
Abstract
Early-onset major depressive disorder (MDD) is a serious and prevalent psychiatric illness in adolescents and young adults. Current treatments are not optimally effective. Biological markers of early-onset MDD could increase diagnostic specificity, but no such biomarker exists. Our innovative approach to biomarker discovery for early-onset MDD combined results from genome-wide transcriptomic profiles in the blood of two animal models of depression, representing the genetic and the environmental, stress-related, etiology of MDD. We carried out unbiased analyses of this combined set of 26 candidate blood transcriptomic markers in a sample of 15–19-year-old subjects with MDD (N=14) and subjects with no disorder (ND, N=14). A panel of 11 blood markers differentiated participants with early-onset MDD from the ND group. Additionally, a separate but partially overlapping panel of 18 transcripts distinguished subjects with MDD with or without comorbid anxiety. Four transcripts, discovered from the chronic stress animal model, correlated with maltreatment scores in youths. These pilot data suggest that our approach can lead to clinically valid diagnostic panels of blood transcripts for early-onset MDD, which could reduce diagnostic heterogeneity in this population and has the potential to advance individualized treatment strategies.
Similar content being viewed by others


Introduction
Early-onset major depressive disorder (MDD) is a serious psychiatric condition occurring in people under 25 years of age.1 Early onset of MDD predicts greater familial risk, suggesting a substantial genetic etiology.2, 3 Approximately 1% of the population of <12 years has MDD, but rates increase to 17–25% by late adolescence and young adulthood.4, 5 Early-onset MDD carries a poorer prognosis than adult onset, characterized by derailment of normal development and, subsequently, increases in the prevalence of substance use, physical illness, social maladjustment and suicidality.6, 7, 8, 9, 10, 11 This dysfunction persists into adulthood with continued mood disturbance, and often presents comorbid anxiety and substance use disorders.9, 12
Treatments for early-onset MDD exist, but medication side effects are unpredictable and adolescents have lower response rates than adults.13, 14 Unmeasured heterogeneity in the etiology of MDD and the presence of symptom cluster subtypes that require unique treatments may explain these problems.15, 16 Current diagnostic practice is limited to patient self-report and clinician observation, methods that cannot adequately characterize this heterogeneity. Biological markers of different etiologic pathways and/or endophenotypes would provide objective data to augment verbal information and improve the accuracy of diagnosis. The diagnosis and classification of early-onset MDD could lead to a larger repertoire of more effective treatments and enhanced individualized care.
The benefit of having valid, reliable, selective and feasible biomarkers for MDD is clear, but despite 30 years of research, we still do not have them.17 Two approaches have been used to search for blood biomarkers. The first approach screens candidate markers suggested by current knowledge of MDD etiology, such as serum levels of cortisol18 or brain-derived neurotrophic factor.19 The other approach takes an exploratory route through the use of omic technologies such as the proteomic profiling for bipolar disorder20 and transcriptomic profiling for psychosis21 or for mood disorders.22 Each approach has its advantages and challenges, but neither has been successful in identifying biomarkers that survive the tests of replication and selectivity.23 In the present paper, we describe a promising novel approach to discover blood biomarkers, and a pilot study focusing on their validity for distinguishing early-onset MDD from no disorder in youth.
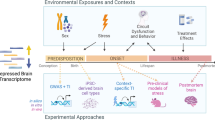
Substantial evidence has accumulated for both the genetic and the environmental stress-related etiologies of MDD. Therefore, in our study we use a theoretical approach that recapitulates these etiologies by utilizing animal models of both. In previous studies, we have shown that the molecular mechanisms through which genetic predisposition and chronic stress precipitate depressive behavior are different.24 Additionally, these molecular pathways can interact, which could hasten the onset of depression or increase the severity. Therefore, if these models are explored in conjunction with one another, a greater part of the spectrum of human MDD, or its endophenotypes, can be accounted for.
The genetic model of depression is based on selective breeding from the Wistar Kyoto (WKY) rat strain, with many behavioral and physiological characteristics mirroring symptoms of patients with MDD.25, 26, 27, 28, 29 The WKY strain is also regarded as the optimal model for juvenile depression, presumably because it is a genetic model and early-onset MDD is associated with a greater familial risk.2, 30 The two substrains of the WKY, identified as WKY More Immobile (WMI, the depression model) and WKY Less Immobile (WLI, the nondepressed control strain), were generated by bidirectional selective breeding from the WKY strain based on depressive behavior in the forced swim test.24, 31 These two inbred strains have maintained clear and significant behavioral differences in measures of depressive behavior over 25 generations; thus, the WMI strain represents a model of genetic, or endogenous, depression. Furthermore, the WMI rats show depressive-like behavior during early adolescence.32 The chronic stress model employed young adult males of four phylogenetically, physiologically and behaviorally different strains of rats as described previously, and half of the animals from each strain were exposed to chronic restraint stress and the other half were not.24 Genetic polymorphisms between the strains ranged from 25.9 to 66%, representing a substantial interstrain variation that aims at mimicking individual variation among human subjects.
By conducting genome-wide expression analyses in the blood of both of these models and their respective controls, we arrived at a set of candidate transcripts whose abundance we measured in the blood of human subjects. We elected to monitor transcript levels, rather than serum protein levels, as a more all-encompassing approach, as not all proteins are secreted and not all transcripts are translated. Moreover, current technology allows the collection of blood for RNA isolation without refrigeration, an additional practical advantage.
The overarching goal of our research is to develop a panel of blood biomarkers for early-onset MDD that are valid, reliable and feasible. The core hypothesis for this study was that the combined list of transcripts that differentiated depressed-like from non-depressed-like rats would also differentiate human subjects with early-onset MDD from those without any disorder. We also explored whether a subset of these transcripts differentiated youths solely with MDD from those with comorbid anxiety, which is one of the best-described endophenotypes.
Materials and methods
Study 1: animal models and procedures
All procedures were approved by the institutional animal care and use committee of Northwestern University. The animals, dissection, RNA isolation, labeling, amplification, hybridization and scanning to identify ‘depression’ markers using the WMI and WLI strains were described previously24 with the following modifications. Whole blood was also collected into PAXgene Blood RNA tubes (PreAnalytiX, Hombrechticon, Switzerland) from three males, derived from different litters, of each strain. Briefly, total RNA isolated from these four tissue sources was reverse-transcribed with an Oligo(dT)-T7 primer (Affymetrix, Santa Clara, CA, USA), and double-stranded complementary DNA (cDNA) was synthesized with the GeneChip Expression 3′-Amplification One-cycle kit (Affymetrix). In an in vitro transcription reaction with T7 RNA polymerase, the cDNA was linearly amplified and labeled with biotinylated nucleotides (Affymetrix). A total of 10 μg of biotin-labeled and fragmented cRNA was then hybridized onto Rat Genome 230 2.0 GeneChip arrays (Affymetrix) for 16 h at 45 °C. Post-hybridization staining and washing were performed according to the manufacturer's protocols using the Fluidics Station 450 instrument (Affymetrix). Finally, the arrays were scanned with a GeneChip Scanner 3000 laser confocal slide scanner.
For the determination of peripheral chronic stress markers, whole blood was collected into PAXgene Blood RNA tubes from nonstressed and chronically stressed Fisher-344 (F344), Brown Norway (BN-SS), Lewis (Lew) and WKY male rats of ∼100 days of age and of different litters (n=3 per strain per experimental condition). The chronically stressed animals were restrained in a breathable decapicone for 2 h per day for a 2-week period. Total RNA was isolated using the PAXgene Blood RNA Kit (Qiagen) according to the manufacturer's instruction, and then reverse transcribed followed by second-strand cDNA synthesis. For each sample, an in vitro transcription reaction was carried out incorporating biotinylated nucleotides according to the manufacturer's protocol for Illumina Totalprep RNA amplification kit (Ambion, Austin, TX, USA). Then, 1.5 μg biotin-labeled cRNA was hybridized onto RatRef-12 Expression BeadChips (Illumina, San Diego CA, USA) for 16 h at 55 °C. Post-hybridization staining and washing were performed according to the manufacturer's protocols (Illumina). Illumina Sentrix RatRef-12 v1.0 BeadChips were scanned using Illumina's BeadStation 500 scanner. Images were checked for grid alignment and then quantified using the BeadStudio software.
The two sets of blood gene expression data came from different microarray platforms. For this reason, we carried out a detailed comparison between the Affymetrix and Illumina platforms used, and validated a number of selected transcripts’ abundances by quantitative real-time PCR (qPCR) as described previously.24
Statistical analyses of microarray data
Analyses to establish significant differences in gene expression between WMI and WLI for the different tissues were carried out as described previously.24 Briefly, probe intensity data from Rat 230v2 Affymetrix GeneChip arrays were read into the R software environment (http://www.R-project.org) directly from .CEL files using the R/affy package.33 The normalization of the Affymetrix data was performed by tissue type using the robust multiarray average method for probe set data.34
Probe intensity data from Illumina Chip arrays were read into the R software environment directly from Bead summary files produced by BeadStudio using the R/beadarray package.35 Quantile normalization was applied to the Illumina bead summary data using the R/preprocessCore package.36 Data quality was assessed using histograms of signal intensities, scatterplots, and hierarchical clustering of samples.
Analysis of variance methods were used to statistically resolve gene expression differences from both studies after data processing and normalization using the R/maanova package.37, 38
Study 2: human subjects: recruitment and procedures
The study was carried out from May through September, 2009 and from July through December, 2010. Recruitment and experimental protocols were exactly the same, except for the addition in 2010 of a questionnaire about maltreatment as a measure of chronic stress.
Male and female subjects, 15–19 years of age, were recruited through community posters, other research studies at the Research Institute at Nationwide Children's Hospital, the Nationwide Children's Hospital (NCH) Adolescent Medicine and Behavioral Health Clinics and private psychologists. Exclusion criteria were: age outside 15–19 years; Tanner Stage <5; major medical illness; antidepressant use in the past 3 months; no MDD, but another psychiatric disorder; MDD with psychosis; pregnancy; or history of mental retardation. An additional exclusion criterion for the no disorder (ND) group was any lifetime psychiatric disorder.
A total of 136 youths underwent telephone screening; 73 were not eligible or refused participation; 10 could not be reached because of a disconnected phone. In total, 53 participants were scheduled for appointments.
The protocol was approved by the NCH Institutional Review Board. Appointments took place at NCH Clinical Research Services (CRS) with data collection standardized to 1600–1700 h. Consent and assent were obtained upon arrival to the CRS. Blood was drawn into two PaxGene RNA (Qiagen) tubes. Trained research assistants conducted concurrent, but separate interviews with parents and youths about the young person's lifetime psychiatric symptoms and medical history. A youth self-report questionnaire about exposure to maltreatment was added in the 2010 subsample. Parents received $30 for time and effort and transportation costs, and youths received $45.
Pubertal stage was assessed with the Tanner Self-Rating Schematic Drawings.39 Demographic characteristics and the youth's lifetime medical history were evaluated through a brief interview with the parent and the Risks and Disorders sub-domains of the Child Health and Illness Profile-Adolescent Edition (CHIP-AE).40 The lifetime history of psychiatric diagnoses was assessed with the computerized version of the Diagnostic Interview Schedule for Children and Adolescents-IV (CDISC-IV; NIMH, 1998), Parent and Youth Versions. The CDISC is a structured psychiatric interview, organized into six Axis I diagnostic sections: Anxiety Disorders, Mood Disorders, Disruptive Behavior Disorders, Schizophrenia, Miscellaneous Disorders (for example, Eating Disorders) and Substance-Use Disorders.41 Psychometric testing of the CDISC has demonstrated good validity and reliability.42, 43 Every participant and parent received the entire diagnostic assessment.
In 2010, we also explored whether exposure to chronic stress in humans, as defined by maltreatment, correlated with the levels of gene transcripts derived from the chronic stress animal model. We used the Childhood Trauma Questionnaire (CTQ), a 28-item self-report questionnaire measuring lifetime exposure to five types of maltreatment: Emotional, Physical and Sexual Abuse and Emotional and Physical Neglect. The instrument has been standardized on over 2200 people, including adolescents, and has excellent psychometric properties.44 We used the total CTQ score.
Determination of blood transcript levels of candidate markers
Blood was drawn on all participants, but the only samples sent to the Redei lab were those of youths suffering from current MDD or those without any disorder (ND). The Redei lab was blind to any information about the participants.
Whole-blood RNA was isolated using the PAXgene Blood RNA Kit (Qiagen) according to the manufacturer's instruction; cDNA was prepared using random primers and the TaqMan RT reagents (Applied Biosystems, Foster City, CA, USA). Specific primers, listed in Supplementary Table S1, were designed to amplify the human ortholog of the rat microarray probe region of the candidate markers using the ABI's Primer Express Software (version 3.0, PE Applied Biosystems). qPCR was carried out using SYBR Green and the ABI 7900 (Applied Biosystems), with 18s rRNA as the internal control. 18s was chosen as the normalizing gene after an extensive comparative pilot study using several housekeeping genes (data not shown). Each sample for each transcript was measured in triplicate using 5 ng of template.
The ΔCT values from qPCR were used to characterize transcript abundance. ΔCT is the cycle threshold difference between the target gene and the housekeeping gene, which in this case is 18s. This method, as opposed to the relative quantification approach in which the ΔCT values are normalized to the control group giving ΔΔCT, will also indicate the variation within the ND samples. ΔΔCT is the ΔCT of the disorder group subtracting ΔCT of the ND group. The relative expression for each transcript and each sample is determined from ΔCT by raising 2 to the power of the negative value of ΔCT. A fold change can then be calculated by raising 2 to the power of the negative value of ΔΔCT for each sample.
Statistical analyses of human data
Descriptive statistics were used to characterize the sample, and nonparametric statistics to analyze gene expression differences between the ND and MDD groups or between the MDD subgroups with and without comorbid anxiety disorders. Spearman's rho correlation coefficients were used to evaluate the relationships between the CTQ scores and gene expression levels. Because this is a pilot study, effect sizes are more informative than statistical significance. We calculated effect sizes for all differences between means, using Hedges’ because of small sample and unequal subgroup sizes. Correlation coefficients themselves can serve as estimates of effect sizes, and hence we report rho values for these analyses. We made an a priori decision to interpret an effect size of ⩾0.50 as clinically significant, which corresponds to Cohen's medium effect size.45 We did not control for multiple comparisons, as the goal of a pilot study is to detect as many potentially clinically significant relationships as possible. Data analyses were performed with SPSS 14.0 (SPSS Inc., Chicago, IL, USA).
Results
Identification of candidate biomarker transcripts
Candidate transcripts were selected with significant (P<0.01) expression differences between WMI and WLI and an absolute fold change >1.2, or 20%. Using these criteria, 238 differentially expressed and annotated transcripts were identified in the hippocampus (Supplementary Table S2). Of these, 60.5% had greater abundance in the WLI hippocampus compared with the WMI. Congruent selection criteria resulted in 396 annotated transcripts from the amygdala (Supplementary Table S3), of which 141 (35.6%) had a greater abundance in WLI amygdala. The analyses of blood microarray data were carried out similarly. These differentially expressed blood transcripts and their human orthologs are listed in Supplementary Table S4. There were 203 transcripts differentially expressed between WMI and WLI blood, and 167 transcripts (82.3%) had greater abundance in WLI as it is clearly visualized in the volcano plot (Figure 1a).

Volcano plots based on microarray analyses depicting individual probe P-value (−log10) versus expression fold change (log2). (a) WMI/WLI (Wistar Kyoto (WKY) More Immobile (WMI) and WKY Less Immobile (WLI)) blood microarray plot, with dotted lines depicting the P-value selection threshold (P<0.01) and the fold change (FC) threshold (absolute FC >1.20). (b) Chronic stress blood microarray plot, with dotted lines depicting the P-value selection threshold (P<0.001) and the fold change threshold (absolute FC >1.20).
Overlap between differentially expressed transcripts in either of the two brain regions and blood was established. The 11 transcripts selected as candidates had to fulfill the criteria that the directional changes had to agree between brain and blood. Seven of these candidates (MARCKS, MAF, FAM46A, NAGA, UBE3A, CD59, ATP11C and RAPH1) fulfilled the selection criteria above and overlapped between brain and blood. Additionally, we selected three genes, SCAPER/ZNF291, TLR7 and IGSF4A/CADM1, whose transcript levels differed between WMI and WLI blood by the same criteria as the others, but were selected from brain regions based on either high fold change (fold change >1.2) or high significance (P<0.01). These transcripts, therefore, could have potential clinical relevance.
The selection criteria for the chronic stress markers differed from those of the genetic model, as they are related to a change induced by environment, rather than individual variation to vulnerability. Chronic restraint stress affected all strains of rats, as described previously.24 To eliminate strain-specific effects in our selection, transcripts were selected based on consistent, significant expression level differences in all four strains, in which significance was set at P<0.001 and absolute fold change at >1.2. We found 125 transcripts in the blood that had differentially expressed transcript levels between the chronic restraint stress and no stress conditions (Figure 1b and Supplementary Table S5). From these, we randomly selected 15 representative transcripts with absolute fold change between 1.28 and 1.95.
During the selection processes we eliminated candidates if they did not have human orthologs, or if the mRNA sequence that the probes were designed for had been retired or retracted in the database. Additionally, we also eliminated candidates that had UniGene EST (expressed sequence tag)-only information, as we could not identify associated mRNAs.
Participant characteristics
Of the 53 participants examined, 28 (14 MDD and 14 with ND) met all criteria for inclusion in testing blood transcript levels of candidate markers. There were no statistically significant differences between the two groups in mean age, proportion of females or proportion of African-Americans and Caucasians. Table 1 displays the characteristics of our final study group.
Transcript-level differences: MDD vs ND
Mean transcript abundance levels for the MDD and ND groups are shown in Table 2. The number of subjects (n) varies by one because some of the qPCR analyses were repeatedly unsuccessful for the particular subject and transcript, possibly because of polymorphisms located in the primer sequence(s). Medium or large effect sizes were found for group mean differences in 11 of the 26 transcripts examined (Table 3). The corresponding genes are ATP11C, CD59, IGSF4/CADM1, MAF and RAPH1 (derived from the genetic model); and AMFR, CAT, CDR2, CMAS, PSME1 and PTP4A3 (derived from the chronic stress model).
Transcript-level differences: MDD-only vs MDD with anxiety disorders
Intragroup analyses of MDD with and without comorbid anxiety disorders revealed medium to large effect size differences in 18 of the 26 candidate biomarkers: CD59, FAM46A, IGSF4A/CADM1, NAGA, TLR7 and ZNF291/SCAPER as well as AHSP, AMFR, CAT, CDR2, CMAS, DGKA, GCLM, GGA3, IRF3, KIAA1539, PSME1 and SLC4A1 (Table 3). This panel diverged in three ways from the panel that differentiated MDD from ND. First, the list of potentially clinical markers was longer for the intra-MDD analyses. Second, the two panels for MDD versus ND and MDD with-versus-without anxiety disorders only overlapped by six genes; thus, the non-overlapping candidates FAM46A, NAGA, TLR7, ZNF291/SCAPER, AHSP DGKA, GCLM, GGA3, IRF3, KIAA1539 and SLC4A1 are specific to MDD comorbid with anxiety. Third, 55% of the potential markers in the MDD/ND panel are from the chronic stress model, whereas nearly 70% of those in the MDD with or without Anxiety Disorders panel are from the chronic stress model.
Chronic stress gene transcripts and chronic stress in youths
Youths with depression had more exposure to maltreatment than did those who did not have any disorder (MeanMDD = 41.1±5.9 (s.d.) vs MeanND = 30.2±5.9 (s.d.), effect size = 1.74). CTQ Total score was correlated with 4 of the 26 gene transcripts at the level of a medium effect size or greater: CMAS, PSME1, PTP4A3 and IRF3. These markers were all derived from the chronic stress model.
Discussion
Despite considerable efforts, there are still no valid, reliable and feasible peripheral/blood biomarkers that can diagnose MDD, classify MDD subtypes and measure treatment response, even in adult-onset MDD.23 Our research is unique in the attempt to discover a biomarker panel for early-onset MDD, a more severe disorder than adult onset. Our approach is also innovative in combining theoretical and atheoretical strategies in animal models of depression to identify a panel of transcripts in the blood that did distinguish subjects with early-onset MDD from those in the ND group. Moreover, a partly overlapping set of transcripts differentiated youths with MDD-only from those having MDD with comorbid Anxiety Disorders, providing the first panel of blood transcripts that might be useful for detecting these endophenotypes.
Previous research to identify panels of blood biomarkers has focused on serum factors46, 47 or on blood expression biomarkers using the complex approach of Convergent Functional Genomics developed by Le-Niculescu et al.22 The strategy recently being used to identify serum-based markers is to select them from biochemical domains previously associated with MDD.47 This approach has the advantage of focusing on the biologically functional protein end points. The Convergent Functional Genomics approach combined brain and blood expression data from a pharmacogenomic animal model with human blood and post-mortem expression data and human genetic linkage/association data. This elegant assemblage of multiple levels of information has the advantage that the markers thus selected could yield information about genetic vulnerability and related transcriptomic changes.
Our approach is not a duplication of these efforts described above, but rather one that is based on the atheoretical, or unbiased, exploratory exploitation of two theoretical animal models of depression. These models comprised the genetic and the environmental (chronic stress) components of MDD etiology, and thereby these candidate biomarkers for MDD highlight genetic vulnerability factors and their transcriptomic consequences, in addition to biological costs of a repeated stressor. The strengths of our approach include the uniqueness of the genetic animal model we employed, and the selection process of candidate markers. Namely, transcripts were selected as candidates when they showed significant same-directional differences in any of the brain regions examined and in the blood of the genetically ‘depressed’ WMI compared with their genetically very close control, the WLI. This allowed us to verify that the candidate transcripts were relevant to brain functions and, therefore, some of these transcripts can be regarded as prospective novel drug targets. Another strength is that our chronic stress blood markers were compiled from a study of four genetically diverse strains, thereby providing a rather narrow, but powerfully informative, set of transcripts. These transcripts represent generalizable responsiveness to stress and signify the notion that stress marks all organisms, regardless of their vulnerability or resilience.
The first biomarker panel we define consists of 11 transcripts that differentiated youths with MDD from those without any disorder. These candidate markers are derived almost equally from the genetic and chronic stress models of depression. The genes expressing these transcripts belong to three broad functional categories: those involved in transcription, neurodevelopment and neurodegeneration. Genes with transcriptional regulatory functions include MAF, which encodes a DNA-binding, leucine zipper-containing transcription factor, and the cytoplasmic cerebellar degeneration-related protein 2 antigen (CDR2), which harbors a helix-leucine zipper motif and interacts specifically with c-Myc.48
Genes that regulate, modify or interfere with neurodevelopment include RAPH1 (Ras association and pleckstrin homology domains 1, also known as LPD), which is intimately involved in proper neuronal migration.49 Tyrosine phosphatase PTP4A3, also called PRL-3, has oncogenic activity, but has also been reported to promote cell migration.50 CMAS encodes an enzyme, cytidine monophosphate N-acetylneuraminic acid synthetase (also known as CMP-Neu5Ac synthetase), which regulates brain sialylation levels and, therefore, very likely affects brain development.51 Cell adhesion molecule 1 (CADM1), also known as immunoglobulin superfamily, member 4 (IGSF4), is a synaptic adhesive molecule involved in neural cell adhesion processes and synaptogenesis.52 CADM1 is thought to contribute to depressive-like behavior in a recent mouse genetic study,53 and has also been linked with social impairments and anxiety-like behavior.54, 55
A group of candidate biomarkers have known functions in processes that mediate neuronal damage. PSME1 (proteasome activator subunit 1, also called PA28a) activates the proteosomal hydrolysis of intracellular proteins. Inadequate glucose supply causes damage of neuronal cells, and PSME1 is highly responsive to hypoglycemic environment in neurons.56 CD59, also called MIRL or protectin, encodes a single-chain, glycosylphosphatidylinositol-anchored cell surface protein structurally homologous to snake venom neurotoxins.57 The protein has complement-inhibitory properties, but its capability to mediate complement-mediated damage to neurons is also recognized.58, 59, 60, 61 The CAT gene encodes catalase, a key antioxidant enzyme that serves as a defense against oxidative stress. Chronic unpredictable stress decreases CAT expression in the mouse cerebral cortex and hippocampus;62 these effects may be mirrored by the decreased expression of this gene in the blood of both the chronic stress model and subjects with MDD. The expression of AMFR (autocrine motility factor receptor), otherwise known as GP78, which encodes an endoplasmic reticulum membrane-anchored ubiquitin ligase, is increased by accumulation of neurodegenerative disease proteins, such as mutant huntingtin,63 SOD1 and ataxin-3.64 This increase in AMFR expression may represent a protective response to enhance the removal of these disease proteins, and suggests that a decrease of AMFR/GP78 expression, as we found in MDD, might make the organism more vulnerable to these diseases.
The second panel consists of 18 transcripts that distinguished youths who had MDD with comorbid anxiety disorders from those with MDD alone. Only six gene transcripts were in both panels, and thus the majority of these markers are unique to this diagnostic category. Furthermore, the panel differentiating MDD youths with and without comorbid anxiety disorders had a substantially higher number of genes derived from the chronic stress model than from the genetic model. These latter observations support the long-standing clinical impression that MDD with comorbid anxiety disorders is a unique phenotype. Moreover, it is possible that there are different etiologic factors involved in this endophenotype, for example, exposure to chronic stress particularly at the highly stressful period of adolescence. Most of these transcripts have no known function relevant to MDD or brain, including three of the four ‘genetic depression model’ markers: FAM46A (family with sequence similarity 46, member A), NAGA (N-acetylgalactosaminidase) and ZNF291/SCAPER (S-phase cyclin A-associated protein in the ER). Among the ‘chronic stress model’ markers, AHSP (α hemoglobin stabilizing protein), DGKA (diacylglycerol kinase, α), SLC4A1 (solute carrier family 4, anion exchanger, member 1) and KIAA1539 have either no brain-related functions to date or no known function.
Genes whose transcripts differentiated MDD with and without comorbid anxiety and which have known functions relevant to MDD or stress encode proteins involved in immunoregulation or neurodegeneration. IRF3 (interferon regulatory factor 3), identified from the chronic stress animal model, has been established previously as a chronic psychological stress-responsive gene in human peripheral blood cells.65 TLR7, an intracellular Toll-like receptor, is an innate immunity receptor that activates inflammation and adaptive immunity. TLR7, or its agonists, induce inflammatory responses in the periphery and in the brain,66 and greater expression of TLR7 are associated with poor functional outcome in ischemic stroke patients.67 Polymorphism in GCLM (glutamate cysteine ligase modulatory subunit), the first rate-limiting enzyme of glutathione synthesis, has been associated with depression and schizophrenia, although this association has not been confirmed.68 Nevertheless, glutathione deficits have been observed in several neurodegenerative and psychiatric disorders including Alzheimer's, Parkinson's or Huntington's diseases69 as well as in schizophrenia. GGA3 (golgi-associated, gamma adaptin ear containing ARF binding protein 3) is involved in the pathogenesis of Alzheimer's disease.70 It is interesting to note that subjects with lower levels of GGA3 may be at risk for developing Alzheimer's disease,70 and that MDD is thought to be a triggering or precipitating factor in Alzheimer's disease. Several of the blood candidate markers whose expression differed between MDD and controls or between MDD subtypes are involved in different neurodegenerative processes, suggesting that MDD, and perhaps other psychiatric illnesses, can lead to neurodegeneration. These markers, therefore, could present a generic ‘neurodegenerative fingerprint’ in the brain and blood. Alternatively, as Alzheimer's, Parkinson's and other neurodegenerative illnesses are known to show symptoms of depression before their regular presentation, depression might be a precipitating factor in these illnesses indicated by the presence of these biomarkers.
When we explored associations between CTQ Total scores and transcript levels, we found that the transcripts with potential clinical significance were all derived from the chronic stress animal model. These four transcripts, CMAS, IRF3, PSME1 and PTP4A3, share no close connections in their functions, but their altered expressions are likely to represent long-term consequences of maltreatment, as these youths may have experienced maltreatment for as long ago as 8–10 years before the time of data collection.
The current study's limitations lie in its relatively small samples sizes and the limited number of target transcripts we were able to pursue. Additionally, we recognize that the animal models only mirror some aspects of early-onset MDD and, therefore, the markers derived from them cannot be all inclusive. Nonetheless, the purpose of this type of study in biomarker research is to determine if there are sufficient data to proceed to a full-size study of the candidate transcripts in a large representative sample of young people with and without MDD.
In summary, we have taken a novel approach to identifying potential peripheral biomarkers for early-onset MDD. The main goal of this pilot study was to determine if blood transcripts from both a genetic and a chronic stress animal model of depression could lead to candidate blood biomarkers for early-onset MDD in human subjects. The pilot data presented here suggest that our approach leads to a clinically valid diagnostic panel of blood transcripts that can differentiate early-onset MDD from controls and MDD with from MDD without anxiety. The next step is to test our findings in a large sample of youths with MDD, comparing them with youths without any psychiatric disorder and youths diagnosed with other psychiatric disorders. Eventually, the effect of treatment on validated biomarkers panels can be established, allowing for further individualization of MDD treatment strategies.
References
Emslie GJ, Ryan ND, Wagner KD . Major depressive disorder in children and adolescents: clinical trial design and antidepressant efficacy. J Clin Psychiatry 2005; 66 (Suppl 7): 14–20.
Shi J, Potash JB, Knowles JA, Weissman MM, Coryell W, Scheftner WA et al. Genome-wide association study of recurrent early-onset major depressive disorder. Mol Psychiatry 2011; 16: 193–201.
Kendler KS, Gatz M, Gardner CO, Pedersen NL . Clinical indices of familial depression in the Swedish Twin Registry. Acta Psychiatr Scand 2007; 115: 214–220.
Kessler RC, Avenevoli S, Ries Merikangas K . Mood disorders in children and adolescents: an epidemiologic perspective. Biol Psychiatry 2001; 49: 1002–1014.
Hankin BL, Abramson LY, Moffitt TE, Silva PA, McGee R, Angell KE . Development of depression from preadolescence to young adulthood: emerging gender differences in a 10-year longitudinal study. J Abnorm Psychol 1998; 107: 128–140.
Harrington R, Fudge H, Rutter M, Pickles A, Hill J . Adult outcomes of childhood and adolescent depression. I. Psychiatric status. Arch Gen Psychiatry 1990; 47: 465–473.
Lewinsohn PM, Rohde P, Klein DN, Seeley JR . Natural course of adolescent major depressive disorder: I. Continuity into young adulthood. J Am Acad Child Adolesc Psychiatry 1999; 38: 56–63.
Lewinsohn PM, Rohde P, Seeley JR, Klein DN, Gotlib IH . Psychosocial functioning of young adults who have experienced and recovered from major depressive disorder during adolescence. J Abnorm Psychol 2003; 112: 353–363.
Weissman MM, Wolk S, Goldstein RB, Moreau D, Adams P, Greenwald S et al. Depressed adolescents grown up. JAMA 1999; 281: 1707–1713.
Keenan-Miller D, Hammen CL, Brennan PA . Health outcomes related to early adolescent depression. J Adolesc Health 2007; 41: 256–262.
Aalto-Setala T, Marttunen M, Tuulio-Henriksson A, Poikolainen K, Lonnqvist J . Depressive symptoms in adolescence as predictors of early adulthood depressive disorders and maladjustment. Am J Psychiatry 2002; 159: 1235–1237.
Rao U, Ryan ND, Birmaher B, Dahl RE, Williamson DE, Kaufman J et al. Unipolar depression in adolescents: clinical outcome in adulthood. J Am Acad Child Adolesc Psychiatry 1995; 34: 566–578.
Brent D, Emslie G, Clarke G, Wagner KD, Asarnow JR, Keller M et al. Switching to another SSRI or to venlafaxine with or without cognitive behavioral therapy for adolescents with SSRI-resistant depression: the TORDIA randomized controlled trial. JAMA 2008; 299: 901–913.
Bridge JA, Iyengar S, Salary CB, Barbe RP, Birmaher B, Pincus HA et al. Clinical response and risk for reported suicidal ideation and suicide attempts in pediatric antidepressant treatment: a meta-analysis of randomized controlled trials. JAMA 2007; 297: 1683–1696.
Gould TD, Gottesman . Psychiatric endophenotypes and the development of valid animal models. Genes Brain Behav 2006; 5: 113–119.
Gottesman II, Gould TD . The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry 2003; 160: 636–645.
Lakhan SE, Vieira K, Hamlat E . Biomarkers in psychiatry: drawbacks and potential for misuse. Int Arch Med 2010; 3: 1.
Carroll BJ, Feinberg M, Greden JF, Tarika J, Albala AA, Haskett RF et al. A specific laboratory test for the diagnosis of melancholia. Standardization, validation, and clinical utility. Arch Gen Psychiatry 1981; 38: 15–22.
Hashimoto K . Brain-derived neurotrophic factor as a biomarker for mood disorders: an historical overview and future directions. Psychiatry Clin Neurosci 2010; 64: 341–357.
Herberth M, Koethe D, Levin Y, Schwarz E, Krzyszton ND, Schoeffmann S et al. Peripheral profiling analysis for bipolar disorder reveals markers associated with reduced cell survival. Proteomics 2011; 11: 94–105.
Kurian SM, Le-Niculescu H, Patel SD, Bertram D, Davis J, Dike C et al. Identification of blood biomarkers for psychosis using convergent functional genomics. Mol Psychiatry 2011; 16: 37–58.
Le-Niculescu H, McFarland MJ, Ogden CA, Balaraman Y, Patel S, Tan J et al. Phenomic, convergent functional genomic, and biomarker studies in a stress-reactive genetic animal model of bipolar disorder and co-morbid alcoholism. Am J Med Genet B Neuropsychiatr Genet 2008; 147B: 134–166.
Schmidt HD, Shelton RC, Duman RS . Functional biomarkers of depression: diagnosis, treatment, and pathophysiology. Neuropsychopharmacology 2011; 36: 2375–2394.
Andrus BM, Blizinsky K, Vedell PT, Dennis K, Shukla PK, Schaffer DJ et al. Gene expression patterns in the hippocampus and amygdala of endogenous depression and chronic stress models. Mol Psychiatry 2012; 17: 49–61.
Rittenhouse PA, Lopez-Rubalcava C, Stanwood GD, Lucki I . Amplified behavioral and endocrine responses to forced swim stress in the Wistar-Kyoto rat. Psychoneuroendocrinology 2002; 27: 303–318.
Redei E, Pare WP, Aird F, Kluczynski J . Strain differences in hypothalamic-pituitary-adrenal activity and stress ulcer. Am J Physiol 1994; 266 (2 Pt 2): R353–R360.
Redei EE, Ahmadiyeh N, Baum A, Sasso D, Slone J, Solberg LC et al. Novel animal models of affective disorders. In: Tucker GJ, McKinney W (eds). Seminars in Clinical Neuropsychiatry 2001; 6: 43–67. WB Saunders Company: Philadelphia, 2001.
Solberg LC, Olson SL, Turek FW, Redei E . Altered hormone levels and circadian rhythm of activity in the WKY rat, a putative animal model of depression. Am J Physiol Regul Integr Comp Physiol 2001; 281: R786–R794.
Solberg LC, Baum AE, Ahmadiyeh N, Shimomura K, Li R, Turek FW et al. Sex- and lineage-specific inheritance of depression-like behavior in the rat. Mamm Genome 2004; 15: 648–662.
Malkesman O, Weller A . Two different putative genetic animal models of childhood depression--a review. Prog Neurobiol 2009; 88: 153–169.
Will CC, Aird F, Redei EE . Selectively bred Wistar-Kyoto rats: an animal model of depression and hyper-responsiveness to antidepressants. Mol Psychiatry 2003; 8: 925–932.
Mehta NS . An Animal Model of Adolescent Depression. Neuroscience 2011: Washington, DC, 2011.
Gautier L, Cope L, Bolstad BM, Irizarry RA . affy--analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004; 20: 307–315.
Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003; 4: 249–264.
Dunning MJ, Smith ML, Ritchie ME, Tavare S . beadarray: R classes and methods for Illumina bead-based data. Bioinformatics 2007; 23: 2183–2184.
Bolstad BM, Irizarry RA, Astrand M, Speed TP . A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003; 19: 185–193.
Churchill GA . Using ANOVA to analyze microarray data. Biotechniques 2004; 37: 173–175, 177.
Wu X, Pang ST, Sahlin L, Blanck A, Norstedt G, Flores-Morales A . Gene expression profiling of the effects of castration and estrogen treatment in the rat uterus. Biol Reprod 2003; 69: 1308–1317.
Taylor SJ, Whincup PH, Hindmarsh PC, Lampe F, Odoki K, Cook DG . Performance of a new pubertal self-assessment questionnaire: a preliminary study. Paediatr Perinat Epidemiol 2001; 15: 88–94.
Starfield B, Riley AW, Green BF, Ensminger ME, Ryan SA, Kelleher K et al. The adolescent child health and illness profile. A population-based measure of health. Med Care 1995; 33: 553–566.
Shaffer D, Fisher P, Lucas CP, Dulcan MK, Schwab-Stone ME . NIMH Diagnostic Interview Schedule for Children Version IV (NIMH DISC-IV): description, differences from previous versions, and reliability of some common diagnoses. J Am Acad Child Adolesc Psychiatry 2000; 39: 28–38.
Fisher PW, Lucas C, Shaffer D . Diagnostic Interview Schedule for Children, Version IV (DISC IV): Test-retest reliability in a clinical sample. Proceedings of the 44th Annual Meeting of the American Academy of Child and Adolescent Psychiatry: Toronto, Canada, 1997.
Schwab-Stone ME, Shaffer D, Dulcan MK, Jensen PS, Fisher P, Bird HR et al. Criterion validity of the NIMH Diagnostic Interview Schedule for Children Version 2.3 (DISC-2.3). J Am Acad Child Adolesc Psychiatry 1996; 35: 878–888.
Bernstein DP, Fink L . Childhood Trauma Questionnaire: A Retrospective Self-Report. Harcourt Brace & Co.: San Antonio, TX, 1998.
Cohen J . Statistical Power Analysis for the Behavioral Sciences. 2nd edn. L Erlbaum Associates: Hillsdale, NJ, 1988.
Domenici E, Wille DR, Tozzi F, Prokopenko I, Miller S, McKeown A et al. Plasma protein biomarkers for depression and schizophrenia by multi analyte profiling of case-control collections. PLoS One 2010; 5: e9166.
Papakostas GI, Shelton RC, Kinrys G, Henry ME, Bakow BR, Lipkin SH et al. Assessment of a multi-assay, serum-based biological diagnostic test for major depressive disorder: a pilot and replication study. Mol Psychiatry; advance online publication, 13 December 2011; doi:10.1038/mp.2011.166.
Okano HJ, Park WY, Corradi JP, Darnell RB . The cytoplasmic Purkinje onconeural antigen cdr2 down-regulates c-Myc function: implications for neuronal and tumor cell survival. Genes Dev 1999; 13: 2087–2097.
Pinheiro EM, Xie Z, Norovich AL, Vidaki M, Tsai LH, Gertler FB . Lpd depletion reveals that SRF specifies radial versus tangential migration of pyramidal neurons. Nat Cell Biol 2011; 13: 989–995.
McParland V, Varsano G, Li X, Thornton J, Baby J, Aravind A et al. The metastasis-promoting phosphatase PRL-3 shows activity toward phosphoinositides. Biochemistry 2011; 50: 7579–7590.
Revilla-Nuin B, Reglero A, Feo JC, Rodriguez-Aparicio LB, Ferrero MA . Identification, expression and tissue distribution of cytidine 5¢-monophosphate N-acetylneuraminic acid synthetase activity in the rat. Glycoconj J 1998; 15: 233–241.
Biederer T, Sara Y, Mozhayeva M, Atasoy D, Liu X, Kavalali ET et al. SynCAM, a synaptic adhesion molecule that drives synapse assembly. Science 2002; 297: 1525–1531.
Benton CS, Miller BH, Skwerer S, Suzuki O, Schultz LE, Cameron MD et al. Evaluating genetic markers and neurobiochemical analytes for fluoxetine response using a panel of mouse inbred strains. Psychopharmacology (Berl); advance online publication, 24 November 2011; doi:10.1007/s00213-011-2574-z.
Takayanagi Y, Fujita E, Yu Z, Yamagata T, Momoi MY, Momoi T et al. Impairment of social and emotional behaviors in Cadm1-knockout mice. Biochem Biophys Res Commun 2010; 396: 703–708.
Zhiling Y, Fujita E, Tanabe Y, Yamagata T, Momoi T, Momoi MY . Mutations in the gene encoding CADM1 are associated with autism spectrum disorder. Biochem Biophys Res Commun 2008; 377: 926–929.
Kobayashi K, Xin Y, Ymer SI, Werther GA, Russo VC . Subtractive hybridisation screen identifies genes regulated by glucose deprivation in human neuroblastoma cells. Brain Res 2007; 1170: 129–139.
Kieffer B, Driscoll PC, Campbell ID, Willis AC, van der Merwe PA, Davis SJ . Three-dimensional solution structure of the extracellular region of the complement regulatory protein CD59, a new cell-surface protein domain related to snake venom neurotoxins. Biochemistry 1994; 33: 4471–4482.
Storch MK, Piddlesden S, Haltia M, Iivanainen M, Morgan P, Lassmann H . Multiple sclerosis: in situ evidence for antibody- and complement-mediated demyelination. Ann Neurol 1998; 43: 465–471.
Kolev MV, Ruseva MM, Morgan BP, Donev RM . Targeting neural-restrictive silencer factor sensitizes tumor cells to antibody-based cancer immunotherapy in vitro via multiple mechanisms. J Immunol 2010; 184: 6035–6042.
Yang LB, Li R, Meri S, Rogers J, Shen Y . Deficiency of complement defense protein CD59 may contribute to neurodegeneration in Alzheimer's disease. J Neurosci 2000; 20: 7505–7509.
Pedersen ED, Froyland E, Kvissel AK, Pharo AM, Skalhegg BS, Rootwelt T et al. Expression of complement regulators and receptors on human NT2-N neurons--effect of hypoxia and reoxygenation. Mol Immunol 2007; 44: 2459–2468.
Moretti M, Colla A, de Oliveira Balen G, Dos Santos DB, Budni J, de Freitas AE et al. Ascorbic acid treatment, similarly to fluoxetine, reverses depressive-like behavior and brain oxidative damage induced by chronic unpredictable stress. J Psychiatr Res 2011; 46: 331–340.
Yang H, Liu C, Zhong Y, Luo S, Monteiro MJ, Fang S . Huntingtin interacts with the cue domain of gp78 and inhibits gp78 binding to ubiquitin and p97/VCP. PLoS One 2010; 5: e8905.
Ying Z, Wang H, Fan H, Zhu X, Zhou J, Fei E et al. Gp78, an ER associated E3, promotes SOD1 and ataxin-3 degradation. Hum Mol Genet 2009; 18: 4268–4281.
Kawai T, Morita K, Masuda K, Nishida K, Shikishima M, Ohta M et al. Gene expression signature in peripheral blood cells from medical students exposed to chronic psychological stress. Biol Psychol 2007; 76: 147–155.
Damm J, Wiegand F, Harden LM, Gerstberger R, Rummel C, Roth J . Fever, sickness behavior, and expression of inflammatory genes in the hypothalamus after systemic and localized subcutaneous stimulation of rats with the toll-like receptor 7 agonist imiquimod. Neuroscience 2011; 201: 166–183.
Brea D, Sobrino T, Rodriguez-Yanez M, Ramos-Cabrer P, Agulla J, Rodriguez-Gonzalez R et al. Toll-like receptors 7 and 8 expression is associated with poor outcome and greater inflammatory response in acute ischemic stroke. Clin Immunol 2011; 139: 193–198.
Berk M, Johansson S, Wray NR, Williams L, Olsson C, Haavik J et al. Glutamate cysteine ligase (GCL) and self reported depression: an association study from the HUNT. J Affect Disord 2011; 131: 207–213.
Ballatori N, Krance SM, Notenboom S, Shi S, Tieu K, Hammond CL . Glutathione dysregulation and the etiology and progression of human diseases. Biol Chem 2009; 390: 191–214.
Tesco G, Koh YH, Kang EL, Cameron AN, Das S, Sena-Esteves M et al. Depletion of GGA3 stabilizes BACE and enhances beta-secretase activity. Neuron 2007; 54: 721–737.
Acknowledgements
We acknowledge the following for their help in this study: Jelena Radulovic, Pradeep Shukla, Claire Will and Fraser Aird. Animal work was supported by grants from the Davee Foundation, the RD Foundation and by the NIH Grant MH077234. Human work was funded by grants from the Research Institute of Nationwide Children's Hospital in Columbus, OH.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Pajer, K., Andrus, B., Gardner, W. et al. Discovery of blood transcriptomic markers for depression in animal models and pilot validation in subjects with early-onset major depression. Transl Psychiatry 2, e101 (2012). https://doi.org/10.1038/tp.2012.26
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2012.26
Keywords
This article is cited by
-
Hypothalamic protein profiling from mice subjected to social defeat stress
Molecular Brain (2024)
-
Whole genome sequencing of nearly isogenic WMI and WLI inbred rats identifies genes potentially involved in depression and stress reactivity
Scientific Reports (2021)
-
Pilot validation of blood-based biomarkers during pregnancy and postpartum in women with prior or current depression
Translational Psychiatry (2021)
-
What do DNA methylation studies tell us about depression? A systematic review
Translational Psychiatry (2019)
-
Animal models of depression: pros and cons
Cell and Tissue Research (2019)
