
Abstract
The genetic basis for bipolar disorder (BPD) is complex with the involvement of multiple genes. As it is well established that cyclic adenosine monophosphate (cAMP) signaling regulates behavior, we tested variants in 29 genes that encode components of this signaling pathway for associations with BPD type I (BPD I) and BPD type II (BPD II). A total of 1172 individuals with BPD I, 516 individuals with BPD II and 1728 controls were analyzed. Single SNP (single-nucleotide polymorphism), haplotype and SNP × SNP interactions were examined for association with BPD. Several statistically significant single-SNP associations were observed between BPD I and variants in the PDE10A gene and between BPD II and variants in the DISC1 and GNAS genes. Haplotype analysis supported the conclusion that variation in these genes is associated with BPD. We followed-up PDE10A’s association with BPD I by sequencing a 23-kb region in 30 subjects homozygous for seven minor allele risk SNPs and discovered eight additional rare variants (minor allele frequency <1%). These single-nucleotide variants were genotyped in 999 BPD cases and 801 controls. We obtained a significant association for these variants in the combined sample using multiple methods for rare variant analysis. After using newly developed methods to account for potential bias from sequencing BPD cases only, the results remained significant. In addition, SNP × SNP interaction studies suggested that variants in several cAMP signaling pathway genes interact to increase the risk of BPD. This report is among the first to use multiple rare variant analysis methods following common tagSNPs associations with BPD.
Similar content being viewed by others


Introduction
Bipolar disorder (BPD) affects ∼2.5% of the United States population aged 18 and older based on data from the population-based National Comorbidity Survey Replication.1 The risk of BPD for a first-degree relative of an affected individual is 9%, and the concordance rate for monozygotic twins is 40–45%.2 Relatives of BPD probands are also at an increased risk of other related psychiatric disorders, such as psychotic, anxiety, substance abuse and impulse control disorders.1, 2, 3, 4 With an overall heritability estimated at 80–90%,2 these and other findings make clear that susceptibility to BPD has a very strong genetic basis that overlaps with the genetic susceptibility for other neuropsychiatric disorders.
The two most common subtypes of BPD are bipolar disorder I (BPD I) and bipolar disorder II (BPD II). The lifetime risks in the US population is 1% for BPD I and 1.1% for BPD II.1 The clinical course of BPD I and BPD II differ in that BPD I patients exhibit one or more manic episodes while BPD II patients are characterized by recurrent depressive episodes.2, 3 Recurrence of manic/depressive episodes and associated disabilities is common5, 6, 7 and there exists no curative treatment. Although BPD I and BPD II have been shown to co-segregate within families,2, 8 the diverse clinical symptoms indicate that subtypes of the BPD phenotype exist and/or that phenotypic modifiers are involved.9 The identification of BPD I and BPD II susceptibility genes may lead to more effective targets for therapy.
Several recent genome-wide genetic association studies and meta-analyses2, 10 have jointly analyzed BPD I and BPD II subjects,10, 11, 12, 13, 14, 15, 16 or have focused only on BPD I subjects.17, 18, 19 One motivation for grouping the subtypes is that it remains possible that BPD II is a milder form of BPD I as opposed to being a distinct disorder. However, genetic heterogeneity can reduce the power to detect susceptibility variants when samples of BPD I and BPD II are combined for analysis. For this study, we analyzed BPD I and BPD II separately to increase the likelihood of genetic homogeneity among cases.
A substantial body of evidence indicates a relationship between BPD and cyclic adenosine monophosphate (cAMP) signaling although the specific nature of the relationship remains obscure (reviewed in Gould and Manji20 and Dwivedi and Pandy21). Several studies on peripheral cells or post-mortem brain have reported that basal and receptor-activated adenylyl cyclase (AC) activities are increased in BPD patients.22, 23 Other studies have reported that the mood stabilizers lithium and carbamazepine interact directly or indirectly to attenuate the receptor-activation of ACs,24, 25, 26, 27, 28, 29 consistent with the possibility that an increased AC activity or activation is a biochemical phenotype associated with BPD. Several studies have shown that mRNA levels for Gαs, the stimulatory G-protein that couples with AC, are increased in peripheral cells of BPD patients.30, 31, 32, 33, 34 Post-mortem analyses on brain have demonstrated an increased abundance of the Gαs protein, but not Gαi or Gαo,32, 30 and lithium has been found to decrease the abundance of mRNAs for several G-proteins.35, 36 Lithium may also decrease the coupling of receptors to G-proteins.22, 26, 37, 38, 39 Post-mortem analyses have demonstrated a decreased level of the regulatory subunits for protein kinase A (PKA) in BPD,40 with increased PKA activity observed in lymphocytes and platelets from BPD subjects.41, 42 Some substrates of PKA have been found to be hyperphosphorylated and increased PKA activity and immunoreactivity have been observed in post-mortem brain43, 44 and in peripheral cells41, 45, 46, 47 of BPD patients. Finally, the antidepressant rolipram inhibits cAMP phosphodiesterases (PDE) of the dunce class of PDEs (PDE4),48 implicating this class of enzymes in the general state of depression. PDE4 expression is also altered in both BPD and schizophrenia.49
Overall, the evidence strongly suggests that aberrant cAMP signaling is part of the spectrum of biochemical phenotypes associated with BPD. Because of the well-established roles of cAMP in regulating behavioral processes, we hypothesize that genetic variation in the cAMP pathway genes is associated BPD etiology. To test this hypothesis, and to identify the specific mutations within the signaling system that confer susceptibility to BPD, we focused our association study on the collection of genes comprising the cAMP signaling system, as opposed to testing a variety of genes representing many different signaling systems with unrelated functions in the central nervous system. These analyses detected associations of PDE10A with BPD I and of DISC1 and GNAS with BPD II. In addition, significant associations were observed for interaction of variants in DISC1 and PDE4 for both BPD I and BPD II.
Materials and methods
Study samples
DNA samples were obtained from the Systemic Treatment Enhancement Program for BPD study (STEP-BPD). STEP-BPD was a national, longitudinal study designed to examine the effectiveness of BPD treatments in over 4000 patients with BPD.50 Assignment of diagnosis was based on information derived from clinical assessment and psychiatric diagnostic interviews using the Affective Disorders Evaluation50 and confirmed by the Mini International Neuropsychiatric Interview.51 The affective disorders evaluation includes a modified version of the mood and psychosis modules of the Structured Clinical Interview from the American Psychiatric Association’s, Diagnostic and Statistical Manual of Mental Disorders Fourth Edition (DSM-IV; 1994). The interviews were administered by trained clinical specialists using DSM-IV criteria to establish diagnoses of BPD I, BPD II, BPD NOS, schizoaffective manic or BP type or cyclothymic disorder. Only subjects with BPD I or II diagnoses were included in the analysis. The National Institute of Mental Health-funded genetics repository (STEP-Genetics Repository) that created anonymous immortalized cell lines from participants in the STEP-BPD study provided genomic DNA samples for this study. Genomic DNA from controls selected to be of European-American descent and free of signs of depression was provided by the National Institute of Mental Health Genetic Repository. Controls in the repository were screened with the Composite International Diagnostic Interview to reduce the chances of inadvertently introducing susceptibility alleles for neuropsychiatric disorders into the control population. All subjects involved in both studies gave written, informed consent. As the subject and control DNA samples were both pre-existing and de-identified, this study was considered exempt human research by the Scripps Institutional and Baylor College of Medicine IRBs.
Single-nucleotide polymorphism (SNP) selection and genotyping
We selected 29 candidate genes representing biochemical functions that have been implicated in cAMP signaling. Cross-talk among signaling pathways is extensive in cellular physiology, which blurs their boundaries, so we selected genes that are widely recognized as comprising the conical cAMP signaling pathway so as to limit our set (Table 1). For these genes, tagSNPs were selected within each gene and flanking 10 kb, using HAPMAP data (European-American Utah residents with ancestry from Northern and Western Europe, which were collected by the Center d’Etude du Polymorphisme Humaine (CEU)) using an r2⩾0.8, as a measure of linkage disequilibrium (LD) to capture variation within each gene and a minor allele frequency (MAF) ⩾0.05. A subset of 238 SNPs were selected to control for European substructure from a panel previously shown to distinguish different European populations.52 One-thousand and fifty tagSNPs and 238 admixture SNPs were genotyped in 1742 cases and 1757 controls in addition to 58 duplicate samples using the Illumina Golden Gate array (Illumina, Inc., San Diego, CA, USA). Beadstudio software (Illumina) was used for allele detection and genotype calling.
Data quality control
Data cleaning and analysis were performed using PLINK software.53 Samples missing >2% of their genotypes were excluded because missingness can be indicative of low-quality DNA and remaining genotypes may have high error rates. SNP markers were excluded if they were not successfully genotyped in >95% of the samples, had a MAF <1% or deviated from Hardy–Weinberg equilibrium with a P-value <1 × 10−6 in controls. SNPs with deviation from Hardy–Weinberg equilibrium with a P-value of <1 × 10−3 were flagged for follow-up and were also removed before SNP marker loci were imputed. Samples were also removed if they exhibited excess homozygosity or excess heterozygosity (>4 s.d. from the median). In addition, identity-by-state statistics were calculated between samples to assess duplicates and to evaluate relatedness among individuals included in the study. Multidimensional scaling (MDS) was used to identify outliers using the population substructure SNPs. Samples were clustered with data from HAPMAP,54 which was collected from individuals from CEU, Yoruba in Ibadan, Nigeria (YRI) and Japanese in Tokyo, Japan (JPT) and Han Chinese in Beijing, China (CBT). Samples were removed from further analysis if they deviated by <3 s.d. from the median of the MDS component 1 and MDS component 2 within the CEU cluster. After data cleaning, 1728 controls, 1172 BPD I and 516 BPD II remained available for analysis.
To evaluate potential population substructure/admixture, λ, the median of the distribution of test statistics over the median expected under the null of no association55 was calculated before and after sex, age and the first MDS component were included in the logistic regression model to control for potential confounding.
Association testing
PLINK was used to perform logistic regression analyses under an additive model and was used to test for associations between single tagSNPs and BPD I and BPD II.53 Haplotype analysis was performed using GenABEL56 and Haplo Stats57 using a sliding window of three SNPs. GenABEL was used to evaluate two-way interactions between tagSNPs in the cAMP signaling pathway. Findings were assessed for confounding with age and sex. The first MDS component calculated from the admixture SNPs for BPD I and for the analysis of BPD II sex was also included in the model to control for population substructure.
Imputation
MACH was used to impute additional SNPs that were not genotyped in genes within the cAMP pathway.58, 59 It was not possible to impute additional SNPs for PRKAR2B and PPP1CA (Table 1) because of the limited number of genotyped SNPs. CEU phase haplotypes from HAPMAP phases 1 and 260 were used as templates to infer SNP markers that were not genotyped. The imputed SNPs with Rsq scores >0.8 were analyzed in PLINK using the probability of each genotype in the analysis, which were analyzed as a dosage.
Single-nucelotide variant discovery
Thirty BPD I patients homozygous for the PDE10A region 1 risk haplotype were randomly selected for single-nucleotide variant (SNV) variant discovery. An ∼23 kb region (chr6: 166063714–166040354) was amplified as three long-range PCR products using the Takara LA polymerase (Fisher Scientific, Pittsburgh, PA, USA). Nested PCR products ranging in size from 2–3 kb were amplified from the long-range PCR products using the Roche high fidelity and GC rich polymerase systems (Branchburg, NJ, USA). The PCR products were then sequenced using Sanger methodology (Genewiz, South Plainfield, NJ, USA), analyzed for variants using the Sequencher v.5 software (Gene Codes Corporation, Ann Arbor, MI, USA), and compared with a reference sequence (UCSC genome browser, Feb 2009 GRCh37/hg19 build). All coding exons of PDE10A isoforms 1 and 2 (Genbank NM001130690.1 and NM 006661.2) were also sequenced and analyzed to rule out any obvious non-synonomous amino acid or splice site changes that could be disease associated.
SNV genotyping and analysis
All variants from the reference sequence were investigated using dbSNP build 135. Any novel variants, not reported in dbSNP, were analyzed using Taqman SNP genotyping assays (Applied Biosystems, Carlsbad, CA, USA). The following numbers of randomly chosen individuals were analyzed for SNV allele frequencies: 807 BD I, 192 BD II and 801 controls. Taqman assays were performed in a 384-well plate format according to manufacturer’s instructions on a 7900HT Fast real-time PCR instrument (Applied Biosystems).
Rare variant analysis was restricted to those SNVs with MAF <1%. We previously demonstrated that directly combining sequence and genotype data can lead to inflated type I errors in two-stage association studies.61 This experimental design involves sequencing a portion of the cases to discover variants followed by genotyping the identified variants in the remaining sample. We therefore developed a likelihood-based method named SEQCHIP, for integrating sequencing and genotype data from two-stage association studies. It corrects for the genotypes of sequenced BPD I and BPD II cases, so that the corrected sequenced genotypes follow the same distribution as that of the genotyped samples.62 The corrected genotypes can then be analyzed by existing rare variant tests, which were shown to have correct type I errors.62 More specifically, the association analyses described here utilized the ANRV (aggregated number of rare variants),63 the WSS (weighted sum statistics)64 and the VT (variable threshold)65 tests.
Results
Nine hundred and eighty-eight tagSNPs that met quality control criteria within 29 candidate genes (Table 1) were tested for associations using 1172 BPD I cases, 516 BPD II cases and 1728 controls. There was evidence of population substructure in the sample comparing the controls to the BPD cases (λ=1.11). The addition of one MDS component calculated from the admixture SNPs decreased λ to 1.03.
None of the association tests of the tagSNPs with either BPD I or BPD II reached genome-wide significance (P<5 × 10−8). Association results for tagSNPs with P<0.01 with BPD I and BPD II are shown in Table 2. Those SNPs that displayed a nominal significance level of P<0.05 are shown in Supplementary Table 1. An additional 2675 tagSNPs were imputed in the cAMP signaling pathway genes using the 988 tagSNPs that passed quality control (Table 1). All imputed SNPs were analyzed and results of imputed SNPs with P<0.05 are included in Supplementary Table 2.
Twelve different tagSNPs in the PDE10A gene were associated with BPD I at P<0.01, including rs2983521 with an odds ratio (OR) of 1.3, a 95% confidence interval ranging from 1.1 to 1.5, and a P=9.3 × 10−5 (Table 2). Seven of the PDE10A tagSNPs, such as rs1033701 (OR=1.2, P=5.4 × 10−4), that associated with BPD I with ORs ranging from 1.2 to 1.3 (P<0.01) were found to be in moderate LD (r2>0.3). An additional 333 SNPs were imputed in PDE10A (Figure 1, panel a). Our analyses of these imputed SNPs indicated that an additional seven SNPs were associated with BPD I at P<0.01 (Supplementary Table 2). One example is rs2983516, with OR of 1.3 and P=5.4 × 10−5.

PDE10A association with bipolar disorder I (BPD I). The points in panel (a) represent genotyped (black circles) and imputed (open triangles) single-nucleotide polymorphisms (SNPs) and their probability of association with BPD I. Panel (b) illustrates probability as a sequential sliding window with three SNPs per window for the genotyped SNPs (empirical haplotype) and all SNPs (haplotype). The results were adjusted for age and population substructure. Panel (c) shows the structure of the PDE10A gene aligned with panels A and B with exons represented by bars. The direction of transcription, 5′→3′ is from right to left in this figure.
The analysis of PDE10A haplotype windows revealed a series of haplotypes that were associated with BPD I (Figure 1, panel b). Two regions in PDE10A (region 1 at 166.1 Mb and region 2 at 165.8 Mb) were associated with BPD I. Region 1 is large non-coding region within intron 1 that may be important for the regulation of PDE10A expression. Because of this possibility and the large number of associated tagSNPs that fell within this region, SNV identification was focused to a 23-kb region on 30 BPD I patients homozygous for the region 1, minor allele risk SNPs (Figure 2).

PDE10A region 1 risk linkage disequilibrium (LD) plot. Shading and values in cells correspond to r2 values between single-nucleotide polymorphism (SNP) pairs. The SNPs within region 1 and with odds ratio (OR) >1.0 were used to define the boundaries for subsequent sequencing.
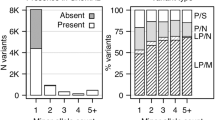
Thirteen SNVs were identified among these BPD I cases. These SNVs were genotyped in 999 cases (807 BPD I and 192 BPD II) in addition to 801 controls. Subsequent analysis was restricted to the eight novel SNVs with a MAF <1% to exclude common polymorphisms (Table 3). Interestingly, the SNVs chr6:166054721 insA and chr6: 166046780_166046778 delATT were found simultaneously in 2 of the 807 BD I cases genotyped. These SNVs are in complete LD in all individuals genotyped. As the newly identified variants are individually rare, they were aggregated and jointly analyzed to determine their collective effect. When only genotyped samples were analyzed, the P-values were P(ANRV)=3.9 × 10−2, P(WSS)=7.1 × 10−2 and P(VT)=8.0 × 10−2. When SEQCHIP was used to integrate both sequence and genotype data, the P-values were P(ANRV)=2.2 × 10−2, P(WSS)=2.0 × 10−3 and P(VT)=3.0 × 10−2. Using SEQCHIP, genotype and sequence data can be more efficiently utilized and a stronger association signal was identified.
One tagSNP in DISC1, rs10495310, was associated with BPD I (Table 2). However, three tagSNPs in DISC1 were associated with BPD II with P<0.01 (Table 2). Two of the DISC1 tagSNPs, rs2812391 and rs10495310, conferred increased risk of BPD II with ORs of 1.2 and 1.4 for each SNP, whereas the third SNP, rs4658954, conferred decreased risk with an OR of 0.8. The tagSNPs rs2812391 and rs10495310 are in mild LD (r2=0.28), whereas both are not in LD with rs4658954 (r2=0.01 and r2=0.02, respectively). Furthermore, an additional 335 SNPs were imputed in DISC1 (Figure 2, panel a) and one SNP, rs3082, was associated with BPD II with an OR of 1.3 (P=5.5 × 10−3). DISC1 haplotype analysis (Figure 2, panel b) indicated that two regions (region 1 at 231.9 Mb and region 2 at 232.1 Mb) were associated with BPD II. The DISC1 region 1 haplotype consisting of rs10495310, rs4658945 and rs16854967 had an empirical global P=1.7 × 10−3 for its association with BPD II. The DISC1 region 2 haplotype consisting of rs12137417, rs9431736 and rs7548491 had an empirical global P=3.9 × 10−2.
Two tagSNPs in GNAS, rs6064714 and rs6026565, were associated with BPD I at P<0.01 (Table 2). However, GNAS appeared to have a stronger association with BPD II in this study with three tagSNPs associated at P<0.01 (Table 2). Two of tagSNPs conferred increased risk (rs35113254 and rs6026565) for BPD II, whereas the third (rs6064714) conferred decreased risk. The SNPs rs35113254 and rs6026565 are in moderate LD (r2=0.35) but are both not in LD with rs6064714 (r2=0.04 and r2=0.01, respectively). The rs35113254 SNP in GNAS was the most significantly associated tagSNP representing the cAMP signaling pathway with P=5.8 × 10−5 with an OR of 1.4 for BPD II. Similarly, an additional 18 SNPs were imputed in GNAS (Figure 3, panel a) and an additional SNP, rs6026560, was associated with BPD II with an OR of 0.7 (P=1.4 × 10−4). Haplotype analysis in GNAS indicated a region around 5.74 Mb that was associated with BPD II (Figure 4, panel b). This region overlaps with the results from imputed and genotyped SNPs.

DISC1 association with bipolar disorder II (BPD II). The points in panel (a) represent genotyped. (black circles) and imputed (open triangles) single-nucleotide polymorphisms (SNPs) and their probability of association with BPD II. Panel (b) illustrates probability as a sequential sliding window with three SNPs per window for the genotyped SNPs (empirical haplotype) and all SNPs (haplotype). The results were adjusted for age and population substructure. Panel (c) shows the structure of the DISC1 gene aligned with panels (a) and (b) with exons represented by bars. The direction of transcription, 5′→3′ is from left to right in this figure.

GNAS association with bipolar disorder II (BPD II). The points in panel (a) represent genotyped. (black circles) and imputed (open triangles) single-nucleotide polymorphisms (SNPs) and their probability of association with BPD II. Panel (b) illustrates probability as a sequential sliding window with three SNPs per window for the genotyped SNPs (empirical haplotype) and all SNPs (haplotype). Panel (c) shows the structure of the GNAS gene aligned with panels (a) and (b) with exons represented by bars. The direction of transcription, 5′→3′ is from right to left in this figure.
Single tagSNPs in PDE4B (rs4384209), PRKACB (rs600674), GNAI2 (rs2282751), PDE4D (rs17741863) and ADCY8 (rs13278912) were also associated with BPD I with P <0.01. Additionally, imputed SNPs in PRKACB (four imputed SNPs), PDE4D (nine imputed SNPs) and ADCY8 (one imputed SNP) were associated with BPD I at P<0.01 (Supplementary Table 2). Numerous haplotype windows had P<0.05, however, only PDE10A, DISC1 and GNAS had notable peaks like those presented in Figures 1, 2, 3, 4 panel (b) for these genes.
Results for all two-way interactions between SNPs in the cAMP signaling pathway with P<0.0001 are presented in Supplementary Table 4. A total of 29 two-way tagSNP interactions with BPD I and 34 two-way tagSNP interactions with BPD II had P<0.0001. For both BPD I and BPD II, several interactions between PDE4 and DISC1 SNPs were significant. PDE4D tagSNP rs4699931 and ADCY8 tagSNP rs928136 interaction was associated with an increased ORinteraction of 3.65 for BPD I (P=1.06 × 10−5), while PDE4B tagSNP rs12404118 and DISC1 tagSNP rs823162 interaction was associated with an increased ORinteraction of 3.53 for BPD II (P=5.47 × 10−5).
Discussion
This study represents the first genetic association study of BPD I and BPD II using dozens of genes comprising the cAMP signaling pathway. Collectively, our results indicate that some of the analyzed SNPs in several genes of the pathway are in LD with causal variants for BPD I and BPD II. Furthermore, the SNV discovery aspect of our study has identified new variants in the PDE10A gene of BPD subjects that may be causally related to the disorder. Our genetic studies are consistent with prior biochemical studies implicating cAMP signaling in BPD, and highlight specific genes and SNVs that could be of major importance for these disorders.
We employed three noteworthy strategies for elucidating the genetic basis for susceptibility to BPD. First, our association study utilized 29 candidate genes representing a single-signaling pathway. The hypothesis underlying this strategy was that if one gene in the signaling pathway is involved, then casual variants in other genes of the same pathway are likely involved as well. We recognize that there exist known and obvious intersections and cross-talk between the cAMP signaling pathway and other signaling pathways, but we selected genes that are widely recognized as comprising the canonical cAMP signaling pathway in order to sample a defined and limited set of genes. A ‘molecular pathway hypothesis’ as contrasted with a ‘single gene hypothesis,’ may account for the genetic heterogeneity of neuropsychiatric disorders in general and the marked difficulty in discovering genetic variants that have prominent roles in these disorders. It can also explain heterogeneity in clinical phenotypes or severity within a disorder, since a variant in an AC gene, for instance, might have largely overlapping but some distinct phenotypic effects compared with a variant in cAMP PDE. The interaction analyses were motivated by the ‘molecular pathway hypothesis’ as functional variants in two molecular functions of a pathway could produce a more pronounced effect than either single functional variant. For instance, the relatively large OR produced by considering an interaction between PDE4D and ADCY8 for BPD I (Supplementary Table 4) could be due to a functional variant that decreases PDE4D activity synergistically interacting with one that increases ADCY8 activity. Similarly, an interaction between variants in PDE4B and DISC1 for BPD II could be because of two separate functional variants that have synergistic effects on signaling. These ideas remain speculative and depend on the sign and magnitude that the functional variants have on the molecular factors involved.
Second, the choice of candidate molecular pathways considered the known and major role of the signaling pathway in the regulation of behavior in model systems. Genetic studies of learning and memory, circadian rhythms and other behaviors in model systems like Drosophila and the mouse have identified hundreds of genes that have behavioral roles. A good example of this is the cAMP PDE encoded by the Drosophila dunce gene, which is involved in learning and memory in Drosophila,66 but whose human counterparts are involved in mood regulation48 and schizoaffective disorders.67 Candidate genes selected from the human homologs of genes with known behavioral roles in model systems should provide an extremely effective filter in gene discovery for neuropsychiatric disorders.
Third, we followed-up the initial association study with SNV discovery in the vicinity of the associated PDE10A tagSNPs that identified additional variants that may be causally involved in BPD. This required the development of a new method for integrating information from sequencing projects in a cost effective way. Using this method, named SEQCHIP, we were able to combine the sequencing data from 30 BPD I patients used for discovery with the data from the follow-up genotyping in a larger subset while accounting for potential bias. This approach was used to identify potentially causal variants that are tagged by the genotyped SNPs. When the data are integrated by SEQCHIP, all rare variant tests have correct type I errors. The genetic information from both sequence and genotype data were efficiently utilized, and therefore, more significant association signals were identified.
Suggestive evidence from other studies is consistent with the possibility that these newly discovered SNVs are involved in BPD. A recent RNA sequencing study employing human prefrontal cortex suggested that two of these SNVs (chr6:166052752 C>G and chr6: 166046780_166046778 delATT) may be associated with novel PDE10A transcripts.68 Another study performing chromatin immunoprecipitation sequencing on neuronal cells from human prefrontal cortex tissue showed that SNVs, chr6:166058713 C>T, chr6:166056798 A>T and chr6: 166046780_166046778 delATT are associated with an enriched H3K4me3 signal, suggesting that they participate in the transcriptional regulation of PDE10A.69 Although this preliminary evidence must be confirmed in human tissue expressing PDE10A, it does suggest that these variants may have a role in PDE10A protein expression.
Our study highlights the association of PDE10A with BPD I. Of related and high interest is the observation that the PDE10A gene resides at 6q26, within one of the four leading linkage regions for BPD.2 The PDEs degrade cAMP and/or cyclic guanosine monophosphate to their 5′-AMP and 5′-GMP counterparts and members of this large enzyme family are known to have broad roles in controlling behavioral output in diverse organisms from flies to humans.66, 70 The family member PDE10A codes for an 89-kDA protein capable of hydrolyzing both cAMP and cyclic guanosine monophosphate that is expressed in the brain and preferentially in the basal ganglia.71, 72, 73, 74 This expression pattern paired with basal ganglia dysfunction in schizophrenia have led to robust efforts in identifying drugs that target PDE10A for the treatment of schizophrenia.75, 76, 77 The association of PDE10A with BPD I expands the potential range over which PDE10A drugs might exhibit efficacy.
The gene disrupted in schizophrenia 1 (DISC1) was first implicated in psychiatric disorders after a t(1;11) translocation was discovered in a large Scottish family cosegregating with schizophrenia, BPD and recurrent major depression.78, 79 The DISC1 region has subsequently been implicated with bipolar spectrum traits in several candidate gene association studies.80, 81, 82, 83, 84, 85 The selection of tagSNPs has varied from study to study so we were unable to compare the precise haplotype windows between this and other studies. In addition, the majority of studies failed to control for population admixture and in most cases termed a P-value of less than nominal significance (P-value <0.05) as significant. Relative to individual tagSNP associations that have been reported, Perlis et al.86 observed an association between rs1934909 and BPD. We observed an association between this tagSNP and BPD II (OR=0.8, P=0.01) but not BPD I cases (Supplementary Table 1). Similarly, Schosser et al.83 observed an increased prevalence of BPD for carriers of the minor allele of rs2492367. We observed an association between this SNP and BPD II but not BPD I (OR=0.8, P=0.03).
It seems highly likely given the complexity of the DISC1 protein and it’s multiple molecular roles87 that there exist multiple functional variants linked to the various tagSNPs used in this and other studies. DISC1 interacts with several key proteins in pathways that have important roles in neuronal migration, dendritic organization and neurogenesis in addition to cAMP signaling.67, 87, 88, 89, 90, 91, 92 The binding sites for interacting proteins are plausible regions for multiple susceptibility regions as are regions of the protein that modify binding. This could underlie the DISC1 and PDE4 tagSNP interactions observed to be significantly associated with BPD I and BPD II in this study. Furthermore, DISC1 variants in intronic regions may influence DISC1 mRNA and subsequent protein levels that also could interfere with key pathways. Indeed, Maeda et al.93 demonstrated reduced DISC1 mRNA levels in lymphoblasts from BPD subjects compared with unaffected family controls. Thus, it is plausible that multiple genetic variants in LD with DISC1 tagSNPs exist that alter protein function or mRNA levels that influence susceptibility to neuropsychiatric disorders. Identifying these variants and characterizing their effect on DISC1 molecular functions may provide useful information for the development of novel therapeutics for bipolar spectrum traits.
The guanine nucleotide-binding protein, α-stimulating polypeptide 1 (GNAS) encodes the Gαs subunit of heterotrimeric G-proteins with Gαs activating AC for increased synthesis of cAMP. Although the exact mode of action of lithium in treating BPD remains elusive, there exists evidence that lithium decreases G-protein subunit mRNA levels.35 Moreover, it has been speculated that lithium functions to stabilize the inactive conformation of G-proteins.94 GNAS variants, like rs6064714 and rs6026565, may be tagging functional variants in BPD I patients that interfere with Gαs’ role in regulating cAMP signaling.
It is of interest that SNPs representing certain genes, such as PDE4B and PDE10A, were found as associated with BPD I but not BPD II. It is possible that this represents an authentic genetic difference between the two BPD subtypes. However, it could also be that the power to detect the association was stronger with the BPD I population, given that the BPD I population was twice the size of the BPD II population. If the data reflect authentic genetic differences between the two subtypes, they could be important to the pharmacogenetics of BPD treatment. For instance, perhaps an altered regulation of PDE10A, specific to BPD I, is most related to full-blown mania associated with BPD I. Drugs with PDE10A action may then have a primary effect on mania.
Overall, this study provides insight into the variation that exists in genes in the cAMP signaling pathway and the association of that variation with BPD I and BPD II. Key regions were identified in PDE10A, GNAS and DISC1 that are associated with BPD I and BPD II. The effect size for these associations was modest, as observed in most other genetic association studies for neuropsychiatric disorders. Our study also illustrates the feasibility of performing large candidate gene or GWAS (genome-wide associated studies) studies followed by targeted direct sequencing to uncover novel, rare SNVs that may be associated with disease.
Accession codes
References
Merikangas KR, Akiskal HS, Angst J, Greenberg PE, Hirschfeld RM, Petukhova M et al. Lifetime and 12-month preva- lence of bipolar spectrum disorder in the national comorbidity survey replication. Arch Gen Psychiatry 2007; 64: 543–552.
Barnett JH, Smoller JW . The genetics of bipolar disorder. Neuroscience 2009; 164: 331–343.
Judd LL, Akiskal HS, Schettler PJ, Coryell W, Maser J, Rice JA et al. The comparative clinical phenotype and long term longitudinal episode course of bipolar I and II: a clinical spectrum or distinct disorders? J Affect Disord 2003; 73: 19–32.
Craddock N, O’Donovan MC, Owen MJ . Psychosis genetics: modeling the relationship between schizophrenia, bipolar disorder, and mixed (or "schizoaffective") psychoses. Schizophr Bull 2009; 35: 482–490.
MacQueen GM, Young LT, Joffe RT . A review of psychosocial outcome in patients with bipolar disorder. Acta Psychiatr Scand 2001; 103: 163–170.
Zarate CA, Tohen M, Land M, Cavanagh S . Functional impairment and cognition in bipolar disorder. Psychiatr Q 2000; 71: 309–329.
Fagiolini A, Kupfer DJ, Masalehdan A, Scott JA, Houck PR, Frank E . Functional impairment in the remission phase of bipolar disorder. Bipolar Disord 2005; 7: 281–285.
McQueen MB, Devlin B, Faraone SV, Nimgaonkar VL, Sklar P, Smoller JW et al. Combined analysis from eleven linkage studies of bipolar disorder provides strong evidence of susceptibility loci on chromosomes 6q and 8q. Am J Hum Genet 2005; 77: 582–595.
Kelsoe JR . Arguments for the genetic basis of the bipolar spectrum. J Affect Disord 2003; 73: 183–197.
Psychiatric GWAS Consortium Bipolar Disorder Working Group. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet 2011; 43: 977–983.
Baum AE, Hamshere M, Green E, Cichon S, Rietschel M, Noethen MM et al. Meta-analysis of two genome-wide association studies of bipolar disorder reveals important points of agreement. Mol Psychiatry 2008; 13: 466–469.
Scotta LJ, Muglia P, Kong XQ, Guan W, Flickinger M, Upmanyu R et al. Genome-wide association and meta-analysis of bipolar disorder in individuals of European ancestry. PNAS 2009; 106: 7501–7506.
Cichon S, Muhleisen TW, Degenhardt FA, Mattheisen M, Miro X, Strohmaier J et al. Genome-wide association study identifies genetic variation in neurocan as a susceptibility factor for bipolar disorder. Am J Hum Genet 2011; 88: 372–381.
Smith EN, Bloss CS, Badner JA, Barrett T, Belmonte PL, Berrettini W et al. Genome-wide association study of bipolar disorder in European American and African American individuals. Mol Psychiatry 2009; 14: 755–763.
Ferreira MAR, O’Donovan MC, Meng YA, Jones IR, Ruderfer DM, Jones L et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet 2008; 40: 1056–1058.
Wellcome Trust Case Control Consortium. Genome-wide association study of 14 000 cases of seven common diseases and 3000 shared controls. Nature 2007; 447: 661–678.
Sklar P, Smoller JW, Fan J, Ferreira MA, Perlis RH, Chambert K et al. Whole-genome association study of bipolar disorder. Mol Psychiatry 2008; 13: 558–569.
Baum AE, Akula N, Cabanero M, Cardona I, Corona W, Klemens B et al. A genome-wide association study implicates diacylglycerol kinase eta (DGKH) and several other genes in the etiology of bipolar disorder. Mol Psychiatry 2008; 13: 197–207.
Lee MT, Chen CH, Lee CS, Chen CC, Chong MY, Ouyang WC et al. Genome-wide association study of bipolar I disorder in the Han Chinese population. Mol Psychiatry 2011; 16: 548–556.
Gould TD, Manji HK . Signaling networks in the pathophysiology and treatment of mood disorders. J Psychosom Res 2002; 53: 687–697.
Dwivedi Y, Pandey GN . Adenylyl cyclase-cyclicAMP signaling in mood disorders: role of the crucial phosphorylating enzyme protein kinase A. Neuropsychiatr Dis Treat 2008; 4: 161–176.
Ebstein RP, Oppenheim G, Ebstein BS, Amiri Z, Stessman J . The cyclic AMP second messenger system in man: the effects of heredity, hormones, drugs, aluminum, age and disease on signal amplification. Prog Neuropsychopharmacol Biol Psychiatry 1986; 10: 323–353.
Extein I, Tallman J, Smith CC, Goodwin FK . Changes in lymphocyte beta-adrenergic receptors in depression and mania. Psychiatry Res 1979; 1: 191–197.
Palmer GC . Interactions of antiepileptic drugs on adenylate cyclase and phosphodiesterases in rat and mouse cerebrum. Exp Neurol 1979; 63: 322–335.
Palmer GC, Jones DJ, Medina MA, Stavinoha WB . Anticonvulsant drug actions on in vitro and in vivo levels of cyclic AMP in the mouse brain. Epilepsia 1979; 20: 95–104.
Risby ED, Hsiao JK, Manji HK, Bitran J, Moses F, Zhou DF et al. The mechanisms of action of lithium. II. Effects on adenylate cyclase activity and beta-adrenergic receptor binding in normal subjects. Arch Gen Psychiatry 1991; 48: 513–524.
Mørk A, Geisler A, Hollund P . Effects of lithium on second messenger systems in the brain. Pharmacol Toxicol 1992; 71 (Suppl 1): 4–17.
Masana MI, Bitran JA, Hsiao JK, Mefford IN, Potter WZ . Lithium effects on noradrenergic-linked adenylate cyclase activity in intact rat brain: an in vivo microdialysis study. Brain Res 1991; 538: 333–336.
Montezinho LP, Mørk A, Duarte CB, Penschuck S, Geraldes CF, Castro MM . Effects of mood stabilizers on the inhibition of adenylate cyclase via dopamine D(2)-like receptors. Bipolar Disord 2007; 9: 290–297.
Young LT, Li PP, Kamble A, Siu KP, Warsh JJ . Mononuclear leukocyte levels of G proteins in depressed patients with bipolar disorder or major depressive disorder. Am J Psychiatry 1994; 151: 594–596.
Spleiss O, van Calker D, Schärer L, Adamovic K, Berger M, Gebicke-Haerter PJ . Abnormal G protein alpha(s) - and alpha(i2)-subunit mRNA expression in bipolar affective disorder. Mol Psychiatry 1998; 3: 512–520.
Warsh JJ, Young LT, Li PP . Guanine nucleotide binding (G) protein disturbances in bipolar affective disorder. In: Manji HK, Bowden CL, Belmaker RH, (eds). Bipolar medications: mechanisms of action 1st edn. American Psychiatric Press: Washington (DC), 2000; 299–329.
Schreiber G, Avissar S, Danon A, Belmaker RH . Hyperfunctional G proteins in mononuclear leukocytes of patients with mania. Biol Psychiatry 1991; 29: 273–280.
Mitchell PB, Manji HK, Chen G, Jolkovsky L, Smith-Jackson E, Denicoff K et al. High levels of Gs alpha in platelets of euthymic patients with bipolar affective disorder. Am J Psychiatry 1997; 154: 218–223.
Jakobsen SN, Wiborg O . Selective effects of long-term lithium and carbamazepine administration on G-protein subunit expression in rat brain. Brain Res 1998; 780: 46–55.
Colin SF, Chang HC, Mollner S, Pfeuffer T, Reed RR, Duman RS et al. Chronic lithium regulates the expression of adenylate cyclase and Gi-protein alpha subunit in rat cerebral cortex. Proc Natl Acad Sci USA 1991; 88: 10634–10637.
Hsiao JK, Manji HK, Chen GA, Bitran JA, Risby ED, Potter WZ . Lithium administration modulates platelet Gi in humans. Life Sci 1992; 50: 227–233.
Lonati-Galligani M, Emrich HM, Raptis C, Pirke KM . Effect of in vivo lithium treatment on (-)isoproterenol-stimulated cAMP accumulation in lymphocytes of healthy subjects and patients with affective psychoses. Pharmacopsychiatry 1989; 22: 241–245.
Garcia-Sevilla JA, Guimon J, Garcia-Vallejo P, Fuster MJ . Biochemical and functional evidence of supersensitive platelet alpha 2-adrenoceptors in major affective disorder. Effect of long-term lithium carbonate treatment. Arch Gen Psychiatry 1986; 43: 51–57.
Rahman S, Li PP, Young LT, Kofman O, Kish SJ, Warsh JJ . Reduced [3H]cyclic AMP binding in postmortem brain from subjects with bipolar affective disorder. J Neurochem 1997; 68: 297–304.
Tardito D, Mori S, Racagni G, Smeraldi E, Zanardi R, Perez J . Protein kinase A activity in platelets from patients with bipolar disorder. J Affect Disord 2003; 76: 249–253.
Karege F, Schwald M, Papadimitriou P, Lachausse C, Cissé M . The cAMP-dependent protein kinase A and brain-derived neurotrophic factor expression in lymphoblast cells of bipolar affective disorder. J Affect Disord 2004; 79: 187–192.
Chang A, Li PP, Warsh JJ . Altered cAMP-dependent protein kinase subunit immunolabeling in post-mortem brain from patients with bipolar affective disorder. J Neurochem 2003; 84: 781–791.
Fields A, Li PP, Kish SJ, Warsh JJ . Increased cyclic AMP-dependent protein kinase activity in postmortem brain from patients with bipolar affective disorder. J Neurochem 1999; 73: 1704–1710.
Karege F, Schwald M, Cisse M . The camp-PKA activity and BDNF expression in lymphoblast cells of bipolar disorder patients. Int J Neuropsychopharmacol 2002; 5 (Suppl 1): S59.
Perez J, Tardito D, Mori S, Racagni G, Smeraldi E, Zanardi R . Abnormalities of cyclic adenosine monophosphate signaling in platelets from untreated patients with bipolar disorder. Arch Gen Psychiatry 1999; 56: 248–253.
Perez J, Tardito D, Racagni G, Smeraldi E, Zanardi R . cAMP signaling pathway in depressed patients with psychotic features. Mol Psychiatry 2002; 7: 208–212.
Henkel-Tigges J, Davis RL . Rat homologs of the Drosophila dunce gene code for cyclic AMP phosphodiesterases sensitive to the antidepressant rolipram. Mole Pharm 1990; 37: 7–10.
Fatemi SH, Reutiman TJ, Folson TD, Lee S . Phosphodiesterase-4A expression is reduced in cerebella of patients with bipolar disorder. Psychiatr Genet 2008; 18: 282–288.
Sachs GS, Thase ME, Otto MW, Bauer M, Miklowitz D, Wisniewski SR et al. Rationale, design, and methods of the systematic treatment enhancement program for bipolar disorder (STEP-BPD). Biol Psychiatry 2003; 53: 1028–1042.
Sheehan DV, Lecrubier Y, Sheehan KH, Amorim P, Janavs J, Weiller E et al. The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry 1998; 59 (Suppl. 20:22-33)Quiz 34–57.
Seldin MF, Shigeta R, Villoslada P, Selmi C, Tuomilehto J, Silva G et al. European population substructure: clustering of northern and southern populations. PLoS Genet 2006; 2: e143.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
International HapMap Consortium. The International HapMap Project. Nature 2003; 426: 789–796.
Devlin B, Roeder K . Genomic control for association studies. Biometrics 1999; 55: 997–1004.
Aulchenko YS, Ripke S, Isaacs A, van Duijn CM . GenABEL: an R library for genome-wide association analysis. Bioinformatics 2007; 23: 1294–1296.
Schaid DJ, Rowland CM, Tines DE, Jacobson RM, Poland GA . Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am J Hum Genet 2002; 70: 425–434.
Li Y, Willer C, Sanna S, Abecasis G . Genotype imputation. Annu Rev Genomics Hum Genet 2009; 10: 387–406.
Li Y, Abecasis G . Mach 1.0: rapid haplotype reconstruction and missing genotype inference. Am J Hum Genet 2006; S79: 2290.
International HapMap Consortium. A haplotype map of the human genome. Nature 2005; 437: 1299–1320.
Li B, Leal SM . Discovery of rare variants via sequencing: implications for the design of complex trait association studies. PLoS Genet 2009; 5: e1000481.
Liu DJ, Leal SM . SEQCHIP: a powerful method to integrate sequence and genotype data for the detection of rare variant associations. Bioinformatics 2012; 28: 1745–1751, in press.
Morris AP, Zeggini E . An evaluation of statistical approaches to rare variant analysis in genetic association studies. Genet Epidemiol 2010; 34: 188–193.
Madsen BE, Browning SRA . Groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet 2009; 5: e1000384.
Price AL, Kryukov GV, de Bakker PI, Purcell SM, Staples J, Wei LJ et al. Pooled association tests for rare variants in exon-resequencing studies. Am J Hum Genet 2010; 86: 832–838.
Davis RL . Olfactory memory formation in Drosophila: from molecular to systems neuroscience. Ann Rev Neurosci 2005; 28: 275–302.
Millar JK, Pickard BS, Mackie S, James R, Christie S, Buchanan SR et al. DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signaling. Science 2005; 310: 1187–1191.
Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D’Souza C, Fouse SD et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010; 466: 253–257.
Cheung I, Shulha HP, Jiang Y, Matevossian A, Wang J, Weng Z et al. Developmental regulation and individual differences of neuronal H3K4me3 epigenomes in the prefrontal cortex. Proc Natl Acad Sci USA 2010; 107: 8824–8829.
Xu Y, Zhang HT, O’Donnell JM . Phosphodiesterases in the central nervous system: implications in mood and cognitive disorders. Handb Exp Pharmacol 2011; 204: 447–485.
Soderling SH, Bayuga SJ, Beavo JA . Isolation and characterization of a dual-substrate phosphodiesterase gene family: PDE10A. Proc Natl Acad Sci USA 1999; 96: 7071–7076.
Loughney K, Snyder PB, Uher L, Rosman GJ, Ferguson K, Florio VA . Isolation and characterization of PDE10A, a novel human 3′, 5′-cyclic nucleotide phosphodiesterase. Gene 1999; 234: 109–117.
Seeger TF, Bartlett B, Coskran TM, Culp JS, James LC, Krull DL et al. Immunohistochemical localization of PDE10A in the rat brain. Brain Res 2003; 985: 113–126.
Fujishige K, Kotera J, Michibata H, Yuasa K, Takebayashi S, Okumura K et al. Cloning and characterization of a novel human phosphodiesterase that hydrolyzes both cAMP and cGMP (PDE10A). J Biol Chem 1999; 274: 18438–18445.
Kehler J, Nielsen J . PDE10A inhibitors: novel therapeutic drugs for schizophrenia. Curr Pharm Des 2011; 17: 137–150.
Grauer SM, Pulito VL, Navarra RL, Kelly MP, Kelley C, Graf R et al. Phosphodiesterase 10A inhibitor activity in preclinical models of the positive, cognitive, and negative symptoms of schizophrenia. J Pharmacol Exp Ther 2009; 331: 574–590.
Schmidt CJ, Chapin DS, Cianfrogna J, Corman ML, Hajos M, Harms FJ et al. Preclinical characterization of selective phosphodiesterase 10A inhibitors: a new therapeutic approach to the treatment of schizophrenia. J Pharmacol Exp Ther 2008; 325: 681–690.
St Clair D, Blackwood D, Muir W, Carothers A, Walker M, Spowart G et al. Association within a family of a balanced autosomal translocation with major mental illness. Lancet 1990; 336: 13–16.
Millar JK, Pickard BS, Mackie S, James R, Christie S, Buchanan SR et al. DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signaling. Science 2005; 310: 1187–1191.
Thomson PA, Wray NR, Millar JK, Evans KL, Hellard SL, Condie A et al. Association between the TRAX/DISC locus and both bipolar disorder and schizophrenia in the Scottish population. Mol Psychiatry 2005; 10: 657–668, 616.
Palo OM, Antila M, Silander K, Hennah W, Kilpinen H, Soronen P et al. Association of distinct allelic haplotypes of DISC1 with psychotic and bipolar spectrum disorders and with underlying cognitive impairments. Hum Mol Genet 2007; 16: 2517–2528.
Hennah W, Varilo T, Kestilä M, Paunio T, Arajärvi R, Haukka J et al. Haplotype transmission analysis provides evidence of association for DISC1 to schizophrenia and suggests sex-dependent effects. Hum Mol Genet 2003; 12: 3151–3159.
Schosser A, Gaysina D, Cohen-Woods S, Chow PC, Martucci L, Craddock N et al. Association of DISC1 and TSNAX genes and affective disorders in the depression case-control (DeCC) and bipolar affective case-control (BACCS) studies. Mol Psychiatry 2009 Available fromhttp://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=19255581.
Hennah W, Tuulio-Henriksson A, Paunio T, Ekelund J, Varilo T, Partonen T et al. A haplotype within the DISC1 gene is associated with visual memory functions in families with a high density of schizophrenia. Mol Psychiatry 2005; 10: 1097–1103.
Hodgkinson CA, Goldman D, Jaeger J, Persaud S, Kane JM, Lipsky RH et al. Disrupted in schizophrenia 1 (DISC1): association with schizophrenia, schizoaffective disorder, and bipolar disorder. Am J Hum Genet 2004; 75: 862–872.
Perlis RH, Purcell S, Fagerness J, Kirby A, Petryshen TL, Fan J et al. Family-based association study of lithium-related and other candidate genes in bipolar disorder. Arch Gen Psychiatry 2008; 65: 53–61.
Porteous DJ, Millar JK, Brandon NJ, Sawa A . DISC1 at 10: connecting psychiatric genetics and neuroscience. Trends Mol Med 2011; 17: 699–706.
Kamiya A, Kubo K, Tomoda T, Takaki M, Youn R, Ozeki Y et al. A schizophrenia-associated mutation of DISC1 perturbs cerebral cortex development. Nat Cell Biol 2005; 7: 1167–1178.
Miyoshi K, Honda A, Baba K, Taniguchi M, Oono K, Fujita T et al. Disrupted-in-schizophrenia 1, a candidate gene for schizophrenia, participates in neurite outgrowth. Mol Psychiatry 2003; 8: 685–694.
Ozeki Y, Tomoda T, Kleiderlein J, Kamiya A, Bord L, Fujii K et al. Disrupted-in-Schizophrenia-1 (DISC-1): mutant truncation prevents binding to NudE-like (NUDEL) and inhibits neurite outgrowth. Proc Natl Acad Sci USA 2003; 100: 289–294.
Sawa A, Snyder SH . Genetics. Two genes link two distinct psychoses. Science 2005; 310: 1128–1129.
Brandon NJ, Handford EJ, Schurov I, Rain J, Pelling M, Duran-Jimeniz B et al. Disrupted in Schizophrenia 1 and Nudel form a neurodevelopmentally regulated protein complex: implications for schizophrenia and other major neurological disorders. Mol Cell Neurosci 2004; 25: 42–55.
Maeda K, Nwulia E, Chang J, Balkissoon R, Ishizuka K, Chen H et al. Differential expression of disrupted-in-schizophrenia (DISC1) in bipolar disorder. Biol Psychiatry 2006; 60: 929–935.
Manji HK, Potter WZ, Lenox RH . Signal transduction pathways. Molecular targets for lithium's actions. Arch Gen Psychiatry 1995; 52: 531–543.
Acknowledgements
This study was supported by grant R01 MH074791 to RLD.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
McDonald, ML., MacMullen, C., Liu, D. et al. Genetic association of cyclic AMP signaling genes with bipolar disorder. Transl Psychiatry 2, e169 (2012). https://doi.org/10.1038/tp.2012.92
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2012.92
Keywords
This article is cited by
-
Lithium rescues dendritic abnormalities in Ank3 deficiency models through the synergic effects of GSK3β and cyclic AMP signaling pathways
Neuropsychopharmacology (2023)
-
Gut microbiome partially mediates and coordinates the effects of genetics on anxiety-like behavior in Collaborative Cross mice
Scientific Reports (2021)
-
Commonality in dysregulated expression of gene sets in cortical brains of individuals with autism, schizophrenia, and bipolar disorder
Translational Psychiatry (2019)
-
Ethanol Exacerbates Manganese-Induced Neurobehavioral Deficits, Striatal Oxidative Stress, and Apoptosis Via Regulation of p53, Caspase-3, and Bax/Bcl-2 Ratio-Dependent Pathway
Biological Trace Element Research (2019)
-
Mitochondrial dysfunction and autism: comprehensive genetic analyses of children with autism and mtDNA deletion
Behavioral and Brain Functions (2018)
