
Abstract
Many scientists are seeking better therapies for treating fibromyalgia (FM) pain. We used a mouse model of FM to determine if ASIC3 and its relevant signaling pathway participated in FM pain. We demonstrated that FM-induced mechanical hyperalgesia was attenuated by electroacupuncture (EA). The decrease in fatigue-induced lower motor function in FM mice was also reversed by EA. These EA-based effects were abolished by the opioid receptor antagonist naloxone and the adenosine A1 receptor antagonist rolofylline. Administration of opioid receptor agonist endomorphin (EM) or adenosine A1 receptor agonist N6-cyclopentyladenosine (CPA) has similar results to EA. Similar results were also observed in ASIC3−/− or ASIC3 antagonist (APETx2) injected mice. Using western blotting, we determined that pPKA, pPI3K, and pERK were increased during a dual acidic injection priming period. Nociceptive receptors, such as ASIC3, Nav1.7, and Nav1.8, were upregulated in the dorsal root ganglion (DRG) and spinal cord (SC) of FM mice. Furthermore, pPKA, pPI3K, and pERK were increased in the central thalamus. These aforementioned mechanisms were completely abolished in ASIC3 knockout mice. Electrophysiological results also indicated that acid potentiated Nav currents through ASIC3 and ERK pathway. Our results highlight the crucial role of ASIC3-mediated mechanisms in the treatment of FM-induced mechanical hyperalgesia.
Similar content being viewed by others

Introduction
Fibromyalgia (FM) pain, which is a crucial worldwide health problem, is characterized by widespread mechanical pain1,2. Pregabalin is a presynaptic voltage-gated calcium channel blocker that reduces FM pain and sleep disturbances3,4. Duloxetine attenuates FM-related pain symptoms but has serious side effects5. Milnacipran is a serotonin-norepinephrine reuptake inhibitor (SNRI) designed to reduce the FM symptom of nausea6. Repeated acidic saline injections into the gastrocnemius muscle (GM) induce widespread chronic mechanical hyperalgesia that can be reversed by electroacupuncture (EA)7,8. FM pain in mice can be attenuated by the administration of μ-/δ-opioid receptor agonists9 or glutamate receptor antagonists in the spinal cord10. The decreased local tissue pH reported in FM activates peripheral nociceptive terminals7, and recordings of peripheral dorsal root ganglion (DRG) neurons have shown that lower pH conditions reliably induce inward currents that regulate voltage-gated sodium channel (Nav) functions11,12,13.
Acid-sensing ion channel 3 (ASIC3), the most sensitive ion channel for acid sensation, is mainly localized in peripheral sensory neurons, especially the DRG, and typically associated with several pain conditions14,15. ASIC3 can be activated by acidic pH (~6.8) and enhanced by lactate16,17. There are four genes that encode at least seven subtypes of receptors: ASIC1a, ASIC1b, ASIC2a, ASIC2b, ASIC3, ASIC4, and ASIC518,19. A recent study showed a reduction in the development of mechanical hypersensitivity after an acid injection in the gastrocnemius muscle of ASIC3-null mice20. Another paper showed that the hypersensitivity induced by an intramuscular acidic saline injection was mediated by ASIC321.
Navs are involved in inflammatory pain, as evidenced by the significant amelioration of pain by sodium channel blockers22,23,24. Nav1.7 and Nav1.8 are expressed in the peripheral DRG and responsible for pain conduction25. Animal studies showed that Nav1.3, Nav1.7, and Nav1.8 play prominent roles in inflammatory pain and are potentiated by the microinjection of carrageenan and CFA23. Hyperalgesia was accompanied by alterations in TTX-sensitive and TTX-resistant Na currents26,27. CFA-induced inflammation was associated with an increase in Nav1.8 expressions in the DRG that was inhibited by ibuprofen28. Persistent hyperalgesia induced by injections of prostaglandin E2 (PGE2) was also accompanied by an increase in Nav1.8 in the peripheral DRG29. Therefore, Nav1.8 is the main target for a diverse array of inflammatory mediators that act through a number of second-messenger pathways, including PKA26,28, PKC26,30, and extracellular signal-regulated kinase26,31.
Several molecules, such as N-methyl-D-aspartate receptors (NMDARs), ASIC3, TRPV1, calcium channels (Cav), and substance P (SP), have been implicated in mouse models of FM pain12,13,21,32,33. Dual acidic saline injections increase the cAMP pathway at the central spinal cord level34. Activation of pERK was detected in the anterior paraventricular nucleus of the thalamus and amygdala33,35. The administration of neurotrophin-3 alleviates acid-induced chronic muscle pain, which is similar to clinical observations36. The calcium channel antagonist pregabalin and the M-type voltage-gated potassium channel activator flupirtine were reported to be effective at treating muscle pain37,38. Acupuncture has been used to control body weight39, reduce epilepsy40, ameliorate learning and memory impairments41, and control pain26,42,43. The analgesic effect of acupuncture activates the release of endogenous opiates44 and adenosine45.
Our rational is that EA could reduce FM symptoms through ASIC3 receptor and related mechanisms. We hypothesized that EA could attenuate mechanical hyperalgesia by releasing endomorphin or adenosine to inhibit ASIC3 receptor in this FM mouse model. Our data showed that pPKA/pPI3K-pERK signaling pathway was crucial for FM priming in mice. EA at acupoint ST36 reliably reduced dual acidic saline-induced mechanical hyperalgesia and motor dysfunction via ASIC3-Nav1.7/Nav1.8 signaling pathways. We also determined that mechanical hyperalgesia and nociceptive signaling were abolished in ASIC3 knockout mice. This study provides novel and detailed molecular mechanisms behind the use of EA to treat FM-related mechanical hyperalgesia.
Results
EA-mediated attenuation of ASIC3 signaling reliably reduces mechanical hyperalgesia through peripheral opioid and adenosine pathways in an FM mouse model
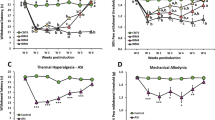
Similar to our previous publication7,12, two normal saline injections in mice did not induce ipsilateral mechanical hyperalgesia (Fig. 1A, n = 10). Dual intramuscular injections of an acidic solution (pH 4.0) into the gastrocnemius muscle (GM) of mice induced bilateral mechanical hyperalgesia on Days 5–8 (Fig. 1B, n = 10). The first acidic saline injection initiated a rapid and transient mechanical hyperalgesia that recovered after 1 day. A second acidic saline injection 5 days after the first injection induced significant and long-lasting mechanical hyperalgesia. EA at acupoint ST36 for 4 continuous days significantly reduced mechanical hyperalgesia (Fig. 1C, n = 10). To elucidate how EA affects neurotransmitters to attenuate mechanical hyperalgesia, we injected opioid and adenosine antagonists into the acupoint. We showed that the analgesic effect of EA was blocked by the opioid antagonist naloxone (Fig. 1D, n = 10) and adenosine antagonist rolofylline (Fig. 1E, n = 10). Injection of endomorphin (EM) or N6-cyclopentyladenosine (CPA) significantly attenuated mechanical hyperalgesia (Fig. 1F and G, n = 10). Mice lacking the ASIC3 gene (Asic3−/−) showed a bilateral decrease in mechanical hyperalgesia (Fig. 1H, n = 10). Similar results were also observed in ASIC3 antagonist (APETx2) injection (Fig. 1H, n = 10). Additionally, we utilized a rotarod to test fatigue behavior. Our data indicated that lower motor function was observed in FM mice (Fig. 1J, 47.8 ± 3.9 s, p < 0.05 compared with Control group, n = 10), and this symptom was alleviated by EA manipulation (Fig. 1J, 73.1 ± 5.6 s, p < 0.05 compared with FM group, n = 10). Furthermore, this phenomenon was blocked by naloxone (Fig. 1J, 47.0 ± 2.1 s, p < 0.05 compared with Con group, n = 10) and rolofylline (Fig. 1J, 48.1 ± 2.4 s, p < 0.05 compared with Con group, n = 10). Injection of endomorphin (EM) (Fig. 1J, 68.3 ± 6.3 s, p > 0.05 compared with Con group, n = 10) or N6-cyclopentyladenosine (CPA) (Fig. 1J, 64.8 ± 7.2 s, p > 0.05 compared with Con group, n = 10) reliably improve motor function. Similar data was also observed in ASIC3−/− or APETx2 injected mice (Fig. 1J, 68.1 ± 4.7 s vs. 66.2 ± 3.9 s, p > 0.05 compared with Con group, n = 10). These data suggest that FM-induced mechanical hyperalgesia and fatigue depend on ASIC3, opioid and adenosine receptors.

(A) Mechanical responses of saline-injected control mice. (B) Mechanical responses of FM group mice. (C) Mechanical responses of FM mice treated with EA (EA group). (D) Mechanical responses of FM mice treated with EA and naloxone (Nal group). (E) Mechanical responses of FM mice treated with EA and rolofyllin (Rol group). (F) Mechanical responses of FM mice treated with endomorphin (EM group). (G) Mechanical responses of FM mice treated with EA and N6-cyclopentyladenosine (CPA group). (H) Mechanical responses of FM in ASIC null mice (ASIC3 null group). (I) Mechanical responses of FM mice treated with APETx2 (APETx2 group). Mice were tested before injection (day 0), 4 hours after injection, day 1 (D1), day 5 (D5), day 6 (D6), and day 8 (D8). *p < 0.05 compared to baseline (n = 10 mice per group).
Acidic saline injections transiently increased the pPKA, pPI3K, and pERK signaling pathway to deliver nociceptive signals, and the effect is attenuated by EA treatment
Much is known about the mechanical hyperalgesia induced by dual acidic saline injections; however, there is no evidence on the detailed mechanisms involved in the transient acid priming period. Thus, we identified which proteins are altered during the first and second acidic saline injections. We showed that ASIC3 was not altered in the DRG 15 min after the first saline injection (Fig. 2A, 99.8 ± 4.7%, p > 0.05, n = 6) or after EA treatment (Fig. 2A, 102.1 ± 5.5%, p > 0.05, n = 6). Interestingly, pPKA expression was increased within this window in FM mice (Fig. 2B, 117.4 ± 2.9%, p < 0.05 compared with Con group, n = 6) and rescued by EA (Fig. 2B, 98.0 ± 4.9%, p < 0.05 compared with FM group, n = 6). A similar pattern was observed for pPI3K signals (Fig. 2C, all p < 0.05, n = 6). There were no significant differences in pPKC among the 3 groups (Fig. 2D, all p > 0.05, n = 6). pERK levels increased after the first acidic saline injection (Fig. 2E, 181.5 ± 15.9%, p < 0.05 compared with Con group, n = 6), which was attenuated by EA treatment (Fig. 2E, 124.9 ± 5.8%, p < 0.05 compared with FM group, n = 6). Both Nav1.7 and Nav1.8 were not affected in any group (Fig. 2F and G, all p > 0.05, n = 6). Next, we assessed the central SC after the first saline injection. ASIC3 was not altered by FM (Fig. 2A, 101.3 ± 3.6%, p > 0.05, n = 6) or EA treatment (Fig. 2A, 98.9 ± 9.8%, p > 0.05, n = 6). Expression of pPKA was transiently increased in FM mice (Fig. 2B, 117.2 ± 2.5%, p < 0.05 compared with Con group, n = 6), and this effect was blocked by EA (Fig. 2B, 98.8 ± 2.9%, p < 0.05 compared with Con group, n = 6). The same results were observed for pPI3K (Fig. 2C, all p < 0.05, n = 6). There were no significant differences in pPKC levels among the 3 groups (Fig. 2D, all p > 0.05, n = 6). The expression of pERK was increased in the SC after the first acidic saline injection (Fig. 2E, 143.2 ± 7.4%, p < 0.05 compared with Con group, n = 6), which was alleviated by EA treatment (Fig. 2E, 112.5 ± 3.2%, p < 0.05 compared with FM group, n = 6). Both Nav1.7 and Nav1.8 levels were similar across all groups (Fig. 2F and G, all p > 0.05, n = 6). We examined if similar mechanisms occurred after a second acidic saline injection. ASIC3 was not changed at 15 min after acid injection in the DRG or SC of FM mice (Fig. 3A, all p > 0.05, n = 6). The FM-induced increase in pPKA was attenuated by EA (Fig. 3B, all p < 0.05, n = 6). A similar result was obtained for pPI3K (Fig. 3C, all p < 0.05, n = 6) but not for pPKC (Fig. 3D, all p > 0.05, n = 6). No changes in pERK (Fig. 3E, all p > 0.05, n = 6) or nociceptive Nav1.7 and Nav1.8 (Fig. 3F and G, all p > 0.05, n = 6) were found. These results provide a novel and detailed mechanism for FM priming.

(A) ASIC3, (B) pPKA, (C) pPI3K, (D) pPKC, (E) pERK, (F) Nav1.7, and (G) Nav1.8 expression levels in tissues from the Con, FM, EA groups (from left to right). Con = Control; FM = Fibromyalgia group; EA = Electroacupuncture. *p < 0.05 compared with the Con group. #p < 0.05 compared with the FM group. The western blot bands at the top show the cropped target protein. The lower bands are cropped internal controls (β-actin or α-tubulin).

(A) ASIC3, (B) pPKA, (C) pPI3K, (D) pPKC, (E) pERK, (F) Nav1.7, and (G) Nav1.8 expression levels in tissues from the Con, FM, EA groups (from left to right). Con = Control; FM = Fibromyalgia group; EA = Electroacupuncture. *p < 0.05 compared with the Con group. #p < 0.05 compared with the FM group. The western blot bands at the top show the cropped target protein. The lower bands are cropped internal controls (β-actin or α-tubulin).
EA attenuates ASIC3-related nociceptive signals upregulated in DRGs 8 days after FM induction via opioid and adenosine pathways
We examined if ASIC3-related nociceptive proteins were altered in our treatment model. ASIC3 showed a normal distribution in the control group (Fig. 4A, 100.1 ± 2.0%, n = 6) and was upregulated at day 8 after FM induction (Fig. 4A, 120.0 ± 4.6%, p < 0.05 compared with Con group, n = 6). EA treatment restored ASIC3 levels (Fig. 4A, 100.2 ± 2.9%, p < 0.05 compared with FM group, n = 6). Interestingly, these EA-mediated effects were blocked by naloxone (Fig. 4A, 114.8 ± 3.2%, p < 0.05 compared with Con group, n = 6), rolofylline (Fig. 4A, 116.4 ± 5.0%, p < 0.05 compared with Con group, n = 6), or not existed in ASIC3−/− mice (Fig. 4A, 1.2 ± 0.3%, p < 0.05 compared with Con group, n = 6). Next, we assessed ASIC3-related downstream nociceptive signaling. Western blot results showed that the levels of pPKA, pPI3K, and pPKC were unchanged in all groups (Fig. 4B–D, all p > 0.05 compared with Con group, n = 6); thus, these molecules are not involved at the time point tested. Based on our previous results, we assessed if pERK-positive neurons were altered7. However, we found that pERK levels were similar in all groups (Fig. 4E, all p > 0.05, n = 6). We next examined nociceptive-relevant Nav channels. Nav1.7 was increased in FM mice (Fig. 4F, 132.2 ± 7.7%, p < 0.05 compared with Con group, n = 6) and restored to normal levels in the EA group (Fig. 4F, 102.7 ± 5.5%, p < 0.05 compared with FM group, n = 6). The EA-mediated Nav1.7 reduction was blocked by opioid, adenosine antagonists, or ASIC3 gene deletion (Fig. 4F, 94.8 ± 5.8%, p > 0.05 compared with Con group, n = 6). Similar effects on Nav1.8 protein levels were found (Fig. 4G, 129.2 ± 7.0%, p < 0.05 compared with Con group, n = 6). Therefore, we speculate that the ASIC3-Nav1.7/Nav1.8 signaling pathway is responsible for persistent FM-induced hyperalgesia in the peripheral DRG.

(A) ASIC3, (B) pPKA, (C) pPI3K, (D) pPKC, (E) pERK, (F) Nav1.7, and (G) Nav1.8 expression levels in tissues from the Con, FM, EA, Nal, Rol, and ASIC3 null groups (from left to right). Con = Control; FM = Fibromyalgia group; EA = Electroacupuncture; Nal = Naloxone group; Rol = Rolofyllin group. ASIC3 null = ASIC3 gene deletion group. *p < 0.05 compared with the Con group. #p < 0.05 compared with the FM group. The western blot bands at the top show the cropped target protein. The lower bands are cropped internal controls (β-actin or α-tubulin).
EA attenuates the upregulation of ASIC3, Nav1.7, and Nav1.8 in the SC of FM mice via opioid and adenosine pathways
ASIC3 was distributed in the SC of control mice (Fig. 5A, 100.6 ± 0.4%, n = 6). After 8 days, FM mice showed an increase in ASIC3 at day 8 after FM induction (Fig. 5A, 127.9 ± 4.9%, p < 0.05 compared with Con group, n = 6). This increase was alleviated by 2 Hz EA stimulation at acupoint ST36 (Fig. 5A, 99.6 ± 3.5%, p < 0.05 compared with FM group, n = 6). Furthermore, naloxone (Fig. 5A, 125.0 ± 5.9%, p < 0.05 compared with Con group, n = 6) and rolofylline (Fig. 5A, 119.4 ± 3.0%, p < 0.05 compared with Con group, n = 6) blocked the effect of EA on ASIC3, and not existed in ASIC3−/− mice (Fig. 5A, 0.9 ± 0.2%, p < 0.05 compared with Con group, n = 6); thus, EA acts through opioid and adenosine receptors. In addition, we assessed the levels of protein kinases, which are involved in several pathways. Proteins pPKA, pPI3K, and pPKC showed no change in the SC of FM mice on Day 8 after modeling (Fig. 5B–D, all p > 0.05, n = 6). Downstream of protein kinase, pERK was also unaltered on Day 8 (Fig. 5E, p > 0.05, n = 6). Thus, all of the tested kinases showed similar levels across all groups. By contrast, we determined that Nav1.7 was potentiated in the SC of mice on Day 8 after FM induction (Fig. 5F, 119.2 ± 3.9%, p < 0.05 compared with Con group, n = 6). EA treatment restored Nav1.7 levels in FM mice (Fig. 5F, 102.7 ± 3.8%, p < 0.05 compared with FM group, n = 6), and this effect was blocked by naloxone (Fig. 5F, 115.7 ± 3.3%, p < 0.05 compared with Con group, n = 6), rolofylline (Fig. 5F, 120.1 ± 5.0%, p < 0.05 compared with Con group, n = 6), and ASIC3 gene deletion (Fig. 5F, 99.7 ± 9.1%, p > 0.05 compared with Con group, n = 6). Nav1.8 is crucial for FM development; therefore, we assessed if EA altered Nav1.8 expression. Nav1.8 protein signals were distributed in DRG neurons (Fig. 5G, 99.9 ± 0.3%, n = 6) and augmented by FM modeling on Day 8 (Fig. 5G, 135.3 ± 9.0%, p < 0.05 compared with Con group, n = 6). Nav1.8 levels were ameliorated by EA at 2 Hz, suggesting that EA alleviates FM-mediated nociception (Fig. 5G, 104.4 ± 3.8%, p < 0.05 compared with FM group, n = 6). The effects of EA were blocked by naloxone (Fig. 5G, 127.7 ± 4.5%, p < 0.05 compared with Con group, n = 6), rolofylline (Fig. 5G, 126.6 ± 4.1%, p < 0.05 compared with Con group, n = 6), or ASIC3 gene deletion (Fig. 5G, 98.3 ± 7.8%, p > 0.05 compared with Con group, n = 6). These data support the hypothesis that EA acts via opioid and adenosine pathways in the SC.

(A) ASIC3, (B) pPKA, (C) pPI3K, (D) pPKC, (E) pERK, (F) Nav1.7, and (G) Nav1.8 expression levels in tissues from the Con, FM, EA, Nal, Rol, and ASIC3 null groups (from left to right). Con = Control; FM = Fibromyalgia group; EA = Electroacupuncture; Nal = Naloxone group; Rol = Rolofyllin group. ASIC3 null = ASIC3 gene deletion group (from left to right). Con = Control; FM = Fibromyalgia group; EA = Electroacupuncture; Nal = Naloxone group; Rol = Rolofyllin group. *p < 0.05 compared with the Con group. #p < 0.05 compared with the FM group. The western blot bands at the top show the cropped target protein. The lower bands are cropped internal controls (β-actin or α-tubulin).
EA blocks the increase in ASIC3, pPKA, pPI3K, and pERK signaling pathway in the thalamus of FM mice via opioid and adenosine pathways
ASIC3 was normally distributed in the thalamus of control mice (Fig. 6A, 100.0 ± 4.5%, n = 6), increased in FM mice at day 8 after FM induction (Fig. 6A, 124.1 ± 2.9%, p < 0.05 compared with Con group, n = 6), and restored by EA (Fig. 6A, 98.4 ± 5.8%, p < 0.05 compared with FM group, n = 6). The rescue effect of EA on ASIC3 was mediated by opioid (Fig. 6A, 114.6 ± 5.2%, p < 0.05 compared with Con group, n = 6), adenosine pathways (Fig. 6A, 113.9 ± 6.2%, p < 0.05 compared with Con group, n = 6), or not existed in ASIC3−/− mice (Fig. 6A, 1.2 ± 0.7%, p < 0.05 compared with Con group, n = 6). An increase of pPKA was found in the thalamus of FM mice (Fig. 6B, 121.5 ± 7.4%, p < 0.05 compared with Con group, n = 6) and rescued by EA (Fig. 6B, 97.5 ± 6.5%, p < 0.05 compared with Con group, n = 6). The effect of EA was blocked by opioid (Fig. 6B, 114.3 ± 6.6%, p < 0.05 compared with Con group, n = 6), adenosine (Fig. 6B, 115.5 ± 5.5%, p < 0.05 compared with Con group, n = 6) antagonists, or ASIC3 gene deletion (Fig. 6B, 99.5 ± 8.4%, p > 0.05 compared with Con group, n = 6) Similar data were obtained for pPI3K (Fig. 6C, n = 6) but not for pPKC (Fig. 6D, all p > 0.05, n = 6). pERK was increased in the thalamus of FM mice, which is consistent with a previous report33. The potentiation of pERK was reversed by EA via opioid and adenosine pathways (Fig. 6E, n = 6). The potentiation of pERK was also reversed in ASIC3−/− mice (Fig. 6E, 99.5 ± 8.4%, p > 0.05 compared with Con group, n = 6). Nav1.7 was increased in the thalamus of FM mice (Fig. 6F, 132.5 ± 6.4%, p < 0.05 compared with Con group, n = 6) and rescued by EA (Fig. 6F, 98.2 ± 5.9%, p < 0.05 compared with FM group, n = 6) via opioid (Fig. 6F, 114.4 ± 6.3%, p < 0.05 compared with Con group, n = 6), adenosine (Fig. 6F, 111.3 ± 6.0%, p < 0.05 compared with Con group, n = 6), and ASIC3 pathways (Fig. 6F, 81.8 ± 7.2%, p > 0.05 compared with Con group, n = 6). A similar pattern was found for Nav1.8 (Fig. 6G, n = 6). These results indicate that ASIC3 in the thalamus is crucial for mechanical hyperalgesia in the FM mouse model.

(A) ASIC3, (B) pPKA, (C) pPI3K, (D) pPKC, (E) pERK, (F) Nav1.7, and (G) Nav1.8 expression levels in tissues from the Con, FM, EA, Nal, Rol, and ASIC3 null groups (from left to right). Con = Control; FM = Fibromyalgia group; EA = Electroacupuncture; Nal = Naloxone group; Rol = Rolofyllin group. ASIC3 null = ASIC3 gene deletion group (from left to right). Con = Control; FM = Fibromyalgia group; EA = Electroacupuncture; Nal = Naloxone group; Rol = Rolofyllin group. *p < 0.05 compared with the Con group. #p < 0.05 compared with the FM group. The western blot bands at the top show the cropped target protein. The lower bands are cropped internal controls (β-actin or α-tubulin).
ASIC3, Nav1.7, and Nav1.8 immunoreactive signals were increased on Day 8 after FM induction and further attenuated by EA using immunohistochemical staining
Immunohistochemical labeling visualized by green fluorescence indicated that ASIC3 was existed in DRG neurons, dramatically increased 8 days after FM induction, and further reversed by EA (Fig. 7A–C). The effect of EA was reduced by naloxone and rolofylline (Fig. 7D and E). Furthermore, DRG neurons showed Nav1.7-positive signals in control mice, increased in FM, and further reversed by EA (Fig. 7F–H). The effect of EA was reversed by naloxone and rolofylline (Fig. 7I and J). Nociceptive Nav1.8-positive DRG neurons were normally distributed in control mice, potentiated in FM, and reversed by EA (Fig. 7K–M). The phenomenon was abolished in naloxone and rolofylline groups (Fig. 7N and O).

ASIC3-positive neurons (green) in the DRG of (A) Con, (B) FM, (C) EA, (D) Nal, and (E) Rol mice. Nav1.7-positive neurons (green) in the DRG of (F) Con, (G) FM, (H) EA, (I) Nal, and (J) Rol mice. Nav1.8-positive neurons (green) in the DRG of (K) Con, (L) FM, (M) EA, (N) Nal, and (O) Rol mice. Scale bar means 100 μm. Arrows identify immunopositive neurons.
Voltage-gated sodium currents in DRG neurons
To investigate whether voltage-gated sodium currents were affected by acid saline injection produced FM mice, we conducted whole-cell patch recording to measure the ASIC3 inward currents or voltage-gated sodium currents. In DRG neurons, acid-induced ASIC3 currents (Fig. 8A) or voltage-gated sodium currents (Fig. 8B) were significantly increased in DRG neurons 8 days after FM induction and further reversed by EA and ASIC3 gene deletion (Fig. 8A and B). Furthermore, acid saline (pH 5.0) significantly potentiated the voltage-gated sodium currents in control DRG (Fig. 8C) that can be abolished by ASIC3 blocker salicylic acid (SA) or ERK antagonist U0126 (Fig. 8C). All data were analyzed and plotted in Fig. 8A–C. These results provide evidences that acid could increase the amplitudes of voltage-gated sodium currents through ASIC3 and ERK pathways.

(A) Representative acid-sensing ion channel 3 currents traces in Con, FM, EA, and ASIC3−/− groups. The acid-sensing ion channel 3 currents were induced by injection of pH5.0 saline directly to DRG neurons. (B) Representative voltage-gated sodium currents traces in Con, FM, EA, and ASIC3−/− groups. The voltage-gated sodium currents were induced by membrane depolarization from −70 to 0 mV. (C) Representative voltage-gated sodium currents traces in pH 7.4, pH 5.0, pH 5.0 + SA, and pH 5.0 + U0126 groups. (A) Mean peak amplitudes of acid-sensing ion channel 3 currents in Con, FM, EA, and ASIC3−/− groups. (B) Mean peak amplitudes of voltage-gated sodium currents in Con, FM, EA, and ASIC3−/− groups. (C) Mean peak amplitudes of voltage-gated sodium currents traces in pH 7.4, pH 5.0, pH 5.0 + SA, and pH 5.0 + U0126 groups. *p < 0.05 compared with Con or pH 7.4 groups. #p < 0.05 compared with FM or pH 5.0 groups.
Discussion
ASIC3 is involved in several types of pain syndromes such as inflammation and FM, that are highly associated with lower local pH13,15. These conditions activate ASIC3 via low pH, which initiates a transient inward current followed by a sustained inward current. The sustained currents from ASIC3 significantly prolong the sensation of acidosis pain in FM, arthritis, and inflammatory pain46,47. In this study, dual acid injections significantly initiated mechanical hyperalgesia via ASIC3, Nav1.7, and Nav1.8 signaling in both the peripheral DRG and the central SC. The potentiated ASIC3 signaling may respond to mechanical hyperalgesia from the peripheral acidosis site and transduce the acid-related pain signaling. Jeong et al. reported that ASIC3 was increased in the dorsal horn of the spinal cord in a spinal nerve ligation model, and this effect was ameliorated by amiloride, an ASIC3 blocker48.
Injections of the non-specific ASIC blocker amiloride, specific ASIC3 blocker APETx2, and artificial miRNA attenuated mechanical hyperalgesia in mice49,50,51. Izuma et al. demonstrated that ASIC3 in knee joint afferents was dramatically increased in an osteoarthritic mouse model. Injection of APETx2 reliably inhibited ASIC3 potentiation and pain behaviors52. Furthermore, in ASIC3−/− mice, mechanical hyperalgesia after the induction of muscle inflammation was abolished53. Our previous results showed that intraplantar inflammation-mediated mechanical hyperalgesia was attenuated in ASIC3−/− mice54. Repeated acidic saline injection-induced FM pain was not observed in ASIC3−/− but was found in ASIC1−/− mice, highlighting the crucial role of ASIC3 in this model21. A recent study showed that both the mechanical and thermal hyperalgesia initiated by two acidic saline injections were significantly reversed by 15 and 100 Hz EA55. Here we further determined that the EA-mediated attenuation of mechanical hyperalgesia was caused by a reduction in ASIC3, Nav1.7, and Nav1.8 proteins in both the peripheral DRG and central SC.
The Nav1.8 sodium channel was increased in rat and mouse DRG neurons after carrageenan and CFA-induced inflammatory pain24,56. Laird et al. showed that visceral pain and referred hyperalgesia were abolished in Nav1.8-null mice57. Intrathecal Nav1.8 antisense injections blocked Nav1.8 currents and attenuated mechanical allodynia after an intraplantar CFA injection58. Recently, A-803467, a novel specific blocker for Nav1.8, reduced nociception in animal models of neuropathic and inflammatory pain59. Our previous study showed that EA attenuated inflammatory pain by reducing Nav1.8 protein expression and functional currents60. Nielsen et al. reported that the sodium channel blocker mexiletine reliably reduce nociception of repeated injections of acidic saline38. We determined that EA attenuated mechanical hyperalgesia in FM mice by reducing ASIC3, Nav1.7, and Nav1.8 protein overexpression.
Conclusion
In this study, EA at acupoint ST36 reliably reduced mechanical hyperalgesia and motor dysfunction in acidic saline injection-induced FM mice. Both nociceptive behavior and priming molecules were abolished in ASIC3-null mice, highlighting the crucial role of this protein in FM hyperalgesia. The pPKA, pPI3K, and pERK signaling pathway was potentiated in the DRG and SC during acid injection-induced hyperalgesia priming. ASIC3, Nav1.7, and Nav1.8 proteins were increased 8 days after FM modeling, and this effect was attenuated in the DRG and SC of FM mice by EA at acupoint ST36. Furthermore, the ASIC, pPKA, pPI3K, pERK, and Nav signaling pathway was increased in the thalamus, and this effect was attenuated by EA. A similar pattern was observed for pERK. We also provide physiological evidences that voltage-gated sodium currents were increased in FM DRGs and reduced in EA or ASIC3−/− mice. Acid saline potentiated sodium currents through ASIC3 receptors and ERK pathway. Our results provide highly valuable data for the investigation of EA-related analgesic mechanisms and can be applied in clinical practice.
Methods
Animals
Experiments were conducted using C57/B6 mice (ages 8 to 12 weeks) purchased from BioLASCO Co. Ltd, Taipei, Taiwan. The sample size required for an alpha of 0.05 and a power of 80% was eight animals per group. After arrival, the mice were housed under a 12/12 h light/dark cycle, where water and food were available ad libitum. All of the procedures were approved by the Institute of Animal Care and Use Committee of China Medical University (permit No. 2016–061) and they were conducted in accordance with the Guide for the use of Laboratory Animals provided by the National Research Council and the ethical guidelines of the International Association for the Study of Pain. The number of animals used and their suffering were minimized.
EA treatment and pharmacological injection
EA was applied using stainless steel needles (0.5″ inch, 32 G, YU KUANG, Taiwan) that were inserted into the muscle layer to a depth of 2–3 mm at ST36 acupoint. EA was administered immediate after the second injection acid saline every day at the same time (10:00–12:00 AM). A Trio-300 (Japan) stimulator delivered electrical square pulses for 15 min with a 100 μs duration and a 2 Hz frequency. The stimulation amplitude was 1 mA. For pharmacological injection, opioid or adenosine A1 receptor antagonist administration, the opioid antagonist naloxone methiodide (Nal) (Sigma, St. Louis, MO, USA) in 100 μl of saline was injected i.p. at a dose of 10 mg/kg. The adenosine A1 receptor antagonist rolofylline (Ro) (Sigma, St. Louis, MO, USA) in 10 μl of saline was injected i.m. at a dose of 3 mg/kg into acupoint ST36. The opioid agonist endomorphin (EM) (Sigma, St. Louis, MO, USA), in 100 μl of saline, was administered intraperitoneally (i.p.) at a dose of 10 mg/kg once a day. Alternatively, the adenosine receptor agonist N6-cyclopentyladenosine (CPA) (Sigma, St. Louis, MO, USA) in 10 μl of saline was administered intramuscularly (i.m.) at a dose of 0.1 mg/kg into acupoint ST36 once a day. A dose of 20 pmole APETx2 (in 20 μL acid saline with a concentration of 1 μM APETx2) was injected into ST36 acupoint under light isoflurane anesthesia (1%).
FM induction and animal behavior of mechanical hyperalgesia
We injected 20 μL of pH 4.0 acid saline into gastrocnemius muscle (GM) while the mice were anesthetized with isoflurane (1%). The second acid saline injection was performed at day 5 after first injection to successfully induce FM mice. All experiments were performed at room temperature (approximately 25 °C) and the stimuli were applied only when the animals were calm but not sleeping or grooming. Mechanical sensitivity was measured by testing the force of responses to stimulation with three applications of electronic von Frey filaments (North Coast Medical, Gilroy, CA, USA). Mice were placed on a metal mesh and adapted to the new environment for at least 30 min. The mechanical hyperalgesia of the hindpaw was measured before, 4 h, 1, 5, 6, 8 days after modeling. The FM mice were further euthanized and the L3-L5 DRG neurons, lumbar SC, and brain thalamus were isolated for analysis.
Rotarod
The mice were put on a rotating machine with different speeds and durations can be tested. When mice fall down, the sensor can record the falling latency. Mice were placed on an accelerating rotarod apparatus (MK-660D, Muromachi Kikai, Tokyo, Japan) for 12 trials (4 trials per day on 3 consecutive days; D1-D3) with 5-min intervals between trials. Each trial lasted for 60 s with a steady speed of 4 rpm. The latency of each mouse falling from the rod was recorded for each trial. On day 4, mice underwent 4 rpm and increase the speeds with a 3 rpm increase with a 10 s duration.
Tissue sampling and western blot analysis
L3-L5 DRG, lumbar SC, and thalamus tissues were excised to extract proteins. The total proteins were prepared by homogenizing the DRG, SC, and thalamus in lysis buffer containing 50 mM Tris-HCl (pH 7.4), 250 mM NaCl, 1% NP-40, 5 mM EDTA, 50 mM NaF, 1 mM Na3VO4, 0.02% NaNO3, and 1 × protease inhibitor cocktail (AMRESCO). The extracted proteins (30 μg per sample according to the BCA protein assay) were subjected to 8% SDS-Tris glycine gel electrophoresis and transferred to a PVDF membrane. The membrane was blocked with 5% non-fat milk in TBS-T buffer (10 mM Tris pH 7.5, 100 mM NaCl, 0.1% Tween 20), incubated with the first antibody in TBS-T and 1% bovine serum albumin, and incubated for 1 h at room temperature. A peroxidase-conjugated anti-rabbit antibody (1:5000) was used as the secondary antibody. The bands were visualized using an enhanced chemiluminescencent substrate kit (PIERCE) with LAS-3000 Fujifilm (Fuji Photo Film Co. Ltd). If appropriate, the image intensities of specific bands were quantified with NIH ImageJ software (Bethesda, MD, USA). The protein ratios were obtained by dividing the target protein intensities by the intensity of α-tubulin in the same sample. The calculated ratios were then adjusted by dividing the ratios from the same comparison group relative to the control.
Immunohistochemical staining
Mice were anesthetized with isoflurane and then perfused transcardially with 4% paraformaldehyde. The tissue samples were cryprotected with 30% sucrose. The tissues were cut to a thickness of 15 μm and were post-fixed briefly with 4% paraformaldehyde and then incubated with blocking solution containing 3% BSA, 0.1% Triton X-100, and 0.02% sodium azide in PBS for 2 h at room temperature. After blocking, the sections were incubated at 4 °C overnight with the primary antibodies prepared in blocking solution. The secondary antibody was goat anti-rabbit (1:500) antibody (Molecular Probes, Carlsbad, CA, USA). We incubated the slices with fluorescence-conjugated secondary antibodies or avidin-biotin horseradish peroxidase complex (1 h), washed them three times with 0.1 M Tris buffer (5 min each), and then developed them in diaminobenzidine tetrahydrochloride (1–2 min), before washing three times with 0.1 M Tris buffer (5 min each). Finally, the sections were incubated with 0.1 M Tris buffer to stop the reaction. The slides were mounted with cover slips and visualized by using a CKX41 microscope with an Olympus U-RFLT50 Power Supply Unit (Olympus, Tokyo, Japan).
Electrophysiology
L3–L5 DRGs were isolated from mice at 8 days after FM injection. For ASIC3 currents recording (checked by SA inhibition), recording cells were superfused in artificial cerebrospinal fluid (ACSF) containing (in mM) 130 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, and 20 Hepes, adjusted to pH 7.4 with NaOH. ACSF solutions were applied by use of gravity. The recording electrodes were filled with (in mM) 100 KCl, 2 Na2-ATP, 0.3 Na3-GTP, 10 EGTA, 5 MgCl2, and 40 Hepes, adjusted to pH 7.4 with KOH. The pH 5.0 ACSF was titrated by 2-[N-morpholino]ethanesulfonic acid (MES). For Nav current recording, the internal solution contained (in mM) 10 NaCl, 110 CsCl, 20 tetraethylammonium-Cl, 2.5 MgCl2, 5 EGTA, 3 Mg2+-ATP, and 5 HEPES, adjusted to pH 7.2 with CsOH. The external solution contained (in mM) 100 NaCl, 5 CsCl, 30 tetraethylammonium-Cl, 1.8 CaCl2, 1 MgCl2, 0.1 CdCl2, 25 glucose, 5 4-aminopyridine, and 5 HEPES, adjusted to pH 7.4 with HCl. Osmolarity was adjusted to 300 mosm. Voltage-gated sodium channel currents were evoked by a 50 ms test pulse from −70 to 0 mV. All recordings were obtained at room temperature (25 °C) and completed within 24 h after plating. Salicylic acid (SA) was prepared from a 1-M stock solution (in 100% ethanol) to a final concentration of 500 μM in ACSF. U0126 was from Tocris-Cookson (Bristol, UK).
Statistical analysis
All of the data were expressed as the mean ± standard error. Significant differences between groups were tested using ANOVA, followed by a post hoc Tukey’s test. p < 0.05 was considered significantly different.
Additional Information
How to cite this article: Yen, L.-T. et al. Targeting ASIC3 for Relieving Mice Fibromyalgia Pain: Roles of Electroacupuncture, Opioid, and Adenosine. Sci. Rep. 7, 46663; doi: 10.1038/srep46663 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Vierck, C. J. Jr. Mechanisms underlying development of spatially distributed chronic pain (fibromyalgia). Pain 124, 242–263 (2006).
DeSantana, J. M. & Sluka, K. A. Central mechanisms in the maintenance of chronic widespread noninflammatory muscle pain. Current pain and headache reports 12, 338–343 (2008).
Roth, T., Arnold, L. M., Garcia-Borreguero, D., Resnick, M. & Clair, A. G. A review of the effects of pregabalin on sleep disturbance across multiple clinical conditions. Sleep medicine reviews 18, 261–271 (2014).
Smith, M. T. & Moore, B. J. Pregabalin for the treatment of fibromyalgia. Expert opinion on pharmacotherapy 13, 1527–1533 (2012).
Hauser, W., Walitt, B., Fitzcharles, M. A. & Sommer, C. Review of pharmacological therapies in fibromyalgia syndrome. Arthritis research & therapy 16, 201 (2014).
Trugman, J. M., Palmer, R. H. & Ma, Y. Milnacipran effects on 24-hour ambulatory blood pressure and heart rate in fibromyalgia patients: a randomized, placebo-controlled, dose-escalation study. Current medical research and opinion 30, 589–597 (2014).
Lin, J. G., Hsieh, C. L. & Lin, Y. W. Analgesic Effect of Electroacupuncture in a Mouse Fibromyalgia Model: Roles of TRPV1, TRPV4, and pERK. PloS One 10, e0128037 (2015).
Sluka, K. A., Kalra, A. & Moore, S. A. Unilateral intramuscular injections of acidic saline produce a bilateral, long-lasting hyperalgesia. Muscle & nerve 24, 37–46 (2001).
Sluka, K. A., Rohlwing, J. J., Bussey, R. A., Eikenberry, S. A. & Wilken, J. M. Chronic muscle pain induced by repeated acid Injection is reversed by spinally administered mu- and delta-, but not kappa-, opioid receptor agonists. The Journal of pharmacology and experimental therapeutics 302, 1146–1150, doi: 10.1124/jpet.102.033167 (2002).
Skyba, D. A., King, E. W. & Sluka, K. A. Effects of NMDA and non-NMDA ionotropic glutamate receptor antagonists on the development and maintenance of hyperalgesia induced by repeated intramuscular injection of acidic saline. Pain 98, 69–78 (2002).
Lin, C. C. et al. An antinociceptive role for substance P in acid-induced chronic muscle pain. Proceedings of the National Academy of Sciences of the United States of America 109, E76–83 (2012).
Chen, W. N. et al. Roles of ASIC3, TRPV1, and NaV1.8 in the transition from acute to chronic pain in a mouse model of fibromyalgia. Molecular pain 10 (2014).
Vilin, Y. Y., Peters, C. H. & Ruben, P. C. Acidosis differentially modulates inactivation in na(v)1.2, na(v)1.4, and na(v)1.5 channels. Frontiers in pharmacology 3, 109 (2012).
Chen, C. C. et al. A role for ASIC3 in the modulation of high-intensity pain stimuli. Proceedings of the National Academy of Sciences of the United States of America 99, 8992–8997 (2002).
Chen, W. H. et al. Acid-sensing ion channel 3 mediates peripheral anti-hyperalgesia effects of acupuncture in mice inflammatory pain. Journal of biomedical science 18, 82 (2011).
Wemmie, J. A., Price, M. P. & Welsh, M. J. Acid-sensing ion channels: advances, questions and therapeutic opportunities. Trends in neurosciences 29, 578–586 (2006).
Deval, E. et al. Acid-sensing ion channels (ASICs): pharmacology and implication in pain. Pharmacology & therapeutics 128, 549–558 (2010).
Waldmann, R. & Lazdunski, M. H(+)-gated cation channels: neuronal acid sensors in the NaC/DEG family of ion channels. Current opinion in neurobiology 8, 418–424 (1998).
Sakai, H., Lingueglia, E., Champigny, G., Mattei, M. G. & Lazdunski, M. Cloning and functional expression of a novel degenerin-like Na+ channel gene in mammals. The Journal of physiology 519 Pt 2, 323–333 (1999).
Price, M. P. et al. The DRASIC cation channel contributes to the detection of cutaneous touch and acid stimuli in mice. Neuron 32, 1071–1083 (2001).
Sluka, K. A. et al. Chronic hyperalgesia induced by repeated acid injections in muscle is abolished by the loss of ASIC3, but not ASIC1. Pain 106, 229–239 (2003).
Crill, W. E. Persistent sodium current in mammalian central neurons. Annual review of physiology 58, 349–362 (1996).
Baker, M. D. & Bostock, H. Low-threshold, persistent sodium current in rat large dorsal root ganglion neurons in culture. Journal of neurophysiology 77, 1503–1513 (1997).
Black, J. A., Liu, S., Tanaka, M., Cummins, T. R. & Waxman, S. G. Changes in the expression of tetrodotoxin-sensitive sodium channels within dorsal root ganglia neurons in inflammatory pain. Pain 108, 237–247 (2004).
Baker, M. D. & Wood, J. N. Involvement of Na+ channels in pain pathways. Trends in pharmacological sciences 22, 27–31 (2001).
Lu, K. W., Hsu, C. K., Hsieh, C. L., Yang, J. & Lin, Y. W. Probing the Effects and Mechanisms of Electroacupuncture at Ipsilateral or Contralateral ST36-ST37 Acupoints on CFA-induced Inflammatory Pain. Scientific reports 6, 22123 (2016).
Waxman, S. G. The molecular pathophysiology of pain: abnormal expression of sodium channel genes and its contributions to hyperexcitability of primary sensory neurons. Pain Suppl 6, S133–140 (1999).
Gold, M. S., Levine, J. D. & Correa, A. M. Modulation of TTX-R INa by PKC and PKA and their role in PGE2-induced sensitization of rat sensory neurons in vitro . The Journal of neuroscience: the official journal of the Society for Neuroscience 18, 10345–10355 (1998).
Villarreal, C. F., Sachs, D., Cunha, F. Q., Parada, C. A. & Ferreira, S. H. The role of Na(V)1.8 sodium channel in the maintenance of chronic inflammatory hypernociception. Neuroscience letters 386, 72–77 (2005).
Khasar, S. G., McCarter, G. & Levine, J. D. Epinephrine produces a beta-adrenergic receptor-mediated mechanical hyperalgesia and in vitro sensitization of rat nociceptors. Journal of neurophysiology 81, 1104–1112 (1999).
Dina, O. A., McCarter, G. C., de Coupade, C. & Levine, J. D. Role of the sensory neuron cytoskeleton in second messenger signaling for inflammatory pain. Neuron 39, 613–624 (2003).
Chen, W. N. & Chen, C. C. Acid mediates a prolonged antinociception via substance P signaling in acid-induced chronic widespread pain. Molecular pain 10, 30 (2014).
Chen, W. K. et al. Ca(v)3.2 T-type Ca2+ channel-dependent activation of ERK in paraventricular thalamus modulates acid-induced chronic muscle pain. The Journal of neuroscience: the official journal of the Society for Neuroscience 30, 10360–10368 (2010).
Hoeger-Bement, M. K. & Sluka, K. A. Phosphorylation of CREB and mechanical hyperalgesia is reversed by blockade of the cAMP pathway in a time-dependent manner after repeated intramuscular acid injections. The Journal of neuroscience 23, 5437–5445 (2003).
Cheng, S. J. et al. Role of extracellular signal-regulated kinase in synaptic transmission and plasticity of a nociceptive input on capsular central amygdaloid neurons in normal and acid-induced muscle pain mice. The Journal of neuroscience: the official journal of the Society for Neuroscience 31, 2258–2270 (2011).
Gandhi, R., Ryals, J. M. & Wright, D. E. Neurotrophin-3 reverses chronic mechanical hyperalgesia induced by intramuscular acid injection. The Journal of neuroscience: the official journal of the Society for Neuroscience 24, 9405–9413 (2004).
Yokoyama, T., Maeda, Y., Audette, K. M. & Sluka, K. A. Pregabalin reduces muscle and cutaneous hyperalgesia in two models of chronic muscle pain in rats. The journal of pain: official journal of the American Pain Society 8, 422–429 (2007).
Nielsen, A. N., Mathiesen, C. & Blackburn-Munro, G. Pharmacological characterisation of acid-induced muscle allodynia in rats. European journal of pharmacology 487, 93–103 (2004).
Choowanthanapakorn, M., Lu, K. W., Yang, J., Hsieh, C. L. & Lin, Y. W. Targeting TRPV1 for Body Weight Control using TRPV1(−/−) Mice and Electroacupuncture. Scientific reports 5, 17366 (2015).
Lin, Y. W. & Hsieh, C. L. Auricular electroacupuncture reduced inflammation-related epilepsy accompanied by altered TRPA1, pPKCalpha, pPKCepsilon, and pERk1/2 signaling pathways in kainic acid-treated rats. Mediators of inflammation 2014, 493480 (2014).
Kuo, C. T., Lin, Y. W., Tang, N. Y., Cheng, C. Y. & Hsieh, C. L. Electric stimulation of the ears ameliorated learning and memory impairment in rats with cerebral ischemia-reperfusion injury. Scientific reports 6, 20381 (2016).
Lu, K. W., Hsieh, C. L., Yang, J. & Lin, Y. W. Effects of electroacupuncture in a mouse model of fibromyalgia: role of N-methyl-D-aspartate receptors and related mechanisms. Acupuncture in medicine (2016).
Wu, S. Y., Chen, W. H., Hsieh, C. L. & Lin, Y. W. Abundant expression and functional participation of TRPV1 at Zusanli acupoint (ST36) in mice: mechanosensitive TRPV1 as an “acupuncture-responding channel”. BMC complementary and alternative medicine 14, 96 (2014).
Han, J. S. Acupuncture: neuropeptide release produced by electrical stimulation of different frequencies. Trends in neurosciences 26, 17–22 (2003).
Goldman, N. et al. Adenosine A1 receptors mediate local anti-nociceptive effects of acupuncture. Nature neuroscience 13, 883–888 (2010).
Sutherland, S. P., Benson, C. J., Adelman, J. P. & McCleskey, E. W. Acid-sensing ion channel 3 matches the acid-gated current in cardiac ischemia-sensing neurons. Proceedings of the National Academy of Sciences of the United States of America 98, 711–716 (2001).
Lin, Y. W. et al. Identification and characterization of a subset of mouse sensory neurons that express acid-sensing ion channel 3. Neuroscience 151, 544–557 (2008).
Jeong, S., Lee, S. H., Kim, Y. O. & Yoon, M. H. Antinociceptive effects of amiloride and benzamil in neuropathic pain model rats. Journal of Korean medical science 28, 1238–1243 (2013).
Karczewski, J. et al. Reversal of acid-induced and inflammatory pain by the selective ASIC3 inhibitor, APETx2. British journal of pharmacology 161, 950–960 (2010).
Walder, R. Y. et al. ASIC1 and ASIC3 play different roles in the development of Hyperalgesia after inflammatory muscle injury. The journal of pain: official journal of the American Pain Society 11, 210–218 (2010).
Walder, R. Y., Gautam, M., Wilson, S. P., Benson, C. J. & Sluka, K. A. Selective targeting of ASIC3 using artificial miRNAs inhibits primary and secondary hyperalgesia after muscle inflammation. Pain 152, 2348–2356 (2011).
Izumi, M., Ikeuchi, M., Ji, Q. & Tani, T. Local ASIC3 modulates pain and disease progression in a rat model of osteoarthritis. Journal of biomedical science 19, 77 (2012).
Sluka, K. A. et al. ASIC3 in muscle mediates mechanical, but not heat, hyperalgesia associated with muscle inflammation. Pain 129, 102–112 (2007).
Yen, Y. T. et al. Role of acid-sensing ion channel 3 in sub-acute-phase inflammation. Molecular pain 5, 1 (2009).
Maciel, L. Y. et al. Electroacupuncture reduces hyperalgesia after injections of acidic saline in rats. Evidence-based complementary and alternative medicine 2014, 485043 (2014).
Strickland, I. T. et al. Changes in the expression of NaV1.7, NaV1.8 and NaV1.9 in a distinct population of dorsal root ganglia innervating the rat knee joint in a model of chronic inflammatory joint pain. European journal of pain 12, 564–572 (2008).
Laird, J. M., Souslova, V., Wood, J. N. & Cervero, F. Deficits in visceral pain and referred hyperalgesia in Nav1.8 (SNS/PN3)-null mice. The Journal of neuroscience 22, 8352–8356 (2002).
Jarvis, M. F. et al. A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proceedings of the National Academy of Sciences of the United States of America 104, 8520–8525 (2007).
Wu, D. F. et al. PKCepsilon phosphorylation of the sodium channel NaV1.8 increases channel function and produces mechanical hyperalgesia in mice. The Journal of clinical investigation 122, 1306–1315 (2012).
Huang, C. P. et al. Electroacupuncture Reduces Carrageenan- and CFA-Induced Inflammatory Pain Accompanied by Changing the Expression of Nav1.7 and Nav1.8, rather than Nav1.9, in Mice Dorsal Root Ganglia. Evidence-based complementary and alternative medicine 2013, 312184 (2013).
Acknowledgements
This study was supported by CMU under the Aim for Top University Plan of the Ministry of Education, Taiwan, MOST 105-2320-B-039-024, CMU105-S-09.
Author information
Authors and Affiliations
Contributions
H.C.L. and L.Y.W. wrote the manuscript, Y.L.T. and H.H.C. employed the experiments. All authors reviewed the manuscript and agreed for submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Yen, LT., Hsieh, CL., Hsu, HC. et al. Targeting ASIC3 for Relieving Mice Fibromyalgia Pain: Roles of Electroacupuncture, Opioid, and Adenosine. Sci Rep 7, 46663 (2017). https://doi.org/10.1038/srep46663
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep46663
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
