1H]MRS studies reported on Glx in ASD. Of these, four investigated children, one reported a widespread decrease in cortical Glx14 and another reported a nonsignificant reduction in Glx in the left thalamic region.15 However, two other studies found no differences in any region studied: one investigated the frontal, temporal and parietal cortex and basal ganglia,16 and the other the frontal cortex and basal ganglia.17
Only two published [1H]MRS studies have measured Glx in adults with ASD. Page et al.18 reported that adults with ASD had a significantly higher concentration of Glx than controls in the right amygdala-hippocampal complex. In contrast, Bernardi et al.19 found a significantly lower Glx in the right anterior cingulate cortex.
These prior investigations suggest that ASD individuals may have differences in brain Glx, but the results are inconsistent. Possible explanations for this mixed picture are that some of these studies investigated relatively small samples, and they examined different age groups and/or brain regions. Also, no study of adults has yet addressed whether any of these putative differences are present across the behavioural spectrum (i.e. in both the ‘core’ disorder, and those with the ‘broader phenotype’).
This is potentially of importance because while, in the past, autism was generally treated as a ‘categorical’ diagnosis, it is now understood to likely cover a spectrum of severity. For example, the biological relatives of people with ASD often show an attenuated ‘broader phenotype’ of mild social, cognitive and neurobiological abnormalities.20
This clinical (and likely aetiological) heterogeneity has led some to suggest that we refer to ‘the autisms’ rather than to ‘autism’ and search for final common pathways through which various causative agents may lead to disorder.21 However, previous [1H]MRS studies of adults with ASD have treated all participants with ASD as a single group (although one study in children did not).22 Thus, it is unclear whether putative abnormalities in Glx are present across the spectrum, that is, whether they are a potential common pathway, and/or relate to particular core symptoms.
Hence, in this study, we used [1H]MRS to investigate differences in brain glutamate and other metabolites in adults with ASD. We compared controls with ASD people diagnosed with the narrow ‘core’ disorder, who scored above cutoff on research diagnostic criteria, and those with a broader phenotype, who only met some of the criteria.
We focused on regions previously implicated in ASD pathology and symptomatology: the basal ganglia and the dorsolateral prefrontal cortex (DLPFC). For example, (1) anatomical and metabolic abnormalities have been reported in both of these interconnected areas in ASD;23, 24 (2) the basal ganglia have been linked with social and emotional differences25 and compulsive and repetitive behaviours26 and (3) the DLPFC has been linked to deficits in executive function27 and theory of mind.28 Hence, we also correlated [1H]MRS measures that differed significantly between groups with scores on the Autism Diagnostic Interview—Revised (ADI-R) interview.
We also included a ‘control’ region, in the medial parietal lobe, which has not been linked to ASD and where no differences were seen in a previous [1H]MRS study.18
Materials and methods
Participants
We recruited 42 adult participants: 28 individuals with ASD and 14 healthy controls matched for age, gender and IQ (see Table 1). All participants had an IQ above 65. We recruited only participants who reported being right-handed, to avoid possible lateralization effects given our use of unilateral [1H]MRS voxels.
The 28 participants in the ASD group were further divided into two subtypes on the basis of their symptom profile. Fifteen were diagnosed with the ‘narrowly defined phenotype’ of autism based on the fact that they met the ADI-R cutoff criteria in all three symptom domains and fulfilled the diagnostic criteria for childhood autism or Asperger’s syndrome according to the ICD-10 Research Classification of Mental and Behavioural Disorders1 (criteria F84.0 and F84.5, respectively). The other 13 individuals were classified as having the ‘broader phenotype’, that is, they did not meet the ADI-R cutoff in one domain (see Table 1), but fulfilled the ICD-10 diagnostic criteria for atypical autism (F84.1).
All individuals with ASD were recruited through London’s Maudsley Hospital Behavioural Genetics Clinic, a specialist diagnostic service.
Potential participants were excluded if they had a comorbid psychiatric or medical disorder affecting brain development (e.g. epilepsy or psychosis), a history of head injury, a genetic disorder associated with ASD, for example, tuberous sclerosis or fragile X syndrome, or an IQ below 65. Participants with ASD suffering from anxiety or depressive disorders were not excluded, given the high frequency of these comorbidities in ASD. According to participant self-report, all participants were medication naive at the time of scanning.
All participants provided written informed consent. Ethical approval for this study was provided by South London and Maudsley/Institute of Psychiatry NHS Research Ethics Committee, study reference 1997/087.
[1H]MRS data acquisition
[1H]MRS data were acquired on a 1.5 T GE HDx magnetic resonance imaging (MRI) scanner (GE Medical Systems, Milwaukee, WI, USA) equipped with TwinSpeed gradients.
The scanning protocol included a structural MRI scan, namely a three-dimensional fast inversion-recovery-prepared gradient echo acquisition (number of slices=146, slice thickness=1.2 mm, inversion time (TI)=300 ms, repetition time (TR)=11 ms, echo time (TE)=5 ms, field of view=310 mm, flip angle=18°, matrix=256 × 160 over a 310 × 194 mm field of view, giving 1.2 × 1.2 × 1.2 mm3 voxels). This structural MRI was used for the localization of the spectroscopy voxels in each participant.
Single voxel [1H]MRS spectra were then acquired, using a point-resolved spectroscopy sequence. Point-resolved spectroscopy parameters were: TR=3000 ms and TE=30 ms. A voxel of interest was positioned in the left basal ganglia (20 × 20 × 15 mm3). This voxel included parts of the head of the caudate, the anterior putamen and the internal capsule. Voxel of interests were also positioned in the left DLPFC (16 × 24 × 20 mm3) and in the left medial parietal lobe (20 × 20 × 20 mm3), using previously described methods.29 See Figure 1 for an illustration of the location of the voxels.

Examples of the location of proton magnetic resonance spectroscopy ([1H]MRS) voxels. Three voxels were positioned in (a) left basal ganglia (20 × 20 × 15 mm3) to include the head of the caudate, putamen and internal capsule, (b) left dorsolateral prefrontal cortex (16 × 24 × 20 mm3) and (c) left medial parietal lobe (20 × 20 × 20 mm3).
Data processing
[1H]MRS spectra were processed using the LCModel software version 6-1-0 (Stephen Provencher Incorporated, Oakville, Canada). LCModel uses a linear combination of model spectra of metabolite solutions in vitro to analyse the major resonances of in vivo spectra. In this case, a basis set of alanine, aspartate, creatine (Cr), γ-aminobutyric acid (GABA), glutamine, glutamate, glycerophosphocholine, mI, lactate, N-acetylaspartate (NAA), N-acetyl-aspartylglutamate, scyllo-inositol and taurine, together with a baseline function, were used for the analysis. Each spectrum was reviewed to ensure adequate signal-to-noise ratio, as well as the absence of artefacts. Note that the NAA resonance at 2 p.p.m. contains both NAA and N-acetyl-aspartylglutamate; we report here results reflecting the combination of NAA+N-acetyl-aspartylglutamate, and use the term NAA for brevity.
Calculation of absolute metabolite concentrations
Metabolite concentrations for NAA, Cr, Glx and choline (Cho) were calculated, in institutional units, as follows. The raw metabolite estimates (LCModel output) were first corrected by reference to calibration data from a phantom, containing an aqueous solution of known NAA concentration. One phantom [1H]MRS spectrum was acquired at the end of each scanning session. The amplitude of the phantom NAA peak was used to derive a correction factor, by which all metabolite values for the scan were multiplied.
Furthermore, partial volume effects (group differences in proportions of gray matter, white matter and cerebrospinal fluid, CSF, in the [1H]MRS voxels) are a potential confound in spectroscopy. This could be especially relevant to the present investigation, given previously reported volumetric differences between ASD individuals and controls, for example, in the basal ganglia.23, 26
Therefore, to guard against such confounds, we determined the percentage of gray matter, white matter and CSF within each [1H]MRS voxel for each participant. We first segmented the T1-weighted structural MRI using an automated procedure, spm_segment, part of the Statistical Parametric Mapping software package (SPM2; http://www.fil.ion.ucl.ac.uk/spm/software/spm2/; Wellcome Trust Centre for Neuroimaging, London, UK).
The position of each individual [1H]MRS voxel relative to the corresponding structural was determined, using positional coordinates embedded in the raw spectra data files. The % grey, white and CSF composition of each voxel was then calculated automatically using in-house software. Finally, all metabolite concentrations were corrected for the amount of CSF in the voxel—under the assumption that CSF only contains negligible quantities of the metabolites of interest—by multiplying values by an individual correction factor=1/(1−ProportionCSF), where ProportionCSF could range from 0 to 1, calculated separately for each voxel from each participant. This was applied after correcting for phantom NAA values (see above).
In summary: Metabolitecorrected=Metaboliteraw × (PhantomNAAknown/PhantomNAAobserved) × (1/(1−ProportionCSF)).
Statistical analysis
Age and IQ were compared using one-way analysis of variance (ANOVA) across the three groups (healthy control, broad ASD and narrow ASD).
Differences in mean metabolite concentrations were calculated using a series of one-way ANOVAs, with group as a between-subjects factor. One such ANOVA was performed for each of the four metabolites, in each of the three voxels, a total of 12 ANOVAs. As this procedure involves multiple (12) comparisons, we applied a Bonferroni correction to guard against Type I errors. We report results both before and after this correction.
Planned post hoc independent sample t-tests were then applied, in metabolites where a significant between-group difference was found on the ANOVA, to evaluate differences between (a) narrow ASD and healthy controls, (b) broader ASD and healthy controls and (c) between the two ASD groups (narrow vs broad).
We examined possible correlations between concentrations of metabolites that differed significantly from controls in these t-tests against ADI-R domain scores, across the whole combined ASD group, using Pearson’s product–moment correlation coefficients.
All analyses were performed using SPSS 15.0 software (SPSS, Chicago, IL, USA).
Results
Demographics
Groups did (Table 1) not differ significantly in age, full-scale IQ, verbal IQ or performance IQ.
Tissue composition and data quality
Groups did not differ significantly in mean voxel % grey matter, white matter or CSF in any of the three voxels (Table 2). This is unsurprising as, although volumetric differences have been observed in ASD in the basal ganglia26 and cortex,30 these were of small magnitude, and would not be expected to materially affect composition of hand-placed voxels.
To verify that the quality of the [1H]MRS data did not vary between groups, we compared the LCModel 6-1-0 Cramer-Rao Lower Bound estimate standard deviations for each metabolite in each voxel, using a one-way ANOVA across the three groups. This revealed no significant differences (all F(2,42)<2.8, all P>0.07.) See Figure 2 for an example of a [1H]MRS spectrum after model fitting.

Example of a proton magnetic resonance spectroscopy ([1H]MRS) spectrum showing LCModel 6-1-0 fit.
Finally, because of the potential risk of ‘drift’ in extended [1H]MRS investigations (in which metabolite estimates on the same scanner change over long periods of time), we compared the timings of scans across the three groups, in terms of days after the first scan of the series. This revealed no significant difference (one-way ANOVA F(2,44)=0.739, P=0.484). Scan date was also not correlated with the value of any metabolite in any voxel (all Pearson’s r<0.23, all P>0.13).
Metabolite differences
Basal ganglia
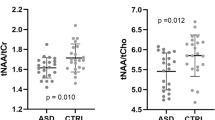
There was a significant group effect in every metabolite concentration we measured in this voxel (ANOVA) (Table 3). Two of these effects—Cr and Glx—survived conservative Bonferroni correction for multiple comparisons over all metabolites and voxels. Post hoc independent sample t-tests showed that Glx concentrations were significantly lower in both the ‘restricted’ and ‘broader’ ASD phenotypes compared with controls (see Figure 3). Both Cr and NAA showed a similar pattern. Cho was also lower in both ASD groups as compared with controls, but this only reached statistical significance in the broader ASD phenotype.

Individual participant data showing basal ganglia glutamate and glutamine (Glx) by group. ASD, autism spectrum disorder.
There were no significant differences between the two ASD subgroups in any metabolite concentration.
To establish whether the finding of lower Glx remained significant after controlling for tissue composition and the other metabolite concentrations, we performed a univariate GLM covarying for basal ganglia grey/white matter, Cr, NAA, Cho, age and IQ. The findings remained significant (F=3.530, P<0.05).
Dorsolateral prefrontal cortex
There was no significant effect of group for Glx or Cho. However, there were significant effects of Cr and NAA, although neither of these differences survived conservative Bonferroni correction. Post hoc tests revealed that Cr was significantly lower in both ASD groups relative to healthy controls, whereas NAA was significantly lower only in the ‘narrow’ ASD phenotype. However, there were no significant differences between the two ASD subgroups.
Parietal region
As predicted, no significant differences were found in any metabolite concentration between any of the groups, even before Bonferroni correction.
Relationship to behavioural variables
Across the combined ASD group (both broader and narrow phenotype), lower basal ganglia concentration of Glx was significantly correlated with worse scores on the ADI-R Communication Scale (i.e. more abnormal Glx concentrations were associated with greater communication impairment (r=−0.465, P=0.013, n=28; see Figure 4). This correlation was specific to this metabolite and this symptom domain: no correlations were seen in other metabolites or domains.

Association between basal ganglia glutamate and glutamine (Glx) and Autism Diagnostic Interview—Revised (ADI-R) Communication Subscale Score in individuals with autism spectrum disorder (ASD).
We further examined this association within each group separately. There was no significant correlation in the narrowly defined autism group (r=−0.224, P=0.422), but there was in those with the broader phenotype (r=−0.805, P=0.001).
Discussion
We found that adults with ASD have a significantly reduced Glx concentration in the basal ganglia as compared with controls. Our preliminary evidence further suggests that that this reduction was (1) regionally specific, that is, there were no significant differences in the other cortical regions we examined; (2) a potential final common pathway in ASD, as it was present in both ‘narrow’ and ‘broadly’ defined ASD; and (3) was associated with some aspects of clinical variation (social communication).
We suggest that it is unlikely that our findings can be fully explained by potential confounds, such as differences in voxel tissue composition, age, IQ or medication. There were no significant between-group differences in voxel grey matter, white matter and CSF, and all metabolite values were corrected for CSF %. Demographic variables such as age, gender and IQ were not different between the groups, all participants were right handed and all of the individuals we studied were psychotropic medication naive according to self-report.
This is the first [1H]MRS study to report on Glx in the basal ganglia of adults with ASD. Our finding of reduced Glx in the basal ganglia (predominantly the lentiform nuclei) agrees with studies finding reductions of Glx in the cingulate cortex and the thalamus,19 but contrasts with our previous finding of increased Glx in the amygdala-hippocampal cortex in adults with ASD.18 Also, similar to prior studies, we found no differences in the parietal cortex.18
Taken together, these results demonstrate that, rather than being a ‘global’ neurobiological abnormality, Glx changes seen in ASD are highly regionally specific, suggesting that the underlying neurobiological cause(s) are also localized. Reductions in Glx could result simply from a local reduced density of glutamatergic synapses and neurons, such as reduced storage capacity and turnover, but could also be the product of alterations in glutamate and glutamine metabolism.
In neurons, glutamate is synthesized from glutamine via glutaminase, but after release in the synapse, glutamate is converted back into glutamine in glial cells, by glutamine synthetase. Glutamate is also converted to GABA by the neuronal enzyme glutamate decarboxylase (GAD).5
Alterations in GAD expression could be a potential explanation for the fact that we observed reduced Glx in the basal ganglia. This hypothesis would also help to reconcile these results with the suggestion that individuals with ASD have an inhibitory signalling deficit and an increased ratio of excitatory glutamate to inhibitory GABA transmission.8, 31 It is possible that, while the Glx signal was reduced, the ratio between glutamate and GABA was still increased. Reduced glutamate would be expected to lead to a corresponding reduction in GABA synthesis, as glutamate is the precursor of GABA. If GAD activity were reduced, one would expect a lower GABA:Glx ratio.
Unfortunately, there have not yet been any studies examining GAD expression in the basal ganglia in ASD, but studies of other brain regions have shown regionally specific differences associated with the disorder. For example, GAD has been reported to be decreased in cerebellar Purkinje cells,32 but increased in cerebellar interneurons,33 in ASD.
A further possibility is that the observed differences in Glx are secondary to alterations in other neurotransmitter systems. For example, the basal ganglia are densely innervated by serotonergic projections, which exert complex modulatory effects on glutamate and GABA release.34 We and others reported reduced density of cortical 5HT2A (serotonin 2A) receptors and serotonin transporter in the cortex and midbrain,35, 36, 37 (although see Girgis et al.38) the same pattern has been found in the parents of children with ASD.39 Further, some have reported that children with ASD have significant differences in serotonin synthesis.40 Hence, it could be that serotonergic abnormalities underlie the differences in Glx we observed—either indirectly via influences on neurodevelopment or through direct action on glutamate metabolism.
Also, in the context of previous findings, our results suggest that within ASD age may be an important moderator of both cortical and subcortical differences in brain Glx. Specifically, prior [1H]MRS studies in children with ASD reported widespread decreases in cortical Glx14—but no differences in the basal ganglia.16, 17 This is the opposite of the pattern we observed in adults, namely no differences in the cortical regions (DLPFC and parietal lobe), but a reduction in the basal ganglia. This is consistent with the idea of autism as a disorder of brain maturation.41 We were unable to address age effects directly, as this study did not include children—but this is a focus of our ongoing studies.
The correlation between basal ganglia (Glx) and the severity of social communication impairments in ASD is consistent with the known involvement of this area in various aspects of language and communication. For example, functional imaging studies demonstrated that the caudate and putamen are involved in ‘higher level’ aspects of language, such as inferring with the implied as opposed to literal meaning of sentences,42 and resolving ambiguous sentences—functions that are characteristically impaired in ASD. This correlation was specific to the basal ganglia, however, with no significant correlation seen in the DLPFC, an area also known to be involved in communication and Theory of Mind.28 One explanation for this could be that alterations in neurotransmitters other than glutamate are responsible for DLPFC dysfunction in autism; serotonin is one possibility, as a previous study found that lower cortical 5HT2A receptor binding is associated with communication impairments in ASD.37
We are only able to report a correlation between ASD in adults (and in particular, Social Communication symptoms as measured by the ADI-R) and reduced basal ganglia Glx levels. Hence, we cannot be certain whether the differences in Glx are the cause of the ASD symptoms. It is possible that they represent a downstream effect of the symptoms if, for example, the lack of social interaction or high-level language use engaged in by people with ASD led, over time, to neuroplastic changes in corticobasal circuits. However, we do not believe that this can explain all our results, because the ADI is a measure childhood (developmental) symptoms in the first years of life.
In addition to the findings related to glutamate, discussed above, we also observed reduced levels of Cho, Cr and NAA in the basal ganglia, and of Cr and NAA in the DLPFC, in individuals with ASD, although only the basal ganglia Cr difference survived conservative Bonferroni correction. The finding of reduced Cr and NAA in both the basal ganglia and in the DLPFC is consistent with previous [1H]MRS studies in this area in ASD.14, 15, 22 However, we have previously29 reported increased NAA, Cr and Cho in the medial prefrontal cortex of adults with Asperger’s syndrome, underlining that the differences seen are regionally specific. Also in line with prior work,18, 29 we did not observe any significant metabolite differences in the parietal cortex control region. This confirms that the effects observed in the basal ganglia and the DLPFC are regionally specific.
As both Cr and NAA are involved in neuronal energy metabolism, our finding of local reductions in these metabolites in the basal ganglia and the DLPFC may indicate either metabolic dysfunction in these areas or a reduced density of metabolically active neurons. If the latter, this may also explain the observed reduction in Cho in the basal ganglia (as this is a component of cell membranes).
Another important implication of our finding of reduced Cr in ASD in the basal ganglia and DLPFC is that it may be invalid to express the concentration of metabolites such as NAA and Glx as ratios to Cr in the same voxel. This approach is commonly used in [1H]MRS studies,22, 43 as it is widely assumed that Cr is constant; our findings suggest the this is not true in adults with ASD.
However, our study does have a number of limitations. We obtained [1H]MRS data on a 1.5 T MRI scanner. At 1.5 T, it is not possible to distinguish between the compounds that contribute to the ‘Glx’ signal, that is, glutamate and glutamine. Future studies at 3 T or higher are needed to distinguish these compounds, but previous studies have cautiously attributed reductions in Glx to glutamate, as glutamate constitutes the most abundant central neurotransmitter.14, 15, 18
Another limitation is that we only recruited people with a normal or above-normal IQ, and excluded those with a history of epilepsy or seizures. This served to increase the homogeneity of the sample and ensure that any differences observed were associated with symptoms of ASD per se, but it means that the results may not be representative of the entire ASD population, as many people with ASD also suffer from a below-normal IQ and/or epilepsy.1 Future studies should examine this population.
In summary, we found preliminary evidence that adults with ASD (both narrowly and broadly defined) have significant differences in brain glutamate and/or glutamine metabolism. This may be a final ‘common pathway’ in the disorder, and underpin some clinical symptoms. Further work is required to determine the cause(s) of this putative abnormality.
References
World Health Organization. ICD-10 International Statistical Classification of Diseases and Related Health Problems 10th edn WHO: Geneva, Switzerland, 1993.
Baron-Cohen S, Scott FJ, Allison C, Williams J, Bolton P, Matthews FE et al. Prevalence of autism-spectrum conditions: UK school-based population study. Br J Psychiatry 2009; 194: 500–509.
Hallahan B, Daly EM, McAlonan G, Loth E, Toal F, O’Brien F et al. Brain morphometry volume in autistic spectrum disorder: a magnetic resonance imaging study of adults. Psychol Med 2009; 39: 337–346.
Barttfeld P, Wicker B, Cukier S, Navarta S, Lew S, Sigman M . A big-world network in ASD: dynamical connectivity analysis reflects a deficit in long-range connections and an excess of short-range connections. Neuropsychologia 2011; 49: 254–263.
Pizzarelli R, Cherubini E . Alterations of GABAergic signaling in autism spectrum disorders. Neural Plast 2011; 2011: 297153.
Coghlan S, Horder J, Inkster B, Mendez MA, Murphy DG, Nutt DJ . GABA system dysfunction in autism and related disorders: from synapse to symptoms. Neurosci Biobehav Rev 2012; 36: 2044–2055.
Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 2010; 468: 263–269.
Gogolla N, Leblanc JJ, Quast KB, Sudhof T, Fagiolini M, Hensch TK . Common circuit defect of excitatory–inhibitory balance in mouse models of autism. J Neurodev Disord 2009; 1: 172–181.
Hussman JP, Chung RH, Griswold AJ, Jaworski JM, Salyakina D, Ma D et al. A noise-reduction GWAS analysis implicates altered regulation of neurite outgrowth and guidance in autism. Mol Autism 2011; 2: 1.
Jamain S, Betancur C, Quach H, Philippe A, Fellous M, Giros B et al. Linkage and association of the glutamate receptor 6 gene with autism. Mol Psychiatry 2002; 7: 302–310.
Jacob S, Brune CW, Badner JA, Ernstrom K, Courchesne E, Lord C et al. Family-based association testing of glutamate transporter genes in autism. Psychiatr Genet 2010; 21: 212–213.
Martin ER, Menold MM, Wolpert CM, Bass MP, Donnelly SL, Ravan SA et al. Analysis of linkage disequilibrium in gamma-aminobutyric acid receptor subunit genes in autistic disorder. Am J Med Genet 2000; 96: 43–48.
Dolen G, Bear MF . Fragile X syndrome and autism: from disease model to therapeutic targets. J Neurodev Disord 2009; 1: 133–140.
DeVito TJ, Drost DJ, Neufeld RW, Rajakumar N, Pavlosky W, Williamson P et al. Evidence for cortical dysfunction in autism: a proton magnetic resonance spectroscopic imaging study. Biol Psychiatry 2007; 61: 465–473.
Hardan AY, Minshew NJ, Melhem NM, Srihari S, Jo B, Bansal R et al. An MRI and proton spectroscopy study of the thalamus in children with autism. Psychiatry Res 2008; 163: 97–105.
Friedman SD, Shaw DW, Artru AA, Dawson G, Petropoulos H, Dager SR . Gray and white matter brain chemistry in young children with autism. Arch Gen Psychiatry 2006; 63: 786–794.
Harada M, Taki MM, Nose A, Kubo H, Mori K, Nishitani H et al. Non-invasive evaluation of the GABAergic/glutamatergic system in autistic patients observed by MEGA-Editing Proton MR Spectroscopy using a Clinical 3 Tesla Instrument. J Autism Dev Disord 2011; 41: 447–454.
Page LA, Daly E, Schmitz N, Simmons A, Toal F, Deeley Q et al. In vivo1H-magnetic resonance spectroscopy study of amygdala-hippocampal and parietal regions in autism. Am J Psychiatry 2006; 163: 2189–2192.
Bernardi S, Anagnostou E, Shen J, Kolevzon A, Buxbaum JD, Hollander E et al. In vivo (1)H-magnetic resonance spectroscopy study of the attentional networks in autism. Brain Res 2011; 1380: 198–205.
Le Couteur A, Bailey A, Goode S, Pickles A, Robertson S, Gottesman I et al. A broader phenotype of autism: the clinical spectrum in twins. J Child Psychol Psychiatry 1996; 37: 785–801.
Geschwind DH, Levitt P . Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol 2007; 17: 103–111.
Endo T, Shioiri T, Kitamura H, Kimura T, Endo S, Masuzawa N et al. Altered chemical metabolites in the amygdala–hippocampus region contribute to autistic symptoms of autism spectrum disorders. Biol Psychiatry 2007; 62: 1030–1037.
Haznedar MM, Buchsbaum MS, Hazlett EA, LiCalzi EM, Cartwright C, Hollander E . Volumetric analysis and three-dimensional glucose metabolic mapping of the striatum and thalamus in patients with autism spectrum disorders. Am J Psychiatry 2006; 163: 1252–1263.
Schmitz N, Daly E, Murphy D . Frontal anatomy and reaction time in autism. Neurosci Lett 2007; 412: 12–17.
Phillips ML, Ladouceur CD, Drevets WC . A neural model of voluntary and automatic emotion regulation: implications for understanding the pathophysiology and neurodevelopment of bipolar disorder. Mol Psychiatry 2008; 13: 833–857.
Hollander E, Anagnostou E, Chaplin W, Esposito K, Haznedar MM, Licalzi E et al. Striatal volume on magnetic resonance imaging and repetitive behaviors in autism. Biol Psychiatry 2005; 58: 226–232.
Alexander GE, DeLong MR, Strick PL . Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu Rev Neurosci 1986; 9: 357–381.
Kalbe E, Schlegel M, Sack AT, Nowak DA, Dafotakis M, Bangard C et al. Dissociating cognitive from affective theory of mind: a TMS study. Cortex 2010; 46: 769–780.
Murphy DG, Critchley HD, Schmitz N, McAlonan G, Van Amelsvoort T, Robertson D et al. Asperger syndrome: a proton magnetic resonance spectroscopy study of brain. Arch Gen Psychiatry 2002; 59: 885–891.
McAlonan GM, Cheung V, Cheung C, Suckling J, Lam GY, Tai KS et al. Mapping the brain in autism. A voxel-based MRI study of volumetric differences and intercorrelations in autism. Brain 2005; 128: 268–276.
Hampson DR, Adusei DC, Pacey LK . The neurochemical basis for the treatment of autism spectrum disorders and fragile X syndrome. Biochem Pharmacol 2011; 81: 1078–1086.
Yip J, Soghomonian JJ, Blatt GJ . Decreased GAD67 mRNA levels in cerebellar Purkinje cells in autism: pathophysiological implications. Acta Neuropathol 2007; 113: 559–568.
Yip J, Soghomonian JJ, Blatt GJ . Increased GAD67 mRNA expression in cerebellar interneurons in autism: implications for Purkinje cell dysfunction. J Neurosci Res 2008; 86: 525–530.
Benarroch EE . Serotonergic modulation of basal ganglia circuits: complexity and therapeutic opportunities. Neurology 2009; 73: 880–886.
Nakamura K, Sekine Y, Ouchi Y, Tsujii M, Yoshikawa E, Futatsubashi M et al. Brain serotonin and dopamine transporter bindings in adults with high-functioning autism. Arch Gen Psychiatry 2010; 67: 59–68.
Makkonen I, Riikonen R, Kokki H, Airaksinen MM, Kuikka JT . Serotonin and dopamine transporter binding in children with autism determined by SPECT. Dev Med Child Neurol 2008; 50: 593–597.
Murphy DG, Daly E, Schmitz N, Toal F, Murphy K, Curran S et al. Cortical serotonin 5-HT2A receptor binding and social communication in adults with Asperger’s syndrome: an in vivo SPECT study. Am J Psychiatry 2006; 163: 934–936.
Girgis RR, Slifstein M, Xu X, Frankle WG, Anagnostou E, Wasserman S et al. The 5-HT(2A) receptor and serotonin transporter in Asperger’s disorder: a PET study with [(11)C]MDL 100907 and [(11)C]DASB. Psychiatry Res 2011; 194: 230–234.
Goldberg J, Anderson GM, Zwaigenbaum L, Hall GB, Nahmias C, Thompson A et al. Cortical serotonin type-2 receptor density in parents of children with autism spectrum disorders. J Autism Dev Disord 2009; 39: 97–104.
Chugani DC, Muzik O, Behen M, Rothermel R, Janisse JJ, Lee J et al. Developmental changes in brain serotonin synthesis capacity in autistic and nonautistic children. Ann Neurol 1999; 45: 287–295.
Hazlett HC, Poe MD, Gerig G, Styner M, Chappell C, Smith RG et al. Early brain overgrowth in autism associated with an increase in cortical surface area before age 2 years. Arch Gen Psychiatry 2011; 68: 467–476.
Uchiyama HT, Saito DN, Tanabe HC, Harada T, Seki A, Ohno K et al. Distinction between the literal and intended meanings of sentences: a functional magnetic resonance imaging study of metaphor and sarcasm. Cortex 2011; 48: 563–583.
Smigielska-Kuzia J, Bockowski L, Sobaniec W, Kulak W, Sendrowski K . Amino acid metabolic processes in the temporal lobes assessed by proton magnetic resonance spectroscopy ((1)H MRS) in children with Down syndrome. Pharmacol Rep 2010; 62: 1070–1077.
Acknowledgements
This work was supported by the National Institute for Health Research (NIHR) Biomedical Research Centre (BRC) for Mental Health at the South London and Maudsley NHS Foundation Trust and the Institute of Psychiatry at King’s College, London, UK (Grant Number RP-PG-0606-1045). The NIHR also supported this study via the program grant ‘Across the Divide’. The Wellcome Trust funded JH (Grant code PCFRAAA). The funders had no role in the study design, data collection, analysis or decision to submit for publication. All of this work was carried out at King’s College London, London, UK. Ethical approval for this study was provided by South London and Maudsley/Institute of Psychiatry NHS Research Ethics Committee, study reference 1997/087.
Disclaimer
The corresponding author (JH) had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Previous Presentation: Parts of the present work were presented in the form of a poster at the International Meeting for Autism Research (IMFAR), 7–9 May 2009, Chicago, IL, USA.
Rights and permissions
This work is licensed under a Creative Commons Attribution 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by/3.0/
About this article
Cite this article
Horder, J., Lavender, T., Mendez, M. et al. Reduced subcortical glutamate/glutamine in adults with autism spectrum disorders: a [1H]MRS study. Transl Psychiatry 3, e279 (2013). https://doi.org/10.1038/tp.2013.53
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2013.53
Keywords
This article is cited by
-
Neurometabolite levels in the brains of patients with autism spectrum disorders: A meta-analysis of proton magnetic resonance spectroscopy studies (N = 1501)
Molecular Psychiatry (2023)
-
16p11.2 deletion mice exhibit compromised fronto-temporal connectivity, GABAergic dysfunction, and enhanced attentional ability
Communications Biology (2023)
-
Topic modeling for multi-omic integration in the human gut microbiome and implications for Autism
Scientific Reports (2023)
-
Visual surround suppression at the neural and perceptual levels
Cognitive Neurodynamics (2023)
-
Decreased in vivo glutamate/GABA ratio correlates with the social behavior deficit in a mouse model of autism spectrum disorder
Molecular Brain (2022)
