
Abstract
Development of a protective and broadly-acting vaccine against the most widely distributed human malaria parasite, Plasmodium vivax, will be a major step towards malaria elimination. However, a P. vivax vaccine has remained elusive by the scarcity of pre-clinical models to test protective efficacy and support further clinical trials. In this study, we report the development of a highly protective CSP-based P. vivax vaccine, a virus-like particle (VLP) known as Rv21, able to provide 100% sterile protection against a stringent sporozoite challenge in rodent models to malaria, where IgG2a antibodies were associated with protection in absence of detectable PvCSP-specific T cell responses. Additionally, we generated two novel transgenic rodent P. berghei parasite lines, where the P. berghei csp gene coding sequence has been replaced with either full-length P. vivax VK210 or the allelic VK247 csp that additionally express GFP-Luciferase. Efficacy of Rv21 surpassed viral-vectored vaccination using ChAd63 and MVA. We show for the first time that a chimeric VK210/247 antigen can elicit high level cross-protection against parasites expressing either CSP allele, which provide accessible and affordable models suitable to support the development of P. vivax vaccines candidates. Rv21 is progressing to GMP production and has entered a path towards clinical evaluation.
Similar content being viewed by others

Introduction
Plasmodium vivax malaria poses a major health risk to 2.85 billion people worldwide living in tropical, sub-tropical and temperate regions1. P. vivax is the most widely distributed human malaria parasite in the world and considered to be the most difficult to control and eliminate from endemic regions. This is largely due to the parasite’s ability to remain latent in the liver of infected people to subsequently reactivate, weeks to years after an initial infection2,3.
Plasmodium sporozoites enter the blood stream through an infectious mosquito bite and quickly migrate to invade the liver. This ‘pre-erythrocytic’ stage is an attractive vaccine target4 used by leading vaccine candidates. This includes the most advanced malaria vaccine RTS,S in the adjuvant AS01. This vaccine aims to prevent a P. falciparum infection by stimulating immune responses against the major Plasmodium sporozoite surface antigen, circumsporozoite protein (CSP). RTS,S/AS01 has undergone extensive clinical testing and has been shown to induce significant protective immunity against P. falciparum in phase III trials5. The homologous CSP protein of P. vivax (PvCSP) is also actively being investigated as a component of a pre-erythrocytic vaccine against P. vivax, with particular emphasis on a vaccine that includes the PvCSP VK210 and VK247 central repeats to protect against the major strains of P. vivax circulating worldwide6,7,8,9,10.
The evaluation of the protective efficacy of P. falciparum vaccine candidates and vaccine formulations has greatly benefited from immunization-challenges studies performed in clinical trials using wild-type parasites in controlled human malaria infection (CHMI) studies6,9,11,12,13,14. A similar direct evaluation of vaccines and protective immunity against P. vivax in CHMI studies has recently been reported7. This has been achieved despite the difficulty of working with P. vivax, as this species is difficult to maintain in long-term continuous in vitro cultures15,16,17,18, and the lack of a safe and effective way to clear latent P. vivax forms (hypnozoites) from the liver. While a non-human primate (Aotus) challenge model exists, these monkeys are not widely used, due to their limited accessibility, ethical issues and the requirement of splenectomy19,20. A transgenic rodent P. berghei parasite has been generated where the type 1 repeat region of PvCSP has been incorporated into the endogenous P. berghei CSP, and these parasites have been used to examine the role of monoclonal and polyclonal antibodies and virus-like particles (VLP)-based immunization in protection against a sporozoite infection in mice21,22. However, only the repeat region of VK210 has been used and the P. vivax CSP epitopes contained in the regions outside of this repeat are absent, thus limiting the use of these parasites in vaccine studies to only those candidates with VK210 epitopes within the repeat region.
Here we report the development of two rodent challenge models suitable to assess the protective efficacy of different PvCSP-based vaccine candidates. In these transgenic P. berghei parasite lines, PbCSP has been replaced with full-length P. vivax VK210 or VK247 CSP. Sporozoites of these transgenic parasites were used to challenge mice that were previously immunized with various clinically relevant vaccine candidates, expressing or presenting PvCSP that contained regions from VK210 or VK247 or both. An effective approach to generate protective immunity against P. falciparum in both animal models and in human volunteers consists of sequential immunization or heterologous ‘prime-boost’ regimes with different viral-vectored vaccines to achieve high frequencies of antigen-specific T-cell responses23,24. A prime-boost regimen for malaria has been well established in both, mouse and humans, involving an initial ‘prime’ immunization with the chimpanzee adenovirus 63 (ChAd63) expressing a Plasmodium antigen followed by a ‘boosting’ immunization with a modified vaccinia virus Ankara (MVA) expressing the same parasite antigen23,24,25. Using this approach, we tested various chimeric forms of PvCSP for efficacy against the transgenic parasites.
We also developed and tested a novel VLP using the Hepatitis B surface antigen platform to present the PvCSP antigen, which we called Rv21. VLPs are known to stimulate B cells to induce high antibody titers; indeed RTS,S/AS01 uses a similar VLP approach5, which presents a hybrid polypeptide consisting of a portion of the P. falciparum CSP repeat and C-terminal regions fused to the amino terminal end of the hepatitis B virus surface (HepB-S) protein26. This VLP has a four-fold excess of HBsAg monomers in the VLP, required to allow expression of the particle in Saccharomyces yeast. Here we report an improved VLP, called Rv21 expressed in Pichia pastoris. This is based on the recent successful use of the corresponding R21 P. falciparum vaccine in pre-clinical (Collins et al. submitted) and clinical trials (Venkatraman et al. unpublished). Both R21 and Rv21 comprise only the hybrid CS-HBsAg protein in the VLP with no additional HBsAg monomers.
Protective immune responses after vaccination with RTS,S are dependent primarily on antibody responses against the CS central repeat region27,28. We tested the efficacy of Rv21 in adjuvant vaccination using the new P. berghei-PvCSP challenge models in mice. The formulated Rv21 presents both VK210 and 247 repeats and the C-terminal portion of PvCSP on its surface. We show that vaccination with a chimeric VK210/247 antigen induces protective immune responses against sporozoites expressing both the PvCSP VK210 or VK247 alleles. Moreover, we found that an immunization protocol using Rv21 induced complete, sterile protection against a stringent sporozoite challenge, surpassing the efficacy of viral-vectored vaccines expressing a similar PvCSP antigen. This work identifies a highly promising new candidate sporozoite vaccine for P. vivax that provides protection against the two major allelic types and highlights the potential and utility of the rodent challenge model to support the development of an efficacious P. vivax malaria vaccine.
Results
Antigen-specific cellular immune responses are not induced in mice immunized with P. vivax circumsporozoite viral-vectored vaccines (PvCSP vv)
We first assessed whether viral-vectored vaccines (vv) expressing the P. vivax circumsporozoite protein (PvCSP) could induce antigen-specific cellular immune responses in inbred or outbred mouse strains. We generated four recombinant adenoviruses from the chimpanzee viral-vectored vaccines (ChAd63) and four Modified Vaccinia Ankara (MVA) vv expressing different regions of PvCSP (PvCSP vv). The resulting recombinant viruses contained a series of PvCSP expression cassettes encoding the PvCSP N- and C-terminal sequences of the Salvador I strain of Plasmodium vivax, including or excluding the most common central repeat sequences present in the two major PvCSP alleles VK210 and VK247 (Fig. 1A)8. First, a set of ChAd63 and MVA vv expressing only the N- and C- terminal sequences and no central repeats were generated (NC); a second set of vv contained a series of the most common VK210 repeats8 flanked by the same NC sequences (N210C); a third set expressed VK247 repeats flanked by NC (N247C). Finally we generated a ChAd63/MVA set expressing a chimeric CSP containing both type of repeats (VK210/247) flanked by NC (N210/247C). Groups of mice (n = 6) of four different strains received a homologous ChAd63-MVA prime-boost at an interval of eight weeks between immunizations. Cellular immune responses in mice were quantified two weeks after the final immunization using an ex vivo IFN-γ ELISpot assay by stimulating peripheral blood mononuclear cells (PBMCs) with a series of peptide pools designed to assess responses against either N210C or N247C (Fig. 1B,C). An initial experiment using the prime-boost regimen in inbred C57Bl/6 mice did not yield any positive responses (Fig. 1B,C; upper panel). This prompted the assessment of responses in additional mouse strains immunized using the same vaccination scheme. None of these other mouse strains (C3H/He, BALB/c or outbred CD1) showed detectable antigen-specific T cell responses (Fig. 1B,C) and we concluded that the PvCSP molecules that we used in our vv do not have immunodominant T cell epitopes in the animal models tested under our experimental conditions. These results are in agreement with earlier observations using a similar vaccine candidate expressing PvCSP9. Nevertheless, splenic follicular T helper cells (Tfh) could be playing a role to support the generation of PvCSP-specific antibody responses and would deserve further exploration.

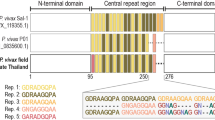
(A) Schematic representation of the CSP sequences expressed by ChAd63 and MVA vectors. NC: the amino (N)- and carboxyl-terminal (C)- sequences without central repeat regions (CR); N210C: a fragment containing the amino (N)- and carboxyl-terminal (C)- sequences flanking the VK210 allele; N247C: a fragment containing the amino (N)- and carboxyl-terminal (C)- sequences flanking the VK247 allele; N210/247CR:. a fragment containing the amino (N)- and carboxyl-terminal (C)- sequences flanking a chimeric fragment with both repeat sequences of the VK210 and VK247 central repeats, all contain an insertion region (IR); (B) Groups of mice of four strains (C57Bl6, C3H/HE, BALB/c, CD1, n = 6–8) were immunized with different vv using an Ad prime- and MVA boost regimen at an interval of 8 weeks and cellular responses were assessed by ex vivo IFN-γ ELISpot on week two after the MVA boost.
Humoral immune responses are induced in mice immunized with the PvCSP vv
We next assessed antibody responses induced by the ChAd63 and MVA vaccination protocols described above, using CD-1 mice. Serum samples were collected fortnightly up until week 16 after the first immunization. We observed that immunization with the vv expressing the chimeric construct N210/247C induced antibody responses to both VK210 and the VK247 PvCSP (Fig. 2, top panel). In contrast responses after an immunization with ChAd63 vv encoding only N210C or N247C, elicited antibodies only against their cognate version of PvCSP (Fig. 2). However, low antibody titres against the heterologous PvCSP allele were detected after a boost with the MVA vv, suggesting that several encounters with the antigen may stimulate low levels of cross-reactive antibody responses, possibly against the conserved N- and C-terminal domains both having a high level of identity of at least 93% in the amino acid sequence. No antibody responses were detected after a ChAd63-MVA immunization with either the NC or with vv expressing rodent P. berghei CSP.

Outbred CD-1 mice (n = 6) were immunized with vv expressing various versions of PvCSP (N210/N247C, N210C, N247C, NC; see Fig. 1) using a prime-boost interval of 8 weeks. Control groups were mock-vaccinated with vv expressing P. berghei CSP and naive mice were injected with PBS, and did not induce any antibodies. Serum was collected every two weeks and antibody titers –total IgG- were quantified using a standardized ELISA against the PvCSP VK210 and VK247 antigens.
P. vivax sporozoites isolated from Mexico (Tapachula, Chiapas) are recognized by mouse antibodies after PvCSP vv vaccination
We investigated whether mouse antibodies raised by immunisation with various PvCSP antigens encoded by the vv could recognize CSP on the surface of P. vivax sporozoites collected from mosquitoes in the endemic region of Tapachula, Chiapas29. We performed immunofluorescence assays (IFAs) using two types of sporozoites in separate slides (e.g. not as a mixing infection), VK210 and VK247. Assays were made using three different sporozoite batches and all yielded similar results. Results shown in Fig. 3 were obtained using sera from C57Bl/6 mice immunized with ChAd63-MVA expressing different regions of the CSP protein as described in Fig. 1a. CD-1 mouse sera yielded similar results (data not shown). Samples were collected at week 2 after the final boost immunization (Fig. 3). Serum from immunized mice reacted with both sporozoites expressing the VK210 allele (Fig. 3 top panel) and sporozoites expressing the VK247 allele (Fig. 3 bottom panel). Mouse sera at 1:500 dilution yielded better results than at 1:100 dilutions and fluorescent pattern was homogeneous with various degrees of intensity: weak, medium and strong. All pre-immune serum and those from primed AdC63 empty vector and boosted with MVA were negative. The monoclonal antibodies 5D3 (from tissue supernatant) and 2E10 (5 μg/ml) were used as positive control for the VK210 and VK247 variants, respectively.

Serum obtained from C57Bl/6 mice (n = 6) was incubated with P. vivax VK210 (top panel) or VK247 (bottom panel) sporozoites collected from the endemic state of Chiapas, Mexico. Each mouse group was immunized with ChAd63-MVA vv expressing homologous PvCSP transgenes (NC, N210C, N247C, N210/247C) and sera obtained on week two after the final immunization was used at 1:500 dilution. PBS or monoclonal antibodies with specificity to VK210 (Mab5D3, 2.5 μg/ml) or VK247 (Mab2E10, 5 μg/ml) were used as negative or positive controls, respectively. Experiment was made using 6 mice/group and three batches of sporozoites for each sample.
Development of a rodent challenge model for assessing protective efficacy of the PvCSP vaccination
In order to assess the in vivo protective efficacy of the P. vivax vaccines, we developed rodent challenge models consisting on transgenic P. berghei sporozoites where pbcsp was replaced with either full-length VK210 pvcsp or VK247 pvcsp. These transgenic parasites were generated using the gene insertion/marker out (GIMO) based transfection technology30,31 to generate two Double-step Replacement (DsR) mutants32,33 that resulted in the coding sequence (CDS) of P. berghei csp being replaced with the CDS of the pvcsp in a two-step GIMO-transfection procedure (Fig. 4). First, the pbcsp coding sequence (CDS) was deleted by replacement with the hdhfr::yfcu selectable marker cassette (SM) (Fig. 4, Step 1). Subsequently, in these pbcsp GIMO-deletion parasites the pvcsp CDS was inserted into the same locus thereby replacing hdhfr::yfcu with the pvcsp CDS (Fig. 4, Step 2). These parasites have the full-length pbcsp CDS replaced with pvcsp and were SM free. In the two transgenic parasite lines, PbANKA-PvCS VK210(r)PbCS (line 2196cl1) and PbANKA-PvCS VK247(r)PbCS (line 2199cl1) the pvcsp gene is under the control of the pbcsp gene promoter (5′UTR) and the transcription terminator (3′UTR) regulatory elements. In addition to expressing PvCSP, these transgenic parasites constitutively express the fusion GFP::luciferase reporter protein, which permits a determination of parasite liver-loads by real-time in vivo imaging (Fig. 4B). Correct replacement of the pbcsp CDS by the pvcsp CDS in the two transgenic lines was confirmed by diagnostic Southern analysis of chromosomes separated by pulsed-field gel electrophoresis and diagnostic PCR on genomic DNA (Fig. 4C,D). Immunofluorescence microscopy of sporozoites using anti-P. vivax CSP monoclonal antibodies to the repeat region of the VK210 or VK247 proteins confirmed the expression of either PvCSP VK210 or VK247 in the two transgenic parasite lines (Fig. 5A,B). The P. berghei CSP-specific monoclonal antibody 3D11 confirmed expression P. berghei CSP in wild-type P. berghei sporozoites but anti-P. berghei CSP staining was absent in both PbANKA-PvCS VK210(r)PbCS and PbANKA-PvCS VK247(r)PbCS sporozoites (Fig. 5C)34,35.

(A) Schematic representation describing the generation of PbANKA-PvCSP(r)PbCSP (2196cl1 and 2199cl1) chimeric lines. The P. berghei csp (Pbcsp) gene coding sequence (CDS) was replaced by the GIMO deletion-construct (construct 1; pL1929) using the positive/negative selectable maker (SM; hdhfr::yfcu) cassette, resulting in the generation of the Pbcsp GIMO line (PbANKA-PbCSP GIMO; 2151cl1) after positive selection with pyrimethamine. Step 2: The GIMO insertion-construct (construct 2; pL1942 or pL1943) replaced the SM in the GIMO line with Pvcsp VK-210 or VK-247 CDS, respectively, after negative selection using 5-fluorocytosine (5-FC). (B) Schematic representation of the reporter PbANKA parasite line PbGFP-Luceef1α (676m1cl1), used to generate the replacement gene [RG] chimeric parasites. (C) Genotype analysis of Replacement Gene [RG] chimeric parasites and their intermediate GIMO mother-line knock out chimeric parasites using Southern analysis of chromosomes (chrs) separated by pulsed-field gel electrophoresis (PFGE). Left panel: PbANKA- ΔCSP GIMO: 2151 cl1. The correct integration of the SM in the right locus in Chr-4 and replacing the endogenous PbCSP gene (PBANKA_040320) was confirmed by using the 3′UTR Pbdhfr/ts probe. Right panel: PbANKA-PvCSP(r)PbCSP: 2196cl1 and 2199cl1. Confirmation of the correct integration of the PvCSP expression construct into the GIMO locus by Southern blot. (D) Genotype analysis by diagnostic PCR analysis of chimeric parasite lines to confirm the correct integration of both PvCSP VK210 and VK247 antigen expression cassettes. Primers sequences used are shown in Table S1, while the expected PCR product sizes and the primer numbers are listed in the table beside the Agarose gel image.

Sporozoites from mosquito salivary-glands were stained with anti-PvCSP-210 (MR4) diluted 200 times, anti-PvCSP-247 (MR4) diluted 200 times, and anti-PbCSP (3D11) diluted 1000 times (3D11 stock (1000X) concentration was at 10 mg/ml and final working concentration was 10 μg/ml). Alexa Fluor® 488-labelled anti-mouse IgG and Hoechst-33342 were also included for the assay. (A) Transgenic parasite line 2196cl1 expressing the PvCSP VK210; (B) Transgenic parasite line 2199cl1 expressing the PvCSP VK247; (C) Wild type P. berghei parasite line 676m1cl1 showing expression of PbCSP.
We next assessed the ability of these two transgenic parasites to produce oocysts and sporozoites since replacing the endogenous csp gene with P. vivax csp could alter these characteristics. Oocyst production in Anopheles stephensi of the two transgenic parasite lines was similar to oocyst production of WT P. berghei (Fig. 6A, p = 0.24). Similarly, the PbANKA-PvCS VK210(r)PbCS parasites produced salivary gland sporozoites comparable to WT parasites. In contrast, the PbANKA-PvCS VK247(r)PbCS parasites showed significant (~30%) reduction in sporozoites production compared to WT P. berghei parasites (Fig. 6B, p < 0.05). The infectivity of sporozoites of both transgenic parasites was assessed by determination of the pre-patent period (i.e. the time to 1% parasitaemia) after intravenous injection of 2,000 sporozoites in the tail vein of outbred CD-1 mice. All sporozoites were capable of a liver infection and, moreover, the time to 1% parasitemia was not significantly different between WT, PbANKA-PvCS VK210(r)PbCS and PbANKA-PvCS VK247(r)PbCS parasites (6.7, 6.5 and 6.7 days, respectively; Fig. 6C,D). Additional direct evidence to confirm that the transgenic sporozoites are as infectious as WT sporozoites was obtained by measuring parasite liver load or burden at 44 hours after infection using in vivo imaging of bioluminescence. Ploemen et al. have reported that the analysis of liver infections by whole body imaging shows a good correlation with quantitative RT-PCR analysis of extracted livers36. Our in vivo imaging results showed no difference in parasite liver loads, i.e. sporozoite infectivity between the transgenic/chimeric lines and wild type parasites (Fig. 6E,F). Therefore, we concluded that the transgenic P. berghei parasites expressing PvCSP alleles in place of PbCSP produce fully infectious sporozoites, which are able to complete liver stage development in mice.

Mean of oocysts numbers (A) and salivary gland sporozoites (B) in A. stephensi mosquitoes at day 10 and 21, respectively (3 independent experiments; 40 mosquitoes per experiment; error bars show standard deviation. (C) Blood-stage parasitaemia (mean + SEM) in mice after intravenous (i.v.) injection of 2000 sporozoites per mouse. (D) The Kaplan-Meier curves illustrate the time to reach 1% parasitaemia (Tto1) in 6 CD1 mice after i.v. injection with 2000 transgenic or wild type sporozoites. Survival analysis was made using a Kaplan-Meier Log-rank (Mantel-Cox) test. n.s.: not significant. (E) Parasite liver load in mice upon intravenous injection of transgenic and wild type parasites, determined by in vivo imaging of bioluminescence. (F) Quantification and comparison of parasite liver load measured as total flux per second.
PvCSP central repeat sequences are necessary to induce partial protective efficacy against P. berghei PvCSP transgenic parasites
We next determined vaccination efficacy by challenging vv-immunized mice with the transgenic P. berghei PvCSP transgenic parasites. These mice were immunized with the four different (ChAd63-MVA) PvCSP vv, as described above (Fig. 1A). Protective efficacy in these challenged mice was determined by measuring the prepatent period after intravenous injection of 2,000 P. berghei PvCSP transgenic sporozoites as described previously25.
Our results indicated that the PvCSP central repeat sequences are necessary to generate protective immunity, as immunisations with NC only (i.e. lacking either VK210 or 247 repeat sequences) failed to induce protective immunity, demonstrated by the prepatent period being similar to mock-immunized mice (Fig. 7A). N210C immunisation resulted in partial protection against a challenge with the transgenic parasites expressing VK210, producing a significant (p = 0.016) prolonged prepatent period. In contrast, N210C immunization did not delay the time to patency of transgenic parasites expressing VK247 (Fig. 7B). Immunisation with the N247C vv did not induce protection against the transgenic parasites expressing either VK247 or VK210 (Fig. 7C). However, immunization with the chimeric N210/247 C induced a significant delay in the prepatent period after challenge with either transgenic parasites expressing either VK210 (p = 0.007) or VK247 (p = 0.0008), while sterile protection in 25% and 16% of mice was achieved after a challenge with VK210 or VK247 transgenic parasites, respectively (Fig. 7D).

Groups of CD-1 mice (n = 6–10) were immunized with vv expressing PvCSP (N210/247C, N210C, N247C, NC; see Fig. 1) using an Ad prime and MVA-boost regimen. A vv expressing PbCSP was used as control. Two (A–D,F) or 12 (E) weeks after the boost, mice were challenged by intravenous injection of 2,000 transgenic sporozoites expressing PvCSP VK210 or VK247. The Kaplan-Meier curves illustrate the time to 1% parasitaemia (A–F). As a control PbCSP or N210/247C immunized mice were challenged 2 weeks post-boost with wild type P. berghei sporozoites (F). Statistical analysis was performed using the Kaplan-Meier survival curves and P values were calculated using a Kaplan-Meier Log-rank (Mantel-Cox) test. Data was obtained from two individual experiments.
While significant protective immunity was maintained 12 weeks after immunisation with the N210/247C vector as determined by a prolonged prepatent period after challenge with either VK210 (p = 0.005) or VK247 (p = 0.016) transgenic sporozoites, durable sterile protection was not induced as all mice eventually developed a blood stage infection (Fig. 7E). We confirmed the absence of unspecific protection induced by N210/247C vv vaccination by challenging mice with wild-type P. berghei sporozoites two weeks after boosting immunization. These mice were not protected; protection against wild-type P. berghei sporozoites was only induced in mice after a prime-boost (ChAd63-MVA) immunisation regimen with a vv expressing P. berghei CS protein (p = 0.024; Fig. 7F). These results demonstrate that immune responses to the central PvCSP repeats are necessary to induce protective immune responses against infection, assuming that N- and C-terminal domains in the NC construct are conformationally correct. Moreover, a chimeric VK210/247 PvCSP vv containing repeats from both forms of PvCSP elicit protective immune responses against sporozoites expressing either VK210 or VK247, which are at least equal to if not better than after immunisation with a vaccine consisting of only one allelic variant of PvCSP.
Vaccination with the Rv21 VLP induces high levels of protective immunity in mice
The ability to induce antibody-mediated protective immunity using the PvCSP vv and the absence of detectable cellular responses led us to develop a Virus-Like Particle (VLP) based vaccine platform that has a greater potential to generate very potent humoral responses against PvCSP. VLPs are known for their ability to induce remarkably rapid, strong and long-lasting high antibody titres, and are a leading vaccine platform not only for malaria5 but also for Human papillomavirus (HPV)37. RTS,S/AS01 is based on the hepatitis B surface antigen virus-like particle (VLP) platform, genetically-engineered to include the central repeats and carboxyl (C-) terminus (amino acids 207–395) of the P. falciparum CSP antigen38. We developed a VLP consisting of the chimeric PvCSP VK210/VK247 central repeats and the CSP C-terminal sequence fused to the Hepatitis B Surface Antigen (HepB-S) gene with a C-terminally placed four amino acid C-tag sequence (Glu-Pro-Glu-Ala). Codon usage of the fusion genes was optimized for expression in Pichia pastoris and production of the intracellular fusion protein (PvCSP-HepB-S) was assessed in three protease knockout strains and protease wild-type P. pastoris strain using a time course study expression. The double knock-out P. pastoris strain for prb1 and pep4 proteases had optimal protein expression levels after 108 hours of methanol induction. The presence of the fusion protein PvCSP-HepB S was confirmed by Western blot analyses using antibodies against PvCSP VK210, PvCSP VK247 and HepB S (Suppl Figure 1A). Presence, size and purity of the Rv21 protein was carried out using a sensitive silver stain technique (Fig. 1A and Suppl Figure 1B).
The protocol used for purification of the fusion protein VLP has been used for R21 and involved two steps (Collins, Brod et al. Scientific Reports, in press). The first consisted of an affinity purification using a capture select C-tag matrix bound to the fusion protein under neutral conditions. In addition to collecting the expected protein band corresponding to the PvCSP-HepB S protein, the sample also contained additional proteins. VLP particle assembly was detected using transmission electron microscopy (TEM) (Suppl Figure 1B). A subsequent purification step was performed by size exclusion chromatography in order to clean up the sample from the other proteins with different molecular size and to remove the high concentration of salts (Elution buffer). A single protein band of expected size of the PvCSP-HepB S protein (75 kDa) was visualized using the silver staining technique and the TEM showed a more homogeneous population of VLPs having a globular shape with some protuberances or spikes on the surface, likely due to the PvCSP protein presence on the surface, as represented in a diagram (Fig. 8A,B). The purified Rv21 was used to immunize mice, using a low dose of 0.5 μg/mouse and employing a homologous prime-boost immunization protocol using an interval of one week between immunizations. Rather than administering the ASO1 adjuvant, standardly used for RTS,S vaccination, Matrix-M adjuvant (Novavax AB Uppsala, Sweden) was used to enhance the immunogenicity and protective efficacy of Rv21. Matrix-M is suitable for human use and consists of saponin-based 40 nm particles that can activate and recruit immune cells to the draining lymph nodes and spleen39,40. This adjuvant functions by inducing high titres of reactive antibodies supported by a balanced Th1/Th2 response41.

The chimeric construct was expressed as a VLP in P. pastoris. (A) Analysis of protein purity using silver staining after two purification rounds, using size exclusion chromatography of 10 ng of elution sample (A and B), and visualization of VLPs, by TEM (right panel). (B) Diagram showing the proposed structure of Rv21. (C) Protective efficacy of Rv21 in CD-1 mice (n = 6) immunized using prime-boost at low doses of 0.5 ug of VLP alone or mixed with Matrix M adjuvant and challenged with transgenic sporozoites expressing P. vivax CSP VK210 or (D) VK247. Kaplan-Meier curves indicate the time to reach 1% parasitaemia. (E) Protective efficacy in C57Bl/6 mice (n = 6) immunized using prime-boost, either a low dose of 0.5 ug or a (F) high dose of 5 ug (right) of Rv21 in presence or absence of Matrix M adjuvant (Adj). Challenge of mice vaccinated with 5 ug was increased to 5,000 spz/mouse to increase stringency. Mice were challenged with transgenic sporozoites expressing P. vivax CSP VK210 or VK247. The Kaplan-Meier curves illustrate the time to 1% parasitaemia. (G) Association and (H) correlation of total IgG antibody titers with protective efficacy using data of sera from VK210-challenged C57Bl/6 mice. (I) Association of titers of IgG2a with efficacy against pre-erythrocytic malaria. (J,G) Kinetic of the antibody responses in presence or absence of the Matrix M adjuvant.
The protective efficacy of each prime-boost regimen was assessed by challenging the Rv21-immunized CD-1 mice with a dose of 2000 PbANKA-PvCS VK210(r)PbCS or PbANKA-PvCS VK247(r)PbCS transgenic sporozoites (Fig. 8C,D). Protective efficacy in these immunized mice was determined by measuring the prepatent period as described above. At the low dose (0.5 μg/mouse) of the Matrix-M-adjuvanted Rv21 prime-boost immunization, protective efficacy against challenge with PbANKA-PvCS VK210(r)PbCS sporozoites was 100% and against PbANKA-PvCS VK247(r)PbCS sporozoites 90%, as confirmed by the absence of a blood infection 20 days after challenge (Fig. 8C,D). In contrast, all mice in the naïve group developed blood stage parasitemia with a prepatent period of 4–5 days. In the absence of the Matrix-M adjuvant, immunisation of mice using the VLP homologous prime-boost regime still generated sterile immunity in 50% of the VLP-immunized mice challenged with PbANKA-PvCS VK210(r)PbCS sporozoites (Fig. 8C) and 43% of immunized mice were protected against a challenge of PbANKA-PvCS VK247(r)PbCS sporozoites (Fig. 8D). These results indicated the importance of both the VLP and the adjuvant in inducing protective immune responses with this vaccination regime. In addition, our results showed that PvCSP VLP-based vaccination induced higher protective efficacy than the viral-vectored vaccine candidates. ELISA analyses of antibody responses to vaccination supported the importance of Matrix-M adjuvant to elicit high antibody titres to PvCSP, even after a single (priming) immunisation with Rv21 (Suppl Figure 2A,B). A prime-boost regimen with Rv21 in the absence of the Matrix-M adjuvant-induced higher titres than those by a single vaccination but still lower compared to the prime-boost regime with the adjuvant. We concluded that the Rv21 antibody responses benefit from the co-administration of an adjuvant, both in a single immunization or a prime-boost regime. Importantly, the antibody titres induced by two doses of Rv21 in matrix-M were over 100-fold higher than those induced by the viral vectored prime-boost regime (Fig. 2 and Suppl Fig 2A).
We tested Rv21 efficacy in C57BL/6, a mouse strain that has previously shown to be highly sensitive to a P. berghei sporozoite infection and for which we have previously failed to induce sterile immunity using a P. vivax TRAP vaccine candidate42. Immunisation of mice with a Rv21 dose as described above (0.5 μg) followed by a challenge with 2,000 transgenic PbANKA-PvCS VK210(r)PbCS sporozoites failed to show protective efficacy (Fig. 8E). However, a 10x increase in the Rv21 dose (5 μg) showed, for the first time in our hands, complete protection in C57BL/6 mice even against a challenge with a higher dose of 5,000 PbANKA-PvCS VK210(r)PbCS sporozoites (Fig. 8F). Antibody titres measured as total IgG were associated with protection (Fig. 8G,H) and from these, only IgG2a was statistically associated with protection after challenging C57Bl/6 mice (Fig. 8H), while none of the other isotypes, such as IgM, IgG1, IgG2b and IgG3 showed any statistical association with protection (Suppl Figure 2D–F). Rv21 immunization (in Matrix M) induced antibody titres that remained consistently high for up to 70 days (Fig. 8J), while the inclusion of Matrix-M adjuvant enhanced antibody avidity with the time, as the addition of urea decreased antibody titres by 26.2% when using adjuvant and 100% in its absence (Suppl Figure 2C).
Our results indicate that immunization with a chimeric VK210/247 vaccine can induce immunity to the two major strains of P. vivax, based on the type of PvCSP central repeats. Moreover, high levels of protective immunity levels can be achieved using a VLP platform in the presence of the Matrix-M adjuvant, even in a mouse strain that is difficult to protect against a sporozoite infection.
Discussion
The circumsporozoite protein (CSP) is a leading vaccine target for both P. falciparum and P. vivax vaccines26,43. For an effective vaccine based on P. vivax PvCSP it has been suggested that immunity must be generated against the two major forms of PvCSP that are circulating around the world8,10,11, encoded by the VK210 and VK247 alleles, thereby generating protective immunity against the majority of P. vivax parasites circulating worldwide. While cellular responses play a role in protective immunity against P. falciparum liver infections23,24,25, the induction of strong humoral responses against both P. falciparum and P. vivax appears to be critical to the induction of protective immunity after immunization with vaccines based on CSP6,9,12. High and long-lasting IgG titers have been reported using a variety of platforms including bacterial-expressed recombinant PvCSP, a hybrid CSP molecule as a polypeptide or PvCSP expressed by the chimpanzee adenovirus ChAd63 and a chimeric circumsporozoite protein6,11,12. The VK210 central repeat region of PvCSP has been expressed as a virus-like particle (VLP) within the loop of the hepatitis B virus core antigen, and both the VLP and a recombinant multi-epitope polypeptide rPvCSP-ME elicit long-lasting antibody titers of high intensity and avidity6. Recently, targeting the CSP antigen of P. vivax with a recombinant soluble protein VMP001 and the particulate antigen CSV-S,S have shown to induce robust humoral and cellular immune responses, especially with the particulate CSV-S,S vaccine10, that induced high levels of sterile protection in Aotus nancymaae monkeys, and for which an association between antibodies to the central repeat region and protection was observed12.
In this study we describe the induction of humoral responses against PvCSP in the absence of detectable ex vivo IFN-γ antigen-specific T cell-mediated immunity, when using either viral vectored (vv) or virus-like particles (VLP)-based vaccine platforms encoding the VK210 and/or VK247 PvCSP repeat regions. Importantly, we demonstrate that antibodies generated after either a VK210 or VK247 based vv-immunization are able to recognize native PvCSP on the surface of P. vivax sporozoites isolated in the field.
In the absence of immune correlates of protection, assessment of the protective efficacy of P. falciparum malaria vaccine candidates has relied on determining protective efficacy directly against a parasite infection in phase IIb field studies or in controlled human malaria infection (CHMI) trials44,45. Due to technical challenges, which include the lack of P. vivax in vitro blood-stage cultivation system, evaluating protective efficacy of P. vivax candidate antigens in CHMI is difficult. Consequently, evaluation of the protective immunity of P. vivax pre-erythrocytic vaccines is more dependent on preclinical evaluation in animal models before proceeding with phase I and II trials. Preclinical evaluation of P. vivax pre-erythrocytic vaccine candidates has been performed in rodents6,9,11,21,42,46 and non-human primates10,12. However, pre-clinical evaluation of protective efficacy in monkeys presents additional difficulties, which include their restricted access, as well as the need to conduct these studies only in regions where both, monkeys and P. vivax parasites coincide19,20. These limitations in evaluating vaccine efficacy very much reduce the rate at which different P. vivax vaccine candidates are ranked and evaluated, and therefore assessed in clinical trials.
In order to evaluate the protective immunogenicity of our PvCSP vaccine candidates and to rationally prioritize vaccine candidates for further clinical development, we developed two novel in vivo challenge models in rodents. We generated two transgenic P. berghei parasite lines in which P. berghei CSP has been replaced with full-length P. vivax VK210 or VK247 CSP. The use of transgenic P. berghei parasites expressing P. vivax or P. falciparum antigens is a practical and affordable option, which can be used in laboratories with access to mouse models. Indeed, such models have been previously used to analyze protective immunity has been reported for several Plasmodium vaccine antigen candidates such as TRAP, CSP and a transmission-blocking vaccine (TBV)21,22,31,47. For example, transgenic P. berghei parasites expressing different forms of P. vivax CSP have been generated by Espinosa et al.21 and Mizutani (2014) and used to analyze protective immunity upon vaccination. The first study used transgenic parasites expressing a chimeric CSP protein consisting of sequences of both P. berghei and P. vivax, whereas the latter study used transgenic parasites expressing the P. vivax CSP corresponding to amino acids His24 to Asp351 of the PvCSP(Sal) gene minus its signal sequence and glycosylphosphatidylinositol (GPI) anchor. As mentioned, it has been suggested that for P. vivax CSP-based vaccines conferring protection against the 2 major isoforms of PvCSP (VK210 and VK247) alleles would be immunologically optimal to recognize and confer protection against most P. vivax parasites circulating worldwide11. However, confirmation of this hypothesis has remained elusive in the absence of challenge experiments using parasites that express either the VK210 or the V247 allele. We therefore decided to replace the complete coding sequence of P. berghei with the coding sequence of the two major alleles of PvCSP. Interestingly, PvCSP appears to be able to complement the function of PbCSP in the transgenic parasites, both in the mosquito and in liver infection. The VK210-expressing parasite produced normal numbers of sporozoites similar to a wild type infection. However, while VK247-expressing sporozoites also exhibited wild type hepatic-infectivity this line produced 30% fewer sporozoites, suggesting that the exact sequence of the CSP repeat region influences sporozoite formation.
The normal in vivo infectivity of the transgenic PvCSP P. berghei sporozoites make these transgenic parasites excellent tools to better analyze protective immune responses induced by novel vaccine platforms that induce immunity against PvCSP, and in turn can be used to prioritize vaccine candidates for clinical assessment. Using these parasites we were able to confirm that epitopes present within the repeat regions are required to confer cross-protection against parasites expressing either the VK247 or the VK210 Pvcsp allele. Consequently, we used these transgenic parasites to compare different PvCSP vaccination approaches, specifically, ChAd63/MVA viral vectored vaccination and a novel Pichia pastoris-expressed Hepatitis B Surface Antigen fusion protein VLP, Rv21. While both approaches were able to generate antibody responses against PvCSP, immunization with Rv21 surpassed antibody responses by viral vectors and demonstrated very high levels of protective immunity, even in hitherto difficult to protect C57BL/6 mice. Prime-boost Rv21 vaccination was able to confer 100% and 90% sterile immunity to CD-1 mice challenged with a dose of 2000 VK210 or VK247 expressing transgenic P. berghei sporozoites, respectively. Recently, Whitacre et al.22 have also shown high protective efficacy in C57BL6 mice using an E. coli derived VLP that expressed VK210 repeat from PvCSP, this VLP-platform was based on the woodchuck hepatitis virus core antigen (WHcAg). However in order to generate protective immunity they required very high doses (100 μg) of the VLP22. When Rv21 vaccination was combined with Matrix-M as an adjuvant we were able to induce very strong protective immunity at VLP doses as low as 0.5 μg or 5 μg in CD-1 or C57Bl/6 mice, respectively. We consider that the use of C57Bl/6 mice is an optimal mouse strain to support vaccine efficacy studies through sporozoite challenge experiments, compared to CD-1 mice that are less suited for these studies. This adjuvant has been successfully and safely used in humans, most recently for P. falciparum malaria (Venkatraman et al. unpublished), and consists of immune-stimulating complexes (ISCOMs) in the form of 40 nm particles made with adjuvant-active saponins (Quillaja saponaria), cholesterol and phospholipids41. Matrix-M has been shown to enhance immune responses in humans when combined with influenza or West Nile vaccines39,48. The observations on the strong adjuvanting properties of Matrix M upon Rv21 is of particular interest as VLPs based on the HepB Surface antigen have previously been considered less immunogenic than those using the HepB core antigen22. Our study shows that HepB-S based VLPs can be highly immunogenic and protective, even at low doses between 0.5 and 5 μg. Future studies should address the contribution of the N- and C- termini sequences to the protective efficacy of VLP vaccines such as Rv21 and CSV-S,S, the latter containing the N-terminus repeat.
In conclusion, the newly developed transgenic P. berghei parasites expressing PvCSP alleles VK210 and VK247 repeats have been highly effective tools that have helped develop and optimize vaccine candidate platforms targeting PvCSP. They have confirmed the value of using immunization strategies that induce immune responses against the two major isoforms of PvCSP and show that they can greatly improve protection against infection. Moreover, using this testing system we have been able to evaluate a novel VLP-based vaccine candidate, Rv21, which consists of a chimeric VK210/247 PvCSP fusion molecule combined with hepatitis B surface antigen, and have demonstrated that this vaccine is highly efficacious, conferring high levels of protective immunity even in mice that have previously been difficult to protect using another leading P. vivax vaccine candidate, TRAP42. Importantly, to our knowledge, this is the first evidence of protection against both sporozoites expressing both isoforms of the PvCS, VK210 and VK247. Rv21 has now received support for GMP production and has entered a path towards clinical trials.
Materials and Methods
Animals
Female inbred BALB/c (H-2d), C57BL/6 (H-2b) and outbred CD1 (ICR) mice were used for the assessment of immunogenicity and protection after challenge. Tuck-ordinary (TO) outbred mice were used for parasite production and transmission. Mice were purchased from Harlan (UK). Transgenic parasites were developed in Leiden University Medical Centre (LUMC) using 6-week old Swiss mice (Charles River).
Immunization of mice
Viral vectors: Mice were primed with simian adenoviral vector 63 (ChAd63) encoding PvCSP at a dose of 1 × 108 infectious units (iu) and 8 weeks later boosted with vaccinia-modified virus strain Ankara (MVA) encoding the same transgene at a concentration of 1 × 106 plaque forming unit (pfu) per mouse. All viral vector vaccines were administered intramuscularly (i.m.) in endotoxin-free PBS and no adjuvant was used for vv immunizations.
Rv21 VLP vaccine: Rv21 was administered in endotoxin-fee PBS mixed with 12 μg of Matrix M Adjuvant (Isconova, Sweden, now Novavax, MD, USA). The Rv21 vaccine was injected intramuscularly at a dose of 0.5 μg/mouse in CD1 mice, while C57Bl/6 mice received two doses, 0.5 μg or 5 ug/mouse in different experiments. The vaccination regimen for Rv21 consisted of homologous prime-boost at an interval of one week between immunizations.
Ethics statement
All animals and procedures were used in accordance with the terms of the UK Home Office Animals Act Project License. Procedures were approved by the University of Oxford Animal Care and Ethical Review Committee (PPL 30/2414). Animal experiments performed at LUMC were approved by the Animal Experiments Committee of the Leiden University Medical Center (DEC 12042). The Dutch Experiments on Animals Act was established under European guidelines (EU directive no. 86/609/EEC regarding the Protection of Animals used for Experimental and Other Scientific Purposes).
Parasite production
Wild type and transgenic parasites used to challenge mice were produced at the insectary of the Jenner Institute. Female Anopheles stephensi mosquitoes were fed on infected TO mice. Briefly, exflagellation was first confirmed and mosquitoes were exposed to anaesthetized infected mice for 10 minutes. Mosquitoes were then maintained for 21 days in a humidified incubator at a temperature of 19–21 °C on a 12 hour day-night cycle and fed with a fructose/PABA solution.
Parasites
The wild type (WT) reference line cl15cy1 of P. berghei ANKA49 and the reporter PbANKA parasite line PbGFP-Luccon (676m1cl1). PbGFP-Luccon parasite expresses a fusion protein of GFP (mutant3) and firefly luciferase (LUC-IAV) under the constitutive eef1a promoter and is SM free49. The reporter-cassette is integrated into the neutral 230p locus (PBANKA_030600). For details of PbGFP-Luccon, see RMgmDB entry #29 (http://www.pberghei.eu/index.php?rmgm=29).
Generation of DNA constructs and genotyping of the transgenic parasites
To generate the transgenic parasites where the P. berghei csp gene (PBANKA_040320) coding sequence (CDS) has been replaced by the CDS of P. vivax csp (PVX_119355), we used a 2-step GIMO transfection protocol31,50. In the first step we deleted the P. berghei csp CDS and replaced it with the positive-negative selectable marker, to create a P. berghei csp deletion GIMO line (PbANKA-CSP GIMO). In order to this we generated pL1929 construct that is based on the standard GIMO DNA construct pL003450. This construct contains the positive-negative (hdhfr::yfcu) selection marker (SM) cassette, and was used to insert both the Pbcsp 5′ and 3′ gene targeting regions (TR), encompassing the full-length promoter and transcription terminators sequences respectively. The linear pL1929 DNA construct was introduced into PbGFP-Luccon parasites using standard methods transfection49. Transfected parasites were selected in mice by applying positive selection by providing pyrimethamine in the drinking water49. Transfected parasites were cloned by limiting dilution51, resulting in the PbANKA-CSP GIMO line (2151cl1). Correct deletion of the P. berghei csp CDS was confirmed by diagnostic PCR-analysis on gDNA and Southern analysis of pulsed field gel (PFG) separated chromosomes as described49. Primers used for PCR genotyping are listed in Table S1.
In the second step we replaced the positive-negative SM in the PbANKA-CSP GIMO genome with the CDS of either P. vivax VK210 or VK247 csp by GIMO transfection to create the two P. berghei transgenic CSP replacement lines. This was achieved by modifying the construct used in the first step (pL1929); specifically, the hdfhr::yfcu SM cassette was removed and replaced with P. vivax csp CDS sequence. The P. vivax csp CDS was ordered from GeneArt (Regensburg, Germany) (i.e. VK210) or cDNA (VK247) Both the P. vivax VK210 and VK247 CSP constructs (pL1942 and pL1943, respectively) were sequenced to ensure there were no mutations in the P. vivax csp CDS. These constructs were linearized using SacI and PacI restriction enzymes outside of the 5′ and 3′ TRs before transfection. These constructs were used to transfect parasites of the PbANKA-CSP GIMO line (2151cl1) using standard methods of GIMO-transfection30. Transfected parasites were selected in mice by applying negative selection by providing 5-fluorocytosine (5-FC) in the drinking water of mice52. Negative selection results in selection of chimeric parasites where the hdhfr::yfcu SM in the csp locus of PbANKA-CSP GIMO line is replaced by the CDS of P. vivax CSP Selected transgenic parasites were cloned by the method of limiting dilution51. Correct integration of the constructs into the genome of chimeric parasites was analysed by diagnostic PCR-analysis on gDNA and Southern analysis of pulsed field gel (PFG) separated chromosomes as described49. Primers used for PCR genotyping are listed in Table S1. This method creates transgenic ‘gene replacement’ P. berghei parasites that do not contain P. berghei csp gene CDS but express either P. vivax VK210 (PbANKA-PvCSP VK210(r)PbCS; 2196cl1) or VK247 csp (PbANKA-PvCSP VK247(r)PbCS; 2199cl1) under the control of the P. berghei csp regulatory sequences.
Phenotyping of reporter and transgenic parasites
The growth of blood stages of the reporter and transgenic P. berghei parasites was determined during the cloning period as described30,49. Feeding of An. stephensi mosquitoes, determination of oocyst production, sporozoite collection were performed as described30. Expression of PvCSP-VK210 and PvCSP-VK247 antigens in sporozoites of the transgenic parasites was analysed by immunofluorescence- staining assay (IFA), using anti-P. vivax antigen monoclonal antibodies (anti-PvCSP-VK210 (MR4) or anti-PvCSP-247 (MR4) antibodies; diluted 200 times) or anti-PbCSP 3D11 antibodies as a control; diluted 1000 times. Purified sporozoites were fixed with 4% paraformaldehyde in PBS for 20 min on ice, then washed three times with PBS and blocked with 20 ul10% FCS + 1% BSA in PBS for 30 min at room temperature. The excess blocking medium was removed, followed by the addition of 20–25 uL primary monoclonal antibody in 10% FCS + 1% BSA in PBS (blocking medium) for 1–2 hours at room temperature or overnight at 4 °C. After incubation the primary antibody was removed and the slides washed three times with PBS, followed by the staining with the secondary antibody (Alexa Fluor® 488 Goat Anti-Mouse IgG from life technologies, Cat# A-11001) diluted 800 times in 10% FCS + 1% BSA in PBS (blocking medium) for 1 hour at room temperature. After washing three times with PBS, nuclei were stained with 2% Hoechst-33342 (Cell Signaling Technology #4082S) in PBS for 10 minutes at room temperature, washed twice with PBS and left to air-dry, this followed by adding Fluorescence Mounting Medium (Dako, code S3023) before complete dry out. Cover slips were mounted onto the slides, and the slides were sealed with nail polish and left to dry overnight in dark. The parasites in both blue and green channels were analyzed using a DMI-300B Leica fluorescence microscope and images processed using ImageJ software.
Immunofluorescence Assay for Wild-Type P. vivax isolates from Mexico
Antibody recognition to native, wild-type P. vivax isolates from Mexico was assessed by IFA using a technique described earlier53. Briefly, sporozoites were produced by infection of laboratory-reared An. albimanus mosquitoes that fed on blood from patients infected with P. vivax. Mosquitoes were maintained for 15–18 days and sporozoites were collected by dissection of the salivary glands and deposited in multiwell IFAT slides at a concentration of 2,000 sporozoites/well to make individual slides containing P. vivax sporozoites VK210 or slides with only VK247, keeping them separate and not as mixed parasites. Confirmation of the VK210 and VK247 variant types was made as described earlier54 using primers to amplify the repeat VK210 or VK247 regions followed by slot blot and hybridization to 32P-labeled oligonucleotide probes for VK210 and VK247 and a final confirmation was made using the monoclonal antibodies 5D3 and 2E10, specific for the VK210 and VK247 variant types. Slides were kept frozen at −70 °C until used. The assay was performed using sera from C57Bl/6 and CD-1 mice, which was incubated with three different batches each of sporozoites, yielding similar results between both strains. Air-dried sporozoites were also incubated with anti-VK210 (monoclonal antibody 5D3 at 2.5 μg/ml) or anti-VK247 (monoclonal antibody 2E10 at 5 μg/ml) monoclonal antibodies as positive controls29. Slides were analyzed using a confocal microscope and titers were calculated using the highest dilution that gave positive fluorescence.
Efficacy studies: Determination of liver parasite liver load by real-time imaging and determination prepatent period (after challenge of immunized mice with transgenic sporozoites)
To determine the efficacy of the liver-stage vaccines, transgenic P. berghei infected A. stephensi mosquitoes were dissected 21 days post-feed and salivary gland sporozoites resuspended in RPMI-1640 media (Sigma-Aldrich). 2000 sporozoites in 100 μl were injected i.v. into the tail vein per mouse, into both vaccinated and naïve controls.
P. vivax CSP DNA sequences
Four versions of ChAd63 and four of MVA were designed and produced to express various versions of the P. vivax CSP protein. One consisted of only the N- and C- terminal regions (NC); a second viral vector expressed VK210 repeats inserted in between the N- and C-terminal sequences (N210C). A third vector expressed VK247 inserted between the N- and C-terminal sequences (N247C) and a final design consisted on a chimeric VK210/247 flanked by similar N- and C-terminal repeats (sequences detailed below).
DNA transgenes were synthesized by GeneArt (Regensburg, Germany) and constructs were previously modified to improve antigen expression within the hosts cells, modifications included codon optimization for mammalian use and replacement of the endogenous PvCSP leading sequence for tPA (human plasminogen activator) (GenBank Accession no. K03021). In addition, the transmembrane GPI-anchor domain was removed from the Plasmodium vivax circumsporozoite protein (PvCSP) genes to allow protein secretion from any virus-transduced cell. The chimeric PvCSP vaccine insert consisted on the N- and C- terminal region from the Salvador I strain (NCBI Reference Sequence XP_001613068.1) flanking the central repeat regions of the circumsporozoite (CSP) VK210 of the Belem strain (GenBank accession number P08677) or VK247 of the Papua New Guinea (PNG) (GenBank accession number M69059.1). The central repeat region was designed using segments of 9 amino acid repeats that are most frequently found in Plasmodium vivax as follows: VK210 (5x(GDRAAGQPA), 4x(GDRADGQPA), 1x(GNGAGGQAA)) and VK247 (2x(ANGAGNQPG/ANGAGGQAA), 1x(ANGAGDQPG/ANGAGDQPG), 1x(ANGADDQPG/ANGAGDQPG), 1x(EDGAGNQPG/ANGAGDQPG))8. A region II-plus was included in the C-terminal sequence of the vaccine (EWTPCSVTCG)55, as well as an insertion region in the C-terminal sequence of PvCSP (GAGGQAAGGNA)56. The VK210 vaccine insert consisted on the Salvador I strain sequence, while the VK247 consisted of the PNG sequence with the accession numbers mentioned above. A control viral vector lacking the expression of any transgene was used as a control in mock-vaccinated mice.
Viral vector construction
All of DNA constructs required for ChAd63 were cloned in two steps. In the first step, unique Acc65I and NotI sites were used to insert the synthetic transgenes into an adenovirus entry plasmid. The transgene was placed upstream of BGH poly(A) transcription termination sequence and under the control of the long cytomegalovirus (CMV) promoter (containing a regulatory element, an enhancer and an intron A). The entry plasmid also contained attachment L (attL) sequences, which were required for site-specific recombination with attachment R (attR) sites located on the destination vector.
In the second step of cloning, an in vitro Gateway reaction was performed mediated by LR Clonase II system (Invitrogen), whereby the transgene of the entry vector was integrated into the destination plasmid by site-specific recombinase through an attL-attR interaction. The diagnostic PCR was performed to confirm the desired integration before completing the production of the recombinant ChAd63.
Similarly, all of the malarial genes were inserted into an MVA shuttle vector using a similar cloning strategy. Unique Acc65I and XhoI sites were used for transgene restriction and ligation and the transgenes were inserted under the control of an endogenous P7.5 promoter. PCR and RFLP were used to verify the correct insertion before linearization of the MVA plasmid.
Rv21 HepB Surface antigen VLP design and development
A gene containing a chimeric CSP sequence comprising the repeat regions VK210 and VK247, followed by the C-terminal sequence (210/247C) was designed to be fused to the Hepatitis B surface antigen (HepBsAg) followed C-terminally by the peptide EPEA (C-Tag) for affinity purification. The sequence was codon-optimised for optimal expression in yeast and purchased from GeneArt® (InVitrogen). DNA sequences were similar to those expressed by recombinant ChAd and MVA viruses, but without the N-terminal CSP region. The construct was cloned into the pPink-HC intracellular Pichia plasmid and amplified in E. coli. Upon plasmid linearization, four strains of Pichia pastoris (knock out for ade2, ade2-pep4, ade2-prb1, ade2-pep4-prb1) were electroporated and white colonies were selected using PAD selection plates. Cultures for protein expression were grown in glycerol containing medium and expression was induced by addition of methanol. Growth kinetics were analysed to determine conditions for optimal protein production The VLPs were purified from yeast lysates by C-Tag affinity chromatography followed by size exclusion chromatography. (Collins et al. manuscript in preparation). Fractions from gel filtration were analyzed by SDS-PAGE and subsequently transferred to nitrocellulose membranes, blocked in 5% skimmed milk/PBS followed by addition of primary and secondary antibodies. VLP-containing fractions from the gel filtration column were detected with anti-HBsAg, anti-PvCSP VK210 and anti-PvCSP VK247 primary mouse antibodies, diluted in 3% BSA/PBS to 1:200, 1:20,000 and 1:20,000 respectively (MR4). Silver staining was used to analyse purity of the fractions from the FPLC column and of progressive stages of the VLP purification process. The samples were run on Mini-PROTEAN 12% gels (BioRad) with the Pierce unstained protein ladder (Thermo Scientific) run for reference. The gels were subjected to silver staining using a Pierce silver stain kit (Thermo Scientific). Direct staining of gels was carried out according to manufacturer’s instructions. The particle-containing fraction from the sephacryl gel filtration column was negatively stained with 2% uranyl acetate as follows: the Rv21 sample was imaged using a FEI Tecnai 12 Transmission Electron Microscope (TEM). The TEM protocol used to evaluate the different Rv21 vaccine purification steps used a FEI Tecnai 12 Transmission Electron Microscope (TEM) at an accelerating voltage of 80 kV. The samples (10 μg) were absorbed for 1 min to a glow-discharged C-Formvar coated Cu-grids (300 mesh) and negatively stained with 2% (w/v) uranyl acetate, pH 4.5. The VLPs were visualized taking under 20,000× magnification, and the photographic records were performed on 4 Megapixel Gatan Ultrascan™ 1000 CCD camera.
Whole IgG Enzyme-linked immunosorbent assay (ELISA)
ELISAs measuring total IgG were carried out as described previously24. Serum antibody endpoint titers were taken as the x-axis intercept of the dilution curve at an absorbance value three standard deviations greater than the OD405 for serum from a naïve mouse. Results were also calculated using a standardized ELISA by including a high-titre reference serum from hyper-immune mice to each ELISA plate to produce a standard curve, which in turn was used to quantify and assign ELISA units to each sample57. Briefly, Nunc Maxisorp Immuno ELISA plates were coated with the antigens diluted in PBS to a final concentration of 1 μg/mL. Each plate contained a standard curve, negative and positive controls, as well as serum samples of mice pre-boost (1:300) or post-boost (1:3000). An anti-mouse IgG coupled to alkaline phosphatase was used as a secondary antibody. Development was made with 4-nitrophenylphosphate diluted in diethanolamine buffer. Data was fitted to a four parameter hyperbolic curve58.
For avidity ELISAs, standard curve ELISAs were performed as described above except following 2 h incubation of serum at room temperature plates were washed 6 times with PBS/Tween and 100 μl 7 M urea added to serum wells for 10 min. Plates were then washed 6 times with PBS/Tween and the standard curve ELISA carried out as before. Avidity index was calculated as the percentage of log10 EU of urea-treated compared untreated serum samples, following the procedure described by Grangeot-Keros, L. et al.59.
For IgG subclass ELISAs, standard curve ELISAs were performed as described above, using the Biorad Mouse Typer Sub-Isotyping kit with these modifications (protocol for mouse typer sub-isotyping kit, Biorad): after serum incubation subclass-specific rabbit anti-mouse IgG was applied at 50 μl per well in triplicate. The standard curve was incubated with kappa-chain specific IgG. After 1 h incubation plates were washed 6 times with PBS/Tween and goat anti-rabbit horseradish peroxidase conjugate applied (50 μl/well, 1:3000). After 1 h plates were washed 6 times with PBS/Tween and developed for 14 min using 100 μl/well peroxidase substrate solution (Biorad). Reactions were stopped by adding 100 μl/well 2% oxalic acid.
Expression and purification of PvCSP VK210 and VK247 proteins
The P. vivax protein CSP210 (Belem; GenBank accession number P08677) and the protein CSP247 (PNG; GenBank accession number M69059) were produced as previously described60. In addition, a chimeric CSP protein containing the repeats from both PvCSP210 and PvCSP247 was produced. Briefly, a codon-optimized synthetic gene (chimeric CSP210/247) was synthesized by GeneArtTM. CSP210/247 was subcloned into the expresion plasmid PHLsec (His6 tag), which is driven by the chicken β-actin/rabbit β-globin hybrid promoter61. The resulting PHLsec CSP210/247 was verified by DNA sequencing.
All secreted proteins were purified from media supernatants from PEI-transfected HEK-293T cells using roller bottles (2,125 cm2) at 37 C and 5% CO2, using immobilised metal ion affinity chromatography (IMAC) medium (resin) precharged with nickel ions (Ni Sepharose™ excel, GE Healthcare). Conditioned media was filtered through a 0.45 μm membrane (Millipore). IMAC purification was performed using a distilled water wash step with 5 column volumes (CV) at a flow velocity of 100 cm/h, equilibration step with 5CV of equilibration buffer (20 mM sodium phosphate, 0.5 M NaCl, pH 7.4/flow velocity: 150 cm/h), load sample step (flow velocity: 150 cm/h), wash step with 20 CV of wash buffer (20 mM sodium phosphate, 0.5 M NaCl, 20 mM imidazole, pH 7.4/flow velocity: 150 cm/h), linear elution step with 2 CV of 7% elution buffer (20 mM sodium phosphate, 0.5 M NaCl, 500 mM imidazole, pH 7.4/flow velocity: 150 cm/h) and 2 CV of 70% elution buffer (flow velocity: 150 cm/h). Elution samples after IMAC purification were analysed using a 12.5% SDS-PAGE under reducing conditions and proteins were visualized with Silver stain and Western Blot analyses using the monoclonal antibody anti PvCSP-VK 210 and VK247. (Obtained through MR4 BEI Resources NIAID, NIH: Monoclonal Antibody Sporozoite ELISA Kit, Anti-Plasmodium vivax Circumsporozoite Proteins, MRA-1028K, contributed by R. A. Wirtz). Samples were concentrated using an Amicon® Ultra centrifugal filter system (Life Technologies) until reaching 10 ml of final volume. Removal of possible contaminant proteins and salts was done by size exclusion purification (SEC). SDS-PAGE electrophoresis and subsequent Coomassie and Silver staining was performed to assess protein purity.
Peptides
Crude 20-mer peptides overlapping by 10 amino acids and representing full-lengths of P. vivax CSP VK210 and VK247 were synthesized by Mimotopes (Victoria, Australia). Individual peptide pools were used at a final concentration of 5 μg/mL per peptide.
Ex-vivo IFN-γ ELISPOT assay
Ex vivo IFN-gamma (IFN-γ) ELISPOTs were carried out using PBMCs isolated from the blood as previously described62,63. The assay was performed using MAIP ELISPOT plates (Millipore) were where PBMCs (1 × 106 cells/well) were co-cultured with splenocytes (2.5 × 105 cells/well) in Minimum Essential Medium Eagle supplemented with 1% L-Glutamine, 10% Foetal Calf Serum (FCS), Penicillin and Streptomycin (100 UI/ml). Overlapping peptide libraries consisting on 20-mer peptides overlapping by 10 were purchased at PEPSCAN Presto, The Netherlands. Peptides spanned the whole sequence of the P. vivax CSP VK210 and VK247 transgenes and were used to stimulate the co-cultures for 16 hours. DMSO, phytohemagglutinin (10 U/mL) and medium controls were used in the same assay plate. Anti-mouse IFN-γ mAb and development reagents were used according to the manufacturer specifications (Mabtech). Spots were counted using an ELISPOT counter (Autoimmun Diagnostika (AID), Germany). Results are expressed as IFN-γ spot-forming units (SFU) per million PBMCs.
Infection of mice
The CD-1 and C57Bl/6 mice that were vaccinated with 0.5 μg of Rv21 were challenged with 2,000 wild type or transgenic P. berghei sporozoites. When C57Bl/6 mice were vaccinated with increased doses of 5 μg/mouse of Rv21, a higher dose of 5,000 sporozoites per mouse was used for a challenge. Infection was monitored from day 5 to 20 by Giemsa staining of blood smears.
Statistical model for parasitaemia prediction
Percent parasitaemia was used to calculate the time required to reach a blood-stage infection of 1% or time to 1% parasitaemia. This was predicted using a linear regression model as described previously25. Briefly, blood parasite counts were obtained for 3–5 consecutive days starting on day 5 after the challenge. Blood smears were stained with Giemsa and percentages of parasitaemia calculated in all animals. The logarithm to base 10 of the calculated percentage of parasitaemia was plotted against the time after challenge and Prism 5 for Mac OS X (GraphPad software) statistical analysis package used for generating a linear regression model on the linear part of the blood-stage growth curve.
In vivo imaging after malaria sporozoite challenge
Bioluminescent luciferase signal was quantified by imaging the whole animals using the in vivo IVIS 200 imaging system (Caliper Life Sciences, USA), as described elsewhere36,64,65. Briefly, 44 hrs after the intravenous injection with 1000 transgenic P. berghei sporozoites the mice were anaesthetized in batches of three using isofluorane, their fronts shaved and D-luciferin (Synchem Laborgemeinschaft OHG, Germany) injected into the neck at a concentration of 100 mg/kg in sterile PBS (Sigma, US). Animals were imaged for 120 seconds at binning value of 8 and FVO of 12.8 cm, 8 minutes after the injection of D-luciferin. Mice were kept anaesthetized throughout the whole procedure. Quantification of bioluminescence signal was performed using Living Image 4.2 software (Caliper Life Sciences, USA). The region of interest (ROI) were set around the liver area of the mouse body and kept constant for all of the animals. The measurements were expressed as a total flux of photons per second of imaging time.
Statistical analysis
For all statistical analyses, GraphPad Prism version 5.0 for Max OS was used unless indicated otherwise. Prior to statistical analysis to compare two or more populations, the Kolmogorov-Smirnov test for normality was used to determine whether the values followed a Gaussian distribution. An unpaired t-test was employed to compare two normally distributed groups, whereas Mann-Whitney rank test was used for comparing two non-parametric groups. If more than two groups were present non-parametric data was compared using Kruskal-Wallis test with Dunn’s multiple comparison post-test, whereas normally distributed data were analyzed by one-way ANOVA with Bonferroni’s multiple comparison post-test. The effect of two variables was explored using two-way ANOVA with Bonferroni’s multiple comparison post-test. Correlation strength was tested using either Pearson’s or Spearman’s tests as indicated in the results chapters. Kaplan-Meier survival curves were used to represent protective efficacy to a challenge with any P. berghei parasite lines. All ELISA titres were also log10 transformed before analysis. The value of p < 0.05 was considered statistically significant (*p < 0.05, **p < 0.01, **p < 0.001, and ***p < 0.001).
Additional Information
How to cite this article: Salman, A. M. et al. Rational development of a protective P. vivax vaccine evaluated with transgenic rodent parasite challenge models. Sci. Rep. 7, 46482; doi: 10.1038/srep46482 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Guerra, C. A. et al. The international limits and population at risk of Plasmodium vivax transmission in 2009. PLoS neglected tropical diseases 4, e774, doi: 10.1371/journal.pntd.0000774 (2010).
White, N. J. Determinants of relapse periodicity in Plasmodium vivax malaria. Malaria journal 10, 297, doi: 10.1186/1475-2875-10-297 (2011).
Markus, M. B. Malaria: origin of the term “hypnozoite”. Journal of the history of biology 44, 781–786, doi: 10.1007/s10739-010-9239-3 (2011).
Duffy, P. E., Sahu, T., Akue, A., Milman, N. & Anderson, C. Pre-erythrocytic malaria vaccines: identifying the targets. Expert review of vaccines 11, 1261–1280, doi: 10.1586/erv.12.92 (2012).
Rts, S. C. T. P. Efficacy and safety of the RTS,S/AS01 malaria vaccine during 18 months after vaccination: a phase 3 randomized, controlled trial in children and young infants at 11 African sites. PLoS medicine 11, e1001685, doi: 10.1371/journal.pmed.1001685 (2014).
Almeida, A. P. et al. Long-lasting humoral and cellular immune responses elicited by immunization with recombinant chimeras of the Plasmodium vivax circumsporozoite protein. Vaccine 32, 2181–2187, doi: 10.1016/j.vaccine.2014.02.053 (2014).
Bennett, J. W. et al. Phase 1/2a Trial of Plasmodium vivax Malaria Vaccine Candidate VMP001/AS01B in Malaria-Naive Adults: Safety, Immunogenicity, and Efficacy. PLoS neglected tropical diseases 10, e0004423, doi: 10.1371/journal.pntd.0004423 (2016).
Lim, C. S., Tazi, L. & Ayala, F. J. Plasmodium vivax: recent world expansion and genetic identity to Plasmodium simium. Proceedings of the National Academy of Sciences of the United States of America 102, 15523–15528, doi: 10.1073/pnas.0507413102 (2005).
Teixeira, L. H. et al. Immunogenicity of a prime-boost vaccine containing the circumsporozoite proteins of Plasmodium vivax in rodents. Infection and immunity 82, 793–807, doi: 10.1128/IAI.01410-13 (2014).
Vanloubbeeck, Y. et al. Comparison of the immune responses induced by soluble and particulate Plasmodium vivax circumsporozoite vaccine candidates formulated in AS01 in rhesus macaques. Vaccine 31, 6216–6224, doi: 10.1016/j.vaccine.2013.10.041 (2013).
Yadava, A. et al. A novel chimeric Plasmodium vivax circumsporozoite protein induces biologically functional antibodies that recognize both VK210 and VK247 sporozoites. Infection and immunity 75, 1177–1185, doi: 10.1128/IAI.01667-06 (2007).
Yadava, A. et al. Protective efficacy of a Plasmodium vivax circumsporozoite protein-based vaccine in Aotus nancymaae is associated with antibodies to the repeat region. PLoS neglected tropical diseases 8, e3268, doi: 10.1371/journal.pntd.0003268 (2014).
Cespedes, N. et al. Antigenicity and immunogenicity of a novel Plasmodium vivax circumsporozoite derived synthetic vaccine construct. Vaccine 32, 3179–3186, doi: 10.1016/j.vaccine.2014.04.007 (2014).
Cespedes, N. et al. Antigenicity and immunogenicity of a novel chimeric peptide antigen based on the P. vivax circumsporozoite protein. Vaccine 31, 4923–4930, doi: 10.1016/j.vaccine.2013.05.082 (2013).
Udomsangpetch, R. et al. Short-term in vitro culture of field isolates of Plasmodium vivax using umbilical cord blood. Parasitology international 56, 65–69, doi: 10.1016/j.parint.2006.12.005 (2007).
Targett, G. A., Moorthy, V. S. & Brown, G. V. Malaria vaccine research and development: the role of the WHO MALVAC committee. Malaria journal 12, 362, doi: 10.1186/1475-2875-12-362 (2013).
Golenda, C. F., Li, J. & Rosenberg, R. Continuous in vitro propagation of the malaria parasite Plasmodium vivax. Proceedings of the National Academy of Sciences of the United States of America 94, 6786–6791 (1997).
Galinski, M. R., Medina, C. C., Ingravallo, P. & Barnwell, J. W. A reticulocyte-binding protein complex of Plasmodium vivax merozoites. Cell 69, 1213–1226 (1992).
Reyes-Sandoval, A. & Bachmann, M. F. Plasmodium vivax malaria vaccines: why are we where we are? Human vaccines & immunotherapeutics 9, 2558–2565, doi: 10.4161/hv.26157 (2013).
Galinski, M. R. & Barnwell, J. W. Plasmodium vivax: who cares? Malaria journal 7 Suppl 1, S9, doi: 10.1186/1475-2875-7-S1-S9 (2008).
Espinosa, D. A. et al. Development of a chimeric Plasmodium berghei strain expressing the repeat region of the P. vivax circumsporozoite protein for in vivo evaluation of vaccine efficacy. Infection and immunity 81, 2882–2887, doi: 10.1128/IAI.00461-13 (2013).
Whitacre, D. C. et al. P. falciparum and P. vivax Epitope-Focused VLPs Elicit Sterile Immunity to Blood Stage Infections. PloS one 10, e0124856, doi: 10.1371/journal.pone.0124856 (2015).
Ewer, K. J. et al. Protective CD8+ T-cell immunity to human malaria induced by chimpanzee adenovirus-MVA immunisation. Nature communications 4, 2836, doi: 10.1038/ncomms3836 (2013).
Reyes-Sandoval, A. et al. Prime-boost immunization with adenoviral and modified vaccinia virus Ankara vectors enhances the durability and polyfunctionality of protective malaria CD8+ T-cell responses. Infection and immunity 78, 145–153, doi: 10.1128/IAI.00740-09 (2010).
Reyes-Sandoval, A. et al. CD8+ T effector memory cells protect against liver-stage malaria. Journal of immunology 187, 1347–1357, doi: 10.4049/jimmunol.1100302 (2011).
Heppner, D. G. The malaria vaccine–status quo 2013. Travel medicine and infectious disease 11, 2–7, doi: 10.1016/j.tmaid.2013.01.006 (2013).
Sun, P. et al. Protective immunity induced with malaria vaccine, RTS,S, is linked to Plasmodium falciparum circumsporozoite protein-specific CD4+ and CD8+ T cells producing IFN-gamma. Journal of immunology 171, 6961–6967 (2003).
Moorthy, V. S. & Ballou, W. R. Immunological mechanisms underlying protection mediated by RTS,S: a review of the available data. Malaria journal 8, 312, doi: 10.1186/1475-2875-8-312 (2009).
Gonzalez-Ceron, L. et al. Plasmodium vivax: a monoclonal antibody recognizes a circumsporozoite protein precursor on the sporozoite surface. Experimental parasitology 90, 203–211, doi: 10.1006/expr.1998.4334 (1998).
Lin, J. W. et al. A novel ‘gene insertion/marker out’ (GIMO) method for transgene expression and gene complementation in rodent malaria parasites. PloS one 6, e29289, doi: 10.1371/journal.pone.0029289 (2011).
Longley, R. J. et al. Comparative assessment of vaccine vectors encoding ten malaria antigens identifies two protective liver-stage candidates. Scientific reports 5, 11820, doi: 10.1038/srep11820 (2015).
Khan, S. M., Kroeze, H., Franke-Fayard, B. & Janse, C. J. Standardization in generating and reporting genetically modified rodent malaria parasites: the RMgmDB database. Methods Mol Biol 923, 139–150, doi: 10.1007/978-1-62703-026-7_9 (2013).
Salman, A. M. et al. Generation of Transgenic Rodent Malaria Parasites Expressing Human Malaria Parasite Proteins. Methods Mol Biol 1325, 257–286, doi: 10.1007/978-1-4939-2815-6_21 (2015).
Rosenberg, R. et al. Circumsporozoite protein heterogeneity in the human malaria parasite Plasmodium vivax. Science 245, 973–976 (1989).
Yoshida, N., Nussenzweig, R. S., Potocnjak, P., Nussenzweig, V. & Aikawa, M. Hybridoma produces protective antibodies directed against the sporozoite stage of malaria parasite. Science 207, 71–73 (1980).
Ploemen, I. H. et al. Visualisation and quantitative analysis of the rodent malaria liver stage by real time imaging. PloS one 4, e7881, doi: 10.1371/journal.pone.0007881 (2009).
Wang, J. W. & Roden, R. B. Virus-like particles for the prevention of human papillomavirus-associated malignancies. Expert review of vaccines 12, 129–141, doi: 10.1586/erv.12.151 (2013).
Garcon, N., Heppner, D. G. & Cohen, J. Development of RTS,S/AS02: a purified subunit-based malaria vaccine candidate formulated with a novel adjuvant. Expert review of vaccines 2, 231–238, doi: 10.1586/14760584.2.2.231 (2003).
Magnusson, S. E. et al. Immune enhancing properties of the novel Matrix-M adjuvant leads to potentiated immune responses to an influenza vaccine in mice. Vaccine 31, 1725–1733, doi: 10.1016/j.vaccine.2013.01.039 (2013).
Reimer, J. M. et al. Matrix-M adjuvant induces local recruitment, activation and maturation of central immune cells in absence of antigen. PloS one 7, e41451, doi: 10.1371/journal.pone.0041451 (2012).
Bengtsson, K. L., Karlsson, K. H., Magnusson, S. E., Reimer, J. M. & Stertman, L. Matrix-M adjuvant: enhancing immune responses by ‘setting the stage’ for the antigen. Expert review of vaccines 12, 821–823, doi: 10.1586/14760584.2013.814822 (2013).
Bauza, K. et al. Efficacy of a Plasmodium vivax malaria vaccine using ChAd63 and modified vaccinia Ankara expressing thrombospondin-related anonymous protein as assessed with transgenic Plasmodium berghei parasites. Infection and immunity 82, 1277–1286, doi: 10.1128/IAI.01187-13 (2014).
Herrera, S., Corradin, G. & Arevalo-Herrera, M. An update on the search for a Plasmodium vivax vaccine. Trends in parasitology 23, 122–128, doi: 10.1016/j.pt.2007.01.008 (2007).
Nahrendorf, W. et al. Memory B-cell and antibody responses induced by Plasmodium falciparum sporozoite immunization. The Journal of infectious diseases 210, 1981–1990, doi: 10.1093/infdis/jiu354 (2014).
Warimwe, G. M. et al. Peripheral blood monocyte-to-lymphocyte ratio at study enrollment predicts efficacy of the RTS,S malaria vaccine: analysis of pooled phase II clinical trial data. BMC Med 11, 184, doi: 10.1186/1741-7015-11-184 (2013).
Cabrera-Mora, M. et al. Induction of Multifunctional Broadly Reactive T Cell Responses by a Plasmodium vivax Circumsporozoite Protein Recombinant Chimera. Infection and immunity 83, 3749–3761, doi: 10.1128/IAI.00480-15 (2015).
Mizutani, M. et al. Baculovirus-vectored multistage Plasmodium vivax vaccine induces both protective and transmission-blocking immunities against transgenic rodent malaria parasites. Infection and immunity 82, 4348–4357, doi: 10.1128/IAI.02040-14 (2014).
Magnusson, S. E. et al. Matrix-M adjuvanted envelope protein vaccine protects against lethal lineage 1 and 2 West Nile virus infection in mice. Vaccine 32, 800–808, doi: 10.1016/j.vaccine.2013.12.030 (2014).
Janse, C. J., Franke-Fayard, B. & Waters, A. P. Selection by flow-sorting of genetically transformed, GFP-expressing blood stages of the rodent malaria parasite, Plasmodium berghei. Nature protocols 1, 614–623, doi: 10.1038/nprot.2006.88 (2006).
Cox, R. J. et al. Evaluation of a virosomal H5N1 vaccine formulated with Matrix M adjuvant in a phase I clinical trial. Vaccine 29, 8049–8059, doi: 10.1016/j.vaccine.2011.08.042 (2011).
Menard, R. & Janse, C. Gene targeting in malaria parasites. Methods 13, 148–157, doi: 10.1006/meth.1997.0507 (1997).
Orr, R. Y., Philip, N. & Waters, A. P. Improved negative selection protocol for Plasmodium berghei in the rodent malarial model. Malaria journal 11, 103, doi: 10.1186/1475-2875-11-103 (2012).
Gonzalez-Ceron, L. et al. Plasmodium vivax: impaired escape of Vk210 phenotype ookinetes from the midgut blood bolus of Anopheles pseudopunctipennis. Experimental parasitology 115, 59–67, doi: 10.1016/j.exppara.2006.06.001 (2007).
Gonzalez-Ceron, L. et al. Differential susceptibilities of Anopheles albimanus and Anopheles pseudopunctipennis to infections with coindigenous Plasmodium vivax variants VK210 and VK247 in southern Mexico. Infection and immunity 67, 410–412 (1999).
Gantt, S. M., Clavijo, P., Bai, X., Esko, J. D. & Sinnis, P. Cell adhesion to a motif shared by the malaria circumsporozoite protein and thrombospondin is mediated by its glycosaminoglycan-binding region and not by CSVTCG. J Biol Chem 272, 19205–19213 (1997).
Hernandez-Martinez, M. A., Escalante, A. A., Arevalo-Herrera, M. & Herrera, S. Antigenic diversity of the Plasmodium vivax circumsporozoite protein in parasite isolates of Western Colombia. The American journal of tropical medicine and hygiene 84, 51–57, doi: 10.4269/ajtmh.2011.09-0785 (2011).
Miura, K. et al. Development and characterization of a standardized ELISA including a reference serum on each plate to detect antibodies induced by experimental malaria vaccines. Vaccine 26, 193–200, doi: 10.1016/j.vaccine.2007.10.064 (2008).
Studnicka, G. M. ELISA assay optimization using hyperbolic regression. Computer applications in the biosciences: CABIOS 7, 217–224 (1991).
Grangeot-Keros, L. et al. Value of cytomegalovirus (CMV) IgG avidity index for the diagnosis of primary CMV infection in pregnant women. The Journal of infectious diseases 175, 944–946 (1997).
Longley, R. J. et al. Acquisition and Longevity of Antibodies to Plasmodium vivax Preerythrocytic Antigens in Western Thailand. Clinical and vaccine immunology: CVI 23, 117–124, doi: 10.1128/CVI.00501-15 (2016).
Aricescu, A. R., Lu, W. & Jones, E. Y. A time- and cost-efficient system for high-level protein production in mammalian cells. Acta crystallographica. Section D, Biological crystallography 62, 1243–1250, doi: 10.1107/S0907444906029799 (2006).
Reyes-Sandoval, A., Pearson, F. E., Todryk, S. & Ewer, K. Potency assays for novel T-cell-inducing vaccines against malaria. Curr Opin Mol Ther 11, 72–80 (2009).
Reyes-Sandoval, A. et al. Single-dose immunogenicity and protective efficacy of simian adenoviral vectors against Plasmodium berghei. Eur J Immunol 38, 732–741, doi: 10.1002/eji.200737672 (2008).
Annoura, T., Chevalley, S., Janse, C. J., Franke-Fayard, B. & Khan, S. M. Quantitative analysis of Plasmodium berghei liver stages by bioluminescence imaging. Methods Mol Biol 923, 429–443, doi: 10.1007/978-1-62703-026-7_30 (2013).
Reyes-Sandoval, A. et al. Mixed vector immunization with recombinant adenovirus and MVA can improve vaccine efficacy while decreasing antivector immunity. Molecular therapy: the journal of the American Society of Gene Therapy 20, 1633–1647, doi: 10.1038/mt.2012.25 (2012).
Acknowledgements
We would like to thank the Jenner Insectary for supplying infected mosquitoes for the studies, we are also grateful to Alex Fyfe for assisting with the production of transgenic P. berghei parasites in the insectary of the University of Oxford. ISCONOVA and Anita Milicic from the adjuvant core facility for supplying the Matrix M adjuvant. The Jenner Vector Core Facility for producing the recombinant viral vectors. The work was funded by a Wellcome Trust Career Development Fellowship award, grant number 097395/Z/11/Z, to A.R.-S, who is also a Jenner Investigator and an Oxford Martin Fellow. Funding was also provided by the Medical Research Council, through a DPFS grant (MR/N019008/1) to A.R.-S. Ahmed M. Salman was funded by EVIMalaR’s Program funding (FP7/2007–2013) under grant agreement N° 242095. Adrian Hill is supported by a Wellcome Trust grant number 095540/Z/11/Z and is a Jenner Investigator and an Oxford Martin Fellow.
Author information
Authors and Affiliations
Contributions
Study concept and design; A.R.-S. Acquisition of data; A.M.S., E.M.-D., H.W., A.L., E.A., C.L.-C., J.R., K.B., K.C., F.B., F.R., L.P., L.G.-C., C.J.J., A.V.S.H., S.M.K. Analyses and interpretation of data; A.M.S., A.L., E.M., H.W., E.A., K.B., F.R., L.P., L.G.-C., C.J.J., A.V.S.H., S.M.K. Drafting of the manuscript; A.R.-S., A.M.S., C.J.J., S.M.K. Critical revision of the manuscript for important intellectual content; A.M.S., C.J.J., S.M.K., A.V.H., L.G.-C., C.L.-C. Statistical analyses; A.M.S., A.L., E.M., H.W., E.A., K.B., F.R., L.P., L.G.C. Obtained funding; A.R.-S. Technical, or material support; A.M.S., C.J.J., S.M.K., A.V.H., K.C., L.G.C. Study supervision: A.R.-S., C.J.J., S.M.K., A.V.H.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Salman, A., Montoya-Díaz, E., West, H. et al. Rational development of a protective P. vivax vaccine evaluated with transgenic rodent parasite challenge models. Sci Rep 7, 46482 (2017). https://doi.org/10.1038/srep46482
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep46482
This article is cited by
-
In search of a vaccine for Plasmodium vivax malaria
Nature (2023)
-
A genetically modified Plasmodium berghei parasite as a surrogate for whole-sporozoite vaccination against P. vivax malaria
npj Vaccines (2022)
-
A universal vaccine candidate against Plasmodium vivax malaria confers protective immunity against the three PvCSP alleles
Scientific Reports (2021)
-
Recombinant Plasmodium vivax circumsporozoite surface protein allelic variants: antibody recognition by individuals from three communities in the Brazilian Amazon
Scientific Reports (2020)
-
Chimeric Plasmodium falciparum parasites expressing Plasmodium vivax circumsporozoite protein fail to produce salivary gland sporozoites
Malaria Journal (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
