
Abstract
Development of castration resistance is a key contributor to mortality in patients with prostate cancer. High expression of RING finger protein 7 (RNF7) in cancer cells is known to play a key role in tumor progression. However, the role of RNF7 in prostate cancer progression is not well elucidated. In this study, we silenced RNF7 by shRNA interference in two castration resistant prostate cancer (CRPC) cell lines, DU145 and PC3. RNF7 knockdown attenuated proliferation and enhanced sensitivity of prostate cancer cells to cisplatin treatment. Invasive property of DU145 and PC3 cells was also attenuated by RNF7 silencing. The underlying mechanisms appear to be associated with accumulation of tumor suppressive proteins p21, p27 and NOXA, while inactivation of ERK1/2 by RNF7 knockdown. We demonstrated that RNF7 knockdown induced growth suppression of prostate cancer cells and inactivated ERK1/2 pathway, which suggested RNF7 might be a potential novel therapeutic target for CRPC.
Similar content being viewed by others


Introduction
Worldwide, an estimated 1.1 million men are diagnosed with prostate cancer (PC) every year1. Prostate cancer progression is amenable to repression at an early stage by androgen deprivation therapy (ADT). However, most patients with PC will eventually become castration resistant, which leads to its relapse. This feature makes prostate cancer the second cause of cancer death in men according to International Agency for Research on Cancer2,3,4. Hence, identification of molecules that are functionally associated with tumor initiation, progression, invasion and metastasis is a key research imperative in cancer therapeutics.
RNF7, also referred to as SAG (sensitive to apoptosis gene), ROC2 (regulator of cullins 2) or Rbx2 (RING-box 2), consists of 113 amino acids and has a molecular weight of 12.6 kDa. RNF7 was originally identified as a redox-inducible anti-oxidant protein5. It is a highly conserved protein and is extensively expressed in human skeletal muscles, heart and testis5. As a redox-inducible anti-oxidant protein, production of RNF7 can be induced by redox agents, which can protect cells from apoptosis caused by oxidation5. RNF7 is also a component of E3 ubiquitin ligases. When combining with other components to assemble E3 ubiquitin ligases, RNF7 has E3 ubiquitin ligase activity6. Although RNF7 is inducible by the transcription factor of activator protein-1 (AP-1), RNF7 inhibits tumor-promoting functionality of AP-1 by ubiquitinylation and degradation of c-Jun7. RNF7 has been shown to mediate ubiquitinylation of various cellular proteins such as, p218, p279,10, pro–caspase-311, IκBα12,13, HIF-1α14, NOXA15 and NF116. It is reported that RNF7 is over-expressed in many human cancers, such as carcinomas of lung, colon, stomach and liver15. Silencing of SAG expression by RNA interference was shown to inhibit proliferation of cancer cells (lung cancer cell line H1299, glioblastoma cell line U87, pancreatic carcinoma cell line PANC-1) in vitro and reverse radiation-resistance15. The mechanisms underlying this effect may include inactivation of NF-κB and mTOR pathway8 and/or accumulation of tumor suppressive proteins, such as NF1, DEPTOR, Procaspase-3, p21, p27, NOXA, and BIM15,17. As a component of E3 ubiquitin ligase, RNF7 may interact with massive cellular proteins. Further investigations are needed to reveal the novel functional aspects of RNF7 and their underlying mechanisms.
The role of RNF7 in prostate cancer, especially in castration resistant prostate cancer (CRPC), is not yet clear. In this study, we found that knockdown of RNF7 in two CRPC cell lines, DU145 and PC3, enhanced the sensitivity of these cells to cisplatin treatment. The underlying mechanisms were likely associated with increased cell apoptosis and inhibited ERK1/2 activity.
Results
RNF7 was efficiently silenced by RNA interference
DU145 and PC3 cell lines were transfected with shRNF7 retroviruse (shRNF7-1 or shRNF7-2) or with negative control retrovirus (shCON). Protein and mRNA levels of RNF7 were determined by Western blot and qRT-PCR, respectively. RNF7 expression was significantly decreased after interference with shRNF7-1 or shRNF7-2 in both DU145 and PC3 cells (Fig. 1a).

Prostate cancer cells were transfected with shRNF7-1, shRNF7-2 retrovirus or shCON negative control retrovirus. RNF7 expression was assessed by western blot (a), and confirmed on qRT-PCR (b). Results were representatives of three independent experiments. Data were mean ± SD. ***P < 0.001.
To measure knockdown efficiency of RNF7, RNF7 mRNA expression level was determined by qRT-PCR. RNF7 transcription was statistically inhibited by shRNF7-1 or shRNF7-2 interference, P < 0.001 (Fig. 1b). RNF7 mRNA level in DU145 cells silenced by shRNF7-1 or shRNF7-2 compared to shCON-negative control was 26.2% ± 4.7% and 12.1% ± 6.4%, respectively. While the RNF7 mRNA level in PC3 cells silenced by shRNF7-1 or shRNF7-2 compared to shCON negative control was 24.9% ± 5.3% and 11.2% ± 2.4%, respectively. The silencing efficiency of shRNF7-2 was significantly greater than that of shRNF7-1, both in DU145 and PC3 cells, P < 0.001 (Fig. 1b). These data were consistent with reduced RNF7 protein expression detected by Western blot assay. Our results demonstrated that both shRNF7-1 and shRNF7-2 efficiently decreased RNF7 production.
RNF7 Knockdown inhibited prostate cancer cell growth
Rapid cell division is one of the properties of cancer cells. To determine the effect of RNF7 knockdown on prostate cancer cell proliferation, we seeded 1 × 105 cells in 12 well plates and cultured them for 7 days. Cell numbers were recorded every day. Cell proliferation in the shRNF7-2 interfered group was significantly attenuated than that in the control (shCON) group from the third day onwards (P < 0.01) (Fig. 2a and b). On the 7th day, the number of DU145 cells decreased to 78.4% by RNF7 interference (Fig. 2a), and 69.6% of PC3 cells (Fig. 2b). The results indicated significant attenuation of DU145 and PC3 cell proliferation after RNF7 knockdown.

Cell numbers of DU145 (a) and PC3 (b) transfected with shCON or shRNF7-2 were monitored for 7 days. Data were mean ± SD. **P < 0.01; ***P < 0.001.
RNF7 Knockdown induced cell cycle arrest in prostate cancer cells
To investigate the underling mechanism of growth suppression caused by RNF7 knockdown, cell cycle distribution of shRNF7 and shCON cells were measured by PI staining on flow cytometer. The mean percentage of negative control cells in G2 phase, in which RNF7 were normally expressed, were about 25.9% and 21.3% (Fig. 3a and b), respectively. However, mean percentage of G2 phase in RNF7 silenced DU145 and PC3 cells were about 41.2% and 38.7% (Fig. 3a and b), respectively. These results showed that RNF7 knockdown induced significantly G2 phase arrest in both DU145 and PC3 cells, P < 0.001.

Cell cycles were detected by PI staining using flow cytometry. (a) representative figures of cell cycle detection. Numbers were percentage of corresponding phases. (b) statistical data of cell cycle assay. G1: G1 phase, S: S phase, G2: G2 phase. shCON: cells transfected with empty vector as negative control, shRNF7: RNF7 knockdown cells.
RNF7 knockdown accelerated prostate cancer cell apoptosis and cell death upon administration of chemotherapy
To investigate whether RNF7 knockdown had synergetic effect with chemotherapy on prostate cancer treatment, DU145 and PC3 cells transfected with shRNF7-2 or shCON were treated with cisplatin. Apoptosis cells were positively stained with Annexin V or double positively stained with Annexin V and PI, while dead cells were only PI positive (Fig. 4a). In DU145 cells, cisplatin combined with shRNF7-2 interference induced significantly higher apoptosis or necrosis indicated by Annexin V and/or PI staining positive (25.7%) as compared to that induced by cisplatin alone (15.3%) (P < 0.0001) (Fig. 4b). Also, cisplatin induced significantly higher cell apoptosis or necrosis (26.3%) in shRNF7-2 silenced PC3 cells compared to shCON control group (19.4%), P < 0.0001 (Fig. 4b). So, RNF7 silencing enhanced the sensitivity of prostate cancer cell lines to cisplatin.

Cells transfected with shRNF7-2 (shRNF7-2 group) or shCON (shCON group) were treated with cisplatin. Untreated cells served as negative controls. Cell apoptosis was detected by Annexin V and PI staining. (a) Representative results of Annexin V and PI staining; (b) Statistical analysis of cell apoptosis. Live: Annexin V and PI double negative cells. Ann+: Annexin V positive but PI negative cells. Ann+ PI+: Annexin V and PI double positive cells. PI+: PI positive but Annexin V negative cells. Data were mean ± SD. ***P < 0.001.
RNF7 Knockdown inhibited prostate cancer cell invasion
Cell invasion plays a critical role in cancer relapse and causes death. The role of RNF7 in DU145 and PC3 cell invasion were analyzed by invasion assay. Our results showed that the invasive ability reduced significantly after RNF7 silencing in DU145 (Fig. 5a) and PC3 (Fig. 5b) cells, P < 0.0001.

Invaded cells of shRNF7-2 interfered DU145 cells (a) and PC3 cells (b) were significantly lower than that of shCON negative controls. Representative figures and statistical data were showed. Data were mean ± SD. ***P < 0.001.
RNF7 Knockdown inhibited prostate cancer cell colonization and tumorigenesis
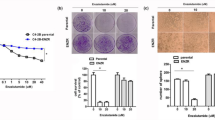
Colony formation indicates the ability of tumorigenesis. Clonogenic survival assay was performed to detect the role of RNF7 in prostate cancer cell colony. As inspected, DU145 (Fig. 6a left panel) and PC3 (Fig. 6b left panel) cells exhibited higher colony forming ability. However, this ability was significantly compromised by RNF7 interference, P < 0.0001 (Fig. 6a and b, middle and right panels). The size and weight of tumors developed from DU145 (Fig. 6c) or PC3 (Fig. 6d) cells in nude mice were significantly decreased by RNF7 silencing, P < 0.0001. These data demonstrated that RNF7 knockdown inhibited tumorigenesis in prostate cancer cells.

Colonies formed by DU145 (a) and PC3 (b) shRNA interfered cells (shRNF7-2 group) were significantly lower than those formed by negative controls (shCON group). DU145 (c) or PC3 cells (d) (both shNRF7 interfered and shCON negative controls) were implanted subcutaneously into the back of nude mice. Each group contained 6 nude mice. Mice were sacrificed six weeks later. Xenografts were collected and their weights were measured. Representative photos and statistical data were showed. Data were mean ± SD. ***P < 0.001.
RNF7 Knockdown induced accumulation of p21, p27 and NOXA, and inhibition of ERK1/2 activity
In both DU145 and PC3 cells, the expression of pro-apoptosis protein NOXA and tumor suppressor proteins p21 and p27 was highly up-regulated by RNF7 silencing (Fig. 7a). ERK plays a critical role in tumor cell survival and proliferation. And phosphorylation indicates activation of ERK. To investigate the effects of RNF7 knockdown on ERK activation, prostate cancer cells were activated by epidermal growth factor (EGF) for 5 to 60 minutes and ERK phosphorylation was detected by Western blot. Our study showed that it was the activity not the expression of ERK that was inhibited by RNF7 knockdown in DU145 and PC3 cells (Fig. 7b). These data demonstrated that RNF7 knockdown up-regulated expression of pro-apoptosis and/or tumor suppressor proteins, while inhibited the activity of ERK pathway.

(a) Expression of pro-apoptosis protein NOXA and tumor suppressor proteins p21 and p27 was detected by western blot assay. shRNF7-1 or shRNF7-2: cells interfered by shRNF7-1 or shRNF7-2 retrovirus. (b) Both shRNF7-2 interfered (shRNF7-2 group) or the negative control (shCON group) prostate cells were stimulated with EGF for 5 to 60 minutes, and expression or phosphorylation of ERK was detected by Western blot assay.
To verify that proliferation suppression was associated with accumulation of p21, p27, and NOXA in RNF7 silenced prostate cancer cell, p21, p27, or NOXA was silenced by siRNA in RNF7 knockdown DU145 and PC3 cells. Four days after siRNA interference, cell proliferation was measured by MTT assay. Our results showed that cell proliferation increased significantly by silencing of p21, p27, or NOXA in both RNF7 knockdown DU145 (Fig. 8a) and PC3 cells (Fig. 8b). These results showed that the suppressive effect of RNF7 knockdown on cell growth was likely attributable to accumulation of P21, P27 and NOXA.

RNF7 silenced (shRNF7 group) DU145 (a) or PC3 (b) cells were interfered by siRNAs targeting p21, p27 or NOXA, respectively. Cell viability was measured by MTT assay 4 days after siRNA interference. Viability of DU145 or PC3 cells normally expressed RNF7 (shCON control group) was set as 100 percent. sip21: cells interfered by siRNA targeting p21. sip27: cells interfered by siRNA targeting p27. siNOXA: cells interfered by siRNA targeting NOXA. Statistical data were mean ± SD. **P < 0.01, ***P < 0.001.
In conclusion, our study demonstrated that RNF7 knockdown could suppress proliferation and inhibit tumor formation of prostate cancer cells. The underlying mechanisms were associated with accumulation of tumor suppressive proteins p21, p27 and NOXA, and inactivation of ERK1/2 pathway.
Discussion
Prostate cancer is one of the leading causes of death in males worldwide. While most prostate cancer patients respond to ADT initially, a vast proportion of them will eventually relapse due to castration resistance2,3,4. Hence, development of treatment regimens that reverse castration resistance is a key priority in CRPC research.
Chemotherapy is one of the main therapeutic modalities for CRPC. It was reported that docetaxel could prolong survival time 2 to 3 months18. Other chemotherapeutic agents used for improving treatment efficacy either singly or in combination with docetaxel include, cisplatin19,20, estramustine21, 153Sm-lexidronam22, prednisolone23, mitoxantrone24,25, epirubicin26, bortezomib27, zoledronic acid28, capecitabine29, and vinorelbine30. Other regimens were used or studied in clinical trials include, combination of estramustine phosphate, ifosfamide and cisplatin20; prednisone plus cabazitaxel and prednisone plus mitoxantrone25; epirubicin and cisplatin31; and cisplatin plus prednisone32. Although these regimens enhanced the 50% survival time up to 15–20 months, more efforts are needed for improving the treatment efficacies. It was reported that cisplatin induced reactive oxygen species (ROS) in prostate cancer cells. And the production of ROS in hormone-sensitive LNCap cells was significantly higher than that of CRPC DU145 and PC3 cells33. Increasing the ROS production in CRPC cells may improve efficiency of chemotherapy especially cisplatin treatment.
RNF7 is highly expressed in various human cancers, such as lung, liver and stomach cancers15. Also we found high expression level of RNF7 in prostate DU145 and PC3 cells. RNF7 was proved to be an antioxidant by Yi Sun and his colleagues5. Then we hypothesized that silencing of RNF7 might have a synergistic effect with cisplatin chemotherapy. So we sought to investigate the effect of RNF7 knockdown on sensitivity of DU145 and PC3 cells to cisplatin treatment. Retrovirus based shRNA were used to silence RNF7 expression. The knockdown efficiency was comparable to that reported in previous studies7,8. RNF7 knockdown significantly inhibited DU145 and PC3 cell proliferation starting from the third day, which may be associated with RNF7 knockdown induced cell cycle arrest in G2 phase. Consistent with our study, Tan et al. recently showed that RNF7 knockdown by lentivirus-based siRNA significantly decreased prostate cancer cell proliferation34. We also investigated the effect of RNF7 knockdown on CRPC cell sensitivity to cisplatin. RNF7 knockdown combined with cisplatin chemotherapy induced significantly higher percentage of Annexin V positively stained apoptosis cells as compared to that observed with cisplatin treatment only. The underlying mechanism might be associated with increased expression of pro-apoptosis protein NOXA in RNF7 silenced cells. We further investigated the effect of RNF7 silencing on cancer cell invasion. RNF7 knockdown significantly inhibited invasive properties of DU145 and PC3 cells. Consistent with our study, Tan et al. also showed that RNF7 knockdown significantly inhibited prostate cancer cell migration and clone formation in vitro34. Based on the above results, we hypothesized that RNF7 silencing might counteract CRPC tumorigenesis. Our study showed that clonogenic forming ability of DU145 and PC3 in vitro, and its tumor formation ability in nude mice were significantly decreased by RNF7 interference. These results were consistent with increased expression of tumor suppressor proteins p21 and p27. Similar findings have been reported from a previous study on lung cancer cells8. We also sought to figure out the potential signal transduction pathway involved in RNF7 knockdown mediated cell growth inhibition. While Tan et al. showed that RNF7 knockdown decreased AKT activation34, we observed inactivation of ERK1/2 activity by RNF7 silencing. Multiple signaling pathways might be involved in the inhibition of cell growth and tumorigenesis mediated by RNF7 knockdown. To address further whether ERK1/2 inactivation is associated with inhibition of RAS activity, we detected RAS activity of shRNF7 and shCON cells, and we did not observe RAS inactivation caused by RNF7 interference (data not shown), indicating ERK1/2 inactivation by RNF7 knockdown might not due to RAS inactivation by potential NF1 accumulation. More investigations are needed to uncover the underlying mechanisms. To our knowledge, this is the first study to demonstrate that ERK1/2 activation is regulated by RNF7 in cancer cells.
In conclusion, our study demonstrated that RNF7 knockdown inhibited prostate cancer cell proliferation and tumorigenesis, suggesting that RNF7 might be a promising target for CRPC treatment. The underlying mechanisms might be associated with accumulation of tumor suppressive proteins p21, p27 and NOXA, and inactivation of ERK1/2 activity.
Materials and Methods
Cell lines
Human prostate cancer cell lines DU145 and PC3, and HEK293T were purchased from American Type Culture Collection (ATCC) and cultured in high glucose DMEM medium (GIBCO, Grand Island, USA) containing 10% fetal bovine serum (FBS) (GIBCO), 100 U/ml penicillin (GIBCO) and 100 μg/ml streptomycin (GIBCO).
shRNA constructs of RNF7
The following two oligos were used for construction of shRNAs against RNF7: shRNF7–1 oligo: TGCTGTTGACAGTGAGCGACCGGCTAATTTTTGTATTTTTTAGTGAAGCCACAGATGTAAAAAATACAAAAATTAGCCGGGTGCCTACTGCCTCGGA. shRNF7-2 oligo TGCTGTTGACAGTGAGCGCCACCTTTATAATTTACCCATTTAGTGAAGCCACAGATGTAAATGGGTAAATTATAAAGGTGATGCCTACTGCCTCGGA. These two oligos were amplified using the following primer pair miR30 XhoI (5′-CAGAAGGCTCGAGAAGGTATATTGCTGTTGACAGTGAGCG-3′) and miR30 EcoRI (5′-CTAAAGTAGCCCCTTGAATTCCGAGGCAGTAGGCA-3′). Thus, each of the RNF7 targeting sequences shRNF7-1 and shRNF7-2 contained miR30 sequences and enzyme sites of XhoI and EcoRI. shRNF7-1 and shRNF7-2 were subcloned into LMP vector by polymerase chain reaction (PCR) and confirmed by DNA sequencing.
Packaging of Retroviruses in HEK293T cells and transfection of prostate cancer cell lines
Recombinant LMP vectors containing shRNF7-1 or shRNF7-2 combined with pCL10A1 retrovirus packaging vector were transfected into HEK293T cells using lipofectamine 2000 (Life Technologies, Carlsbad, CA, USA), according to manufacturer’s instructions. After 48 h incubation, retroviruses containing shRNF7-1 or shRNF7-2 were collected from culture supernatant. pCL10A1 retrovirus and LMP vector without shRNF7 sequences served as the negative control (shCON).
Prostate cancer cells were transfected with retroviruses in the presence of 10 μg/ml polybrene and selected by 1 μg/ml puromycin. Total RNA of prostate cancer cells was extracted using Trizol reagent (Invitrogen, Madison, WI, USA). To determine the efficiency of RNA interference, RNF7 mRNA levels were assessed by quantitative RT-PCR (qRT-PCR), and RNF7 protein levels were assessed by Western blot analysis.
qRT-PCR analysis
Total RNA was reverse transcribed into cDNA using Superscript III Reverse transcriptase kit (Life Technologies). mRNA levels were determined using SYBR® Green Real-time PCR master mix (Takara, Shiga, Japan) on 7900HT qRT-PCR machine (Applied Biosystems, Foster City, CA, USA). The following pair of primers of RNF7 were used for amplification: 5′-TGGAAGACGGAGAGGAAACCT-3′, 5′-TCCCCAGACCACAACACAGT-3′. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was used as an internal control. The primers of GAPDH were 5′-GACAGTCAGCCGCATCTTCT-3′ and 5′-TTAAAAGCAGCCCTGGTGAC-3′35. Relative mRNA levels were analyzed as the ratio of RNF7 to GAPDH using ΔΔCt method.
Cell proliferation assay
DU145 and PC3cell lines transfected with or without RNF7 shRNA were seeded into 12 well plates at a concentration of 2 × 105 cells/ml with a total volume of 0.5 ml/well. Cell numbers were recorded every day using Bio-Rad cell counter (Bio-Rad Laboratories, Hercules, CA, USA). Growth curves of cells (number of cells versus time) were plotted for up to seven days. RNF7 knockdown DU145 and PC3cells interfered by siRNA targeting p21 (SASI_Hs01_00025255, sigma), p27 (SASI_Hs01_00113637) or NOXA (SASI_Hs01_00136187) were seeded to 96 wells with 2 × 104/ml in a total volumn of 200 μl. Cell viability was detected by Methylthiazolyldiphenyl-tetrazolium bromide (MTT, M5655, sigma) after 4 days culturing.
Cell cycle assay
Both RNF7 knockdown and the negative control DU145 and PC3 cells were harvested and fixed by 70% ethanol at 4 °C for 24 h. Cells were incubated with 50 μg/ml propidium iodide (PI) containing 400 U/ml RNase A for 30 min at room temperature in the dark. Cell cycle distribution was measured by FACSCalibur flow cytometer.
Apoptosis assay
DU145 and PC3 cells transfected with or without RNF7 shRNA were seeded into 24-well plates in 0.5 ml culture medium (2 × 105 cells/ml). Cells were treated with or without 10 μg/ml cisplatin (P4394, Sigma) for 24 h. Cells were harvested by centrifugation and stained with Annexin V and 1 μl propidium iodide (PI) (V13242, ThemoFisher Scientific) according to the manufacturer’s instruction. Cells were detected by FACSCalibur C6 (BD Biosciences, San Jose, CA, USA) and analyzed using FlowJo software (FlowJo LLC, Ashland, OR, USA).
Cell invasion assay
Cell invasion was assessed using 24-well Boyden chamber plates which contained polycarbonate membrane filter inserts with 8-μm pore size (Costar Group, Washington, DC, USA). Diluted Matrigel (BD Biosciences) was added to the interior of the transwell inserts for mimicking of basement membrane. The upper chambers were seeded with 1 × 105 cells and the lower chambers were filled with 1 ml DMEM containing 10% FBS, and cells were cultured for 24 h. Subsequent to removal of non-migrating cells present on the upper chamber surface, invaded cells at the bottom of the membrane were fixed with methanol and stained with crystal violet. Invaded cell numbers were recorded under microscope.
Clonogenic survival assay
Ten thousand prostate cancer cells transfected with shRNF7 retrovirus or negative controls were resuspended in DMEM medium containing 10% FBS and 0.3% agarose. Cells were plated onto a solidified bottom layer in DMEM medium containing 10% FBS and 0.5% agarose in 60-mm culture dishes. The colonies that formed at day14 were recorded following fixation with methanol, and stained with 0.05% methylene blue.
Tumor formation assays in nude mice
One million DU145 or PC3 cells transfected with or without RNF7 shRNA in 100 μl phosphate buffer saline (PBS) were injected subcutaneously (s.c.) into the flanks of 6-week-old female nude mice. Tumor growth was monitored for 6 weeks. Tumors were weighted and imaged after surgical removal.
Western blot analysis
Cells were lysed using RIPA lysis buffer (Life Technologies) and protein amount were determined by BCA protein assay kit (ThermoFisher Scientific). Equal amount (10 μg) of proteins were loaded and resolved onto 8–12% SDS-PAGE gel and blotted onto polyvinylidene fluoride (PVDF) membrane by electrophoresis. Following antibodies were used to detect the corresponding proteins: rabbit anti-RNF7 antibody (Ab181986, Abcam, Cambridge, MA, USA); rabbit anti-p21 antibody (#2947, CST, cell signaling technology, Danvers, MA, USA); rabbit anti-p27 antibody (#3686, CST); rabbit anti-NOXA antibody (#14766, CST); rabbit anti-ERK1/2 antibody (#4695, CST); rabbit anti-pERK1/2 antibody (#4370, CST); rabbit anti-GAPDH antibody (#5174, CST); HRP conjugated goat anti-rabbit IgG (#7074, CST). HRP Chemiluminescent substrate (ECL) kit (# 32106, ThermoFisher Scientific) was used for color development.
Statistical analysis
Data were presented as mean ± Standard Deviation (SD). All experiments were repeated at least three times independently with three or more replicates. Statistical differences were determined by GraphPad Prism 5.0 software (GraphPad Software Inc., CA, USA). Gaussian distribution data were analyzed by two-tailed Student’s t test, while non-Gaussian distribution data by Mann-Whitney of nonparametric test.
Ethic statements
All animal procedures were performed in accordance with Guidelines of Shanghai JiaoTong University School of Medicine, and approved by the Animal Care and Use committee of Shanghai Jiaotong University School of Medicine.
Additional Information
How to cite this article: Xiao, Y. et al. RNF7 knockdown inhibits prostate cancer tumorigenesis by inactivation of ERK1/2 pathway. Sci. Rep. 7, 43683; doi: 10.1038/srep43683 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Endzelins, E. et al. Diagnostic, prognostic and predictive value of cell-free miRNAs in prostate cancer: a systematic review. Molecular cancer 15, 41, doi: 10.1186/s12943-016-0523-5 (2016).
Nakazawa, M. & Kyprianou, N. Epithelial-Mesenchymal-Transition Regulators in Prostate Cancer: Androgens and Beyond. The Journal of steroid biochemistry and molecular biology, doi: 10.1016/j.jsbmb.2016.05.007 (2016).
Zuo, M., Xu, X., Li, T., Ge, R. & Li, Z. Progress in the mechanism and drug development of castration-resistant prostate cancer. Future medicinal chemistry 8, 765–788, doi: 10.4155/fmc.16.12 (2016).
Ferlay J., S. I., Ervik M., Dikshit R., Eser S., Mathers C., Rebelo M., Parkin D. M., Forman D., Bray & F. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11 [Internet], doi: Available at: http://globocan.iarc.fr (2013).
Duan, H. et al. SAG, a novel zinc RING finger protein that protects cells from apoptosis induced by redox agents. Molecular and cellular biology 19, 3145–3155 (1999).
Swaroop, M., Gosink, M. & Sun, Y. SAG/ROC2/Rbx2/Hrt2, a component of SCF E3 ubiquitin ligase: genomic structure, a splicing variant, and two family pseudogenes. DNA and cell biology 20, 425–434, doi: 10.1089/104454901750361488 (2001).
Gu, Q., Tan, M. & Sun, Y. SAG/ROC2/Rbx2 is a novel activator protein-1 target that promotes c-Jun degradation and inhibits 12-O-tetradecanoylphorbol-13-acetate-induced neoplastic transformation. Cancer research 67, 3616–3625, doi: 10.1158/0008-5472.CAN-06-4020 (2007).
Li, H. et al. Inactivation of SAG/RBX2 E3 ubiquitin ligase suppresses KrasG12D-driven lung tumorigenesis. The Journal of clinical investigation 124, 835–846, doi: 10.1172/JCI70297 (2014).
Duan, H. et al. Promotion of S-phase entry and cell growth under serum starvation by SAG/ROC2/Rbx2/Hrt2, an E3 ubiquitin ligase component: association with inhibition of p27 accumulation. Molecular carcinogenesis 30, 37–46 (2001).
He, H., Gu, Q., Zheng, M., Normolle, D. & Sun, Y. SAG/ROC2/RBX2 E3 ligase promotes UVB-induced skin hyperplasia, but not skin tumors, by simultaneously targeting c-Jun/AP-1 and p27. Carcinogenesis 29, 858–865, doi: 10.1093/carcin/bgn021 (2008).
Tan, M. et al. SAG/ROC-SCF beta-TrCP E3 ubiquitin ligase promotes pro-caspase-3 degradation as a mechanism of apoptosis protection. Neoplasia 8, 1042–1054, doi: 10.1593/neo.06568 (2006).
Tan, M. et al. Disruption of Sag/Rbx2/Roc2 induces radiosensitization by increasing ROS levels and blocking NF-kappaB activation in mouse embryonic stem cells. Free radical biology & medicine 49, 976–983, doi: 10.1016/j.freeradbiomed.2010.05.030 (2010).
Gu, Q., Bowden, G. T., Normolle, D. & Sun, Y. SAG/ROC2 E3 ligase regulates skin carcinogenesis by stage-dependent targeting of c-Jun/AP1 and IkappaB-alpha/NF-kappaB. The Journal of cell biology 178, 1009–1023, doi: 10.1083/jcb.200612067 (2007).
Tan, M. et al. SAG/ROC2/RBX2 is a HIF-1 target gene that promotes HIF-1 alpha ubiquitination and degradation. Oncogene 27, 1404–1411, doi: 10.1038/sj.onc.1210780 (2008).
Jia, L. et al. Validation of SAG/RBX2/ROC2 E3 ubiquitin ligase as an anticancer and radiosensitizing target. Clinical cancer research: an official journal of the American Association for Cancer Research 16, 814–824, doi: 10.1158/1078-0432.CCR-09-1592 (2010).
Tan, M. et al. SAG/RBX2/ROC2 E3 ubiquitin ligase is essential for vascular and neural development by targeting NF1 for degradation. Developmental cell 21, 1062–1076, doi: 10.1016/j.devcel.2011.09.014 (2011).
Sun, Y. & Li, H. Functional characterization of SAG/RBX2/ROC2/RNF7, an antioxidant protein and an E3 ubiquitin ligase. Protein & cell 4, 103–116, doi: 10.1007/s13238-012-2105-7 (2013).
Petrylak, D. P. Docetaxel for the treatment of hormone-refractory prostate cancer. Reviews in urology 5 Suppl 2, S14–21 (2003).
Chao, D., von Schlippe, M. & Harland, S. J. A phase II study of continuous infusion 5-fluorouracil (5-FU) with epirubicin and cisplatin in metastatic, hormone-resistant prostate cancer: an active new regimen. European journal of cancer 33, 1230–1233 (1997).
Kaku, H. et al. Combination chemotherapy with estramustine phosphate, ifosfamide and cisplatin for hormone-refractory prostate cancer. Acta medica Okayama 60, 43–49 (2006).
Matsumoto, A. et al. Evaluation of docetaxel plus estramustine in the treatment of patients with hormone-refractory prostate cancer. International journal of urology: official journal of the Japanese Urological Association 16, 687–691, doi: 10.1111/j.1442-2042.2009.02341.x (2009).
Suttmann, H. et al. Combining 153Sm-lexidronam and docetaxel for the treatment of patients with hormone-refractory prostate cancer: first experience. Cancer biotherapy & radiopharmaceuticals 23, 609–618, doi: 10.1089/cbr.2008.0487 (2008).
Naito, S. et al. Docetaxel plus prednisolone for the treatment of metastatic hormone-refractory prostate cancer: a multicenter Phase II trial in Japan. Japanese journal of clinical oncology 38, 365–372, doi: 10.1093/jjco/hyn029 (2008).
Berthold, D. R. et al. Treatment of hormone-refractory prostate cancer with docetaxel or mitoxantrone: relationships between prostate-specific antigen, pain, and quality of life response and survival in the TAX-327 study. Clinical cancer research: an official journal of the American Association for Cancer Research 14, 2763–2767, doi: 10.1158/1078-0432.CCR-07-0944 (2008).
de Bono, J. S. et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet 376, 1147–1154, doi: 10.1016/S0140-6736(10)61389-X (2010).
Petrioli, R. et al. Weekly docetaxel and epirubicin in treatment of advanced hormone-refractory prostate cancer. Urology 69, 142–146, doi: 10.1016/j.urology.2006.09.015 (2007).
Hainsworth, J. D. et al. Weekly docetaxel and bortezomib as first-line treatment for patients with hormone-refractory prostate cancer: a Minnie Pearl Cancer Research Network phase II trial. Clinical genitourinary cancer 5, 278–283, doi: 10.3816/CGC.2007.n.004 (2007).
Di Lorenzo, G. et al. Docetaxel, vinorelbine, and zoledronic acid as first-line treatment in patients with hormone refractory prostate cancer: a phase II study. European urology 52, 1020–1027, doi: 10.1016/j.eururo.2007.02.060 (2007).
Ferrero, J. M. et al. Phase II trial evaluating a docetaxel-capecitabine combination as treatment for hormone-refractory prostate cancer. Cancer 107, 738–745, doi: 10.1002/cncr.22070 (2006).
Di Lorenzo, G. et al. Weekly docetaxel and vinorelbine (VIN-DOX) as first line treatment in patients with hormone refractory prostate cancer. European urology 46, 712–716, doi: 10.1016/j.eururo.2004.08.022 (2004).
Huan, S. D., Stewart, D. J., Aitken, S. E., Segal, R. & Yau, J. C. Combination of epirubicin and cisplatin in hormone-refractory metastatic prostate cancer. American journal of clinical oncology 22, 471–474 (1999).
Buonerba, C. et al. Phase II trial of cisplatin plus prednisone in docetaxel-refractory castration-resistant prostate cancer patients. Cancer chemotherapy and pharmacology 67, 1455–1461, doi: 10.1007/s00280-011-1594-z (2011).
Itoh, T. et al. Cisplatin induces production of reactive oxygen species via NADPH oxidase activation in human prostate cancer cells. Free radical research 45, 1033–1039, doi: 10.3109/10715762.2011.591391 (2011).
Tan et al. Depletion of SAG/RBX2 E3 ubiquitin ligase suppresses prostate tumorigenesis via inactivation of the PI3K/AKT/mTOR axis. Molecular Cancer 15, 81–94, doi: 10.1186/s12943-016-0567-6 (2016).
Liu, L. L. et al. Identification of valid reference genes for the normalization of RT-qPCR expression studies in human breast cancer cell lines treated with and without transient transfection. PLoS One 10, e0117058, doi: 10.1371/journal.pone.0117058 (2015).
Acknowledgements
This work was supported by SUMHS seed foundation project (No. HMSF-16-21-016) and Shanghai Pudong new District Science and Technology Development Funding (No. PKJ2014-Y07) and Hundred talent pool of Shanghai university of medicine & health Sciences.
Author information
Authors and Affiliations
Contributions
The study was conceived by Z.W.L., and designed by Z.W.L., Y.J.X., Y.J. and H.M.S. Experiments were performed by Y.J., Y.J.X., H.M.S., T.L., Y.H.L., D.L.Y. and Q.F., and data analyzed by Y.J., Y.J.X., H.M.S. and Z.W.L. The manuscript was drafted and edited by Y.J.X., Y.J., H.M.S. and Z.W.L.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Xiao, Y., Jiang, Y., Song, H. et al. RNF7 knockdown inhibits prostate cancer tumorigenesis by inactivation of ERK1/2 pathway. Sci Rep 7, 43683 (2017). https://doi.org/10.1038/srep43683
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep43683
This article is cited by
-
RNF7 inhibits apoptosis and sunitinib sensitivity and promotes glycolysis in renal cell carcinoma via the SOCS1/JAK/STAT3 feedback loop
Cellular & Molecular Biology Letters (2022)
-
Functional roles of E3 ubiquitin ligases in prostate cancer
Journal of Molecular Medicine (2022)
-
Clinical Significance and Effect of lncRNA HOXA11-AS in NSCLC: A Study Based on Bioinformatics, In Vitro and in Vivo Verification
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
