
Abstract
Stellera chamaejasme L. (Thymelaeaceae) is a perennial herb that is widely used in traditional Chinese medicine to treat tumours, tuberculosis and psoriasis. S. chamaejasme extract (SCE) possesses anti-inflammatory, analgesic and wound healing activities; however, the effect of S. chamaejasme and its active compounds on cutaneous wound healing has not been investigated. We assessed full-thickness wounds of Sprague-Dawley (SD) rats and topically applied SCE for 2 weeks. In vitro studies were performed using HaCaT keratinocytes, Hs68 dermal fibroblasts and RAW 264.7 macrophages to determine cell viability (MTT assay), cell migration, collagen expression, nitric oxide (NO) production, prostaglandin E2 (PGE2) production, inflammatory cytokine expression and β-catenin activation. In vivo, wound size was reduced and epithelisation was improved in SCE-treated SD rats. In vitro, SCE and its active compounds induced keratinocyte migration by regulating the β-catenin, extracellular signal-regulated kinase and Akt signalling pathways. Furthermore, SCE and its active compounds increased mRNA expression of type I and III collagen in Hs68 fibroblasts. SCE and chamechromone inhibited NO and PGE2 release and mRNA expression of inflammatory mediators in RAW 264.7 macrophages. SCE enhances the motility of HaCaT keratinocytes and improves cutaneous wound healing in SD rats.
Similar content being viewed by others


Introduction
Cutaneous wound healing is a dynamic process involving intricate interactions among a variety of inflammatory cells, extracellular matrix (ECM) molecules, parenchymal cells and soluble mediators1. The wound healing process consists of three steps: inflammatory, proliferative and remodelling2. Representative mechanisms associated with cutaneous wound healing are the β-catenin, extracellular signal-regulated kinase (ERK) and Akt signalling pathways3,4,5.
β-catenin signalling pathways play pivotal roles in embryonic development, cell proliferation and cell migration6,7,8. The β-catenin-dependent canonical pathway and β-catenin-independent noncanonical pathways9, are both relevant to wound healing2. During the re-epithelisation stage of wound healing10, β-catenin is activated, modulates cell migration and proliferation, and induces fibromatosis in hyperplastic wounds11.
The ERK and Akt signalling pathways are associated with regulation of cutaneous wound healing12,13. Activation and initiation of ERK and Akt signaling pathways by growth factors such as fibroblast growth factor and epidermal growth factor may induce the motility of keratinocytes and re-epithelialisation in wounded skin14,15. A study reports that suppressing these factors may delay corneal wound healing, suggesting that the wound healing process may be accelerated by stimulating the β-catenin, ERK and Akt signalling pathways5,10.
The transforming growth factor (TGF)-β1 signalling pathway recruits and phosphorylates Smad2 and Smad3 through a complex of type I and type II receptors. Phosphorylated receptor-regulated Smads (Smad2 and Smad3) then form a heteromeric complex with coSmad (Smad4) and accumulate in the nucleus, where they act as transcription factors and participate in the regulation of TGF-β1-responsive gene expression16,17. Administration of exogenous TGF-β1 accelerates the wound healing process through the increased accumulation of ECM molecules, such as type I and III collagen18.
Nitric oxide (NO) is a highly reactive free radical and is an essential signalling factor in various physiological processes and cell types19. Cyclooxygenase (COX) is essential for the conversion of arachidonic acid into prostaglandin H2, a precursor of various biologically active mediators including thromboxane A2, prostacyclin and prostaglandin E2 (PGE2)20. Production and levels of nitric oxide synthase (NOS)-derived NO and COX-2-derived PGE2 were increased in a model of inflammatory response that is induced by proinflammatory mediators such as tumour necrosis factor (TNF)-α, interleukin (IL)-1β, and lipopolysaccharide (LPS)21. This induced inflammatory response may trigger further damage to adjacent cells and tissues around the wound site, thus delaying the wound healing process22. Previous studies have reported that suppression of NO and PGE2 production by treatment with NOS and COX-2 inhibitors may prevent some types of injury23,24.
Stellera chamaejasme L. (Thymelaeaceae) is a famous toxic herb that is widely distributed in China, Mongolia, Russia and Korea. This herb has both toxic and therapeutic effects. It has been used as a pesticide and as a remedy for stubborn skin ulcers25 with antiviral26,27, antitumour28, antibacterial29, immunomodulatory30 and insecticidal31,32 activities. Several previous studies that performed chemical analysis of this plant reported the isolation and purification of various compounds including diterpenes, flavones, lignans and coumarins. Although various physiological activities have been reported, its effects on wound healing activity and its underlying mechanism have not been investigated.
This present study investigated the effect of S. chamaejasme extract (SCE) and its constituents on cutaneous wound healing in vitro and in vivo. The effects of SCE on the wound healing process were evaluated by measuring neoepidermis formation in the wounds. Furthermore, we assessed the effect of SCE and its constituents on cell migration and β-catenin activation in keratinocytes, collagen production in fibroblasts, and PGE2 inhibition in macrophages to confirm the role of SCE and its active compounds in wound healing in an in vitro system.
Results
Identification of seven compounds from S. chamaejasme
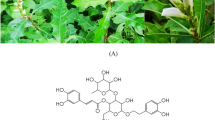
Seven compounds were identified from SCE using LC-NMR/MS: daphnin (compound 1, purity >98%), daphnetin-8-O-glucoside (compound 2, >97%), daphnetin (compound 3, >98%), rutarensin (compound 4, >98%), isoquercitrin (compound 5, >98%), chamechromone (compound 6, >98%), and daphnoretin (>98%). Compound purity was determined by HPLC (Fig. 1).

(A) Structure of compounds isolated from S. chamaejasme. (B) Chromatogram of SCE analyzed by HPLC detected at 330 nm and its assigned peaks; daphnin (peak 1), daphnetin-8-glucoside (peak 2), daphnetin (peak 3), isoquercitrin (peak 4), rutarensin (peak 5), chamechromone (peak 6), and daphnoretin (peak 7). The gradient profile was as follows: 0–5 min, initial mobile phase of 0.1% formic acid prepared in acetonitrile and 0.1% formic acid prepared in water (10:90); 5–45 min, linear gradient of 70:30; and 45–50 min, isocratic 70:30. The flow rate was 1.0 ml/min, and detection was at 254 nm. The sample concentration was 10 mg/min in MeOH, and the injection volume was 10 μl.
SCE and its active compounds enhance the motility and differentiation of HaCaT cells by activating the β-catenin, ERK and Akt signalling pathways
To evaluate whether SCE and its constituents can influence keratinocyte migration and differentiation, we assessed the effect of SCE and its constituents on HaCaT cell motility and expression of keratinocyte differentiation markers. Treatment of HaCaT cell with SCE and its constituents for 24 hours increased the migration of keratinocytes (Fig. 2A and B). In addition, protein expression of filaggrin, loricrin and involucrin that are important keratinocyte differentiation markers, was increased by SCE and its active compounds (Fig. 2C and D).

(A,B) HaCaT keratinocytes were maintained in DMEM supplemented with 10% heat-inactivated FBS, streptomycin (100 mg/ml), and penicillin (100 mg/ml) in 5% CO2 at 37 °C. After scratch wounding with sterile pipette tips, HaCaT keratinocytes were incubated with medium containing 2% serum with or without SCE (1,5, or 10 μg/ml) or various compounds (20 μM) for 24 h. Cells were stained with crystal violet for 24 h (original magnification ×40). (C,D) Western blot analysis of filaggrin, loricrin, and involucrin in control and sample (SCE or various compounds)-treated HaCaT cells. (E,F) SCE (1,5, or 10 μg/ml) or various compounds (20 μM) were added to HaCaT cells for 24 h. Luciferase activity of HaCaT cells transfected with TOPflash/FOPflash was measured. (G,H) Western blot analysis of β-catenin, p-ERK, and p-Akt in control and sample (SCE or various compounds)-treated HaCaT cells. Results are expressed as the mean ± SD of three independent experiments. (##p < 0.01, *p < 0.05 and **p < 0.01).
Considering the correlation between wound healing and the β-catenin signalling pathway3, we investigated the effect of SCE and its constituents on β-catenin activation. SCE and its constituents increased nuclear translocation and protein expression of β-catenin (Fig. 2E–H). The ERK and Akt signalling pathways enhance keratinocyte motility14,15. Therefore, we assessed the effects of SCE and its constituents on phosphorylation of ERK and Akt. Treatment with SCE and its constituents increased the phosphorylation of ERK and Akt (Fig. 2G and H).
SCE and its active compounds induce collagen expression in Hs68 cells
Fibroblast-to-myofibroblast transition plays a critical role in cutaneous wound healing33. mRNA levels of COL1A1 and COL3A1 and production of type І procollagen were markedly augmented by SCE and several compounds (especially daphnin and daphnetin-8-O-glucoside) in Hs68 cells, as shown by RT-PCR and ELISA analysis (Fig. 3A–D).

Human dermal fibroblasts (Hs68) were incubated for 48 h with SCE (1,5, or 10 μg/ml) or various compounds (10 or 20 μM). (A,B) mRNA levels of COL1A1, COL3A1 were determined with RT-PCR. (C,D) Production of type I procollagen was determined with ELISA. Results are expressed as the mean ± SD of three independent experiments. (##p < 0.01 and **p < 0.01).
SCE and chamechromone inhibit NO and PGE2 production and expression of inflammatory mediators in RAW 264.7 cells
The production of NO and PGE2 in LPS-stimulated RAW 264.7 cells was assayed after treatment with SCE or isolated compounds. SCE and chamechromone significantly suppressed LPS-induced NO production (Fig. 4A and B). In addition, SCE and chamechromone pretreatment reduced LPS-induced PGE2 production in a dose-dependent manner in RAW 264.7 cells (Fig. 4C and D). These data indicate that SCE and chamechromone significantly inhibit production of the inflammatory mediators NO and PGE2 in LPS-stimulated macrophages. Macrophages initiate an immune response by identifying and phagocytosing pathogens, inducing the secretion of inflammatory mediators such as iNOS, COX-2, TNF-α, IL-1β and p6534. Thus, we examined the expression of proinflammatory mediators that have a nuclear factor-κB-binding site in their promoter region35. To investigate the effects of SCE and chamechromone on the LPS-induced mRNA expression of proinflammatory cytokines, the mRNA levels of iNOS, COX-2, TNF-α, IL-1β and p65 in RAW 264.7 cells were measured using RT-PCR. Treatment with SCE and chamechromone significantly inhibited the production of iNOS, COX-2, TNF-α, IL-1β and p65 (Fig. 4E and F). These results suggest that SCE and chamechromone suppress the LPS-induced expression of proinflammatory cytokines.

RAW 264.7 cells were pretreated with different concentrations of SCE (25, 50, and 100 μg/ml) or various compounds (20 or 40 μM) for 1 h and then stimulated with LPS (1 μg/ml) for 24 h. (A,B) Nitrite levels were measured by the Griess reaction. (C,D) PGE2 levels were measured by the ELISA kit. (E,F) mRNA levels of inflammatory mediators were determined with RT-PCR. Results are expressed as the mean ± SD of three independent experiments. (##p < 0.01, *p < 0.05 and **p < 0.01).
SCE promotes cutaneous wound healing
We assessed the effect of SCE on cutaneous wound healing. After topical application of SCE (1%, 2% or 3%) to the wound areas of SD rats, SCE significantly reduced the wound size and re-epithelialised skin lesions (Fig. 5A, Tables 1 and 2). Histological analysis (H&E staining) also revealed restoration of normal tissue structure after SCE treatment (Fig. 5B).

A full-thickness skin excision (diameter = 1.5 cm) was made on the backs of 8-week-old SD rat, and SCE (1, 2, or 3%) or Centella asiatica extract (CAE, 1 or 3%) was topically applied to the wounds daily. Tissues were excised from the wounded area and fixed in paraformaldehyde for immunohistochemistry. (A) Representative gross images of wounded skin treated by SCE or CAE for 15 day. (B) Representative H&E stained tissues of wounded skin treated with or without SCE or CAE (original magnification ×100).
Discussion
The proliferative phase of wound healing is associated with the re-epithelialisation process including collagen production and ECM remodelling36. During wound re-epithelialisation stage, skin lesions closes the wound and remodels the cytoskeleton by enhancing the proliferation and migration of keratinocytes37. In this study, SCE and its constituents augmented the expression of keratinocyte differentiation markers, which are influenced by the β-catenin signalling pathway38, in HaCaT keratinocytes. The β-catenin signalling pathway enhances the proliferation and motility of keratinocytes10,11. Although dysregulation of the β-catenin signalling pathway induces hypertrophic keloids and scars39, adequate activation of this pathway in wounds is crucial to improve wound healing. It was reported that the proliferation of keratinocytes is regulated by inhibition of apoptosis during early wound healing10. Our results indicate that SCE and its constituents enhance the proliferation of keratinocytes by activating ERK and Akt signalling.
During the early stage of wound healing, collagen production is promoted to increase scar formation, and then it is gradually decreased to normal levels in the final stage of wound healing40. Several studies verified that TGF-β1 plays a significant role in the re-epithelisation process by increasing ECM production and inhibiting inflammation41. Furthermore, the TGF-β1 signalling pathway also interacts with the β-catenin signalling pathway during the wound healing process42,43. It was reported that Wnt3a treatment increases the levels of differentiation markers in myofibroblasts by activating the TGF-β1 signalling pathway44. Our results suggest that SCE and its constituents increased the expression of COL1A1, COL3A1 and TGF-β1 in dermal fibroblasts by activating the β-catenin signalling pathway.
Many bioactive mediators, such as eicosanoids, cytokines and growth factors, regulate each stage of the wound healing process. During infection and inflammation, activated macrophages that have infiltrated the site of inflammation produce a large amount of NO around the wounded tissues45. NO is a highly reactive radical that is generated by the activation of iNOS and contributes to various biological processes including inflammation. NO is thought to be a key disruptive factor in the wound healing process46, although it was reported that small amounts of NO may enhance wound healing during the early stage47. Excessive generation of PGE2 by COX-2 is a crucial physiological factor accelerating inflammation48. PGE2 is related to keratinocyte proliferation49, angiogenesis50 and mediation of the inflammatory response51. Our results suggest that SCE and chamechromone inhibit LPS-induced NO and PGE2 generation through inhibition of iNOS and COX-2 mRNA expression in RAW 264.7 macrophages. However, further investigation of the underlying mechanism is necessary. Macrophages are key factors in inflammation and reduce various harmful stimuli. Furthermore, they trigger the LPS-induced inflammatory response by producing proinflammatory mediators such as TNF-α, IL-1β and p6552. Overproduction of these mediators leads to excessive inflammatory responses53. Therefore, suppression of proinflammatory mediator production may be helpful in reducing the inflammatory response. In this study, SCE and chamechromone suppressed production of TNF-α, IL-1β and p65 in RAW 264.7 macrophages stimulated by LPS. Our results demonstrate that SCE and chamechromone suppress production of inflammatory mediators in RAW 264.7 cells.
We found that SCE promoted the epithelialization and wound closure rate in SD rat model. CAE, which is a famous traditional medicines proven to be effective in wound healing treatment54,55, was used as a positive control. Based on various studies, the biologically active constituents of CAE are known to be madecassic acid, asiatic acid, madecassoside and asiaticoside56,57. In our study, SCE group (SCE 3%) showed the enhanced cutaneous wound healing activity compared with CAE group (CAE 3%).
According to our data, all of eight tested constituents of SCE play a role at different molecular targets for exerting various biological effects in the wound healing system. For example, most of the constituents increase the keratinocyte migration, proliferation, and differentiation and β-catenin activation in HaCaT keratinocytes. In addition, daphnin and daphnetin-8-O-glucoside augmented collagen production and expression in Hs68 fibroblasts. Chamechromone inhibited the production of inflammatory mediators in Raw 264.7 macrophages. Therefore, in addition to the results that shows the positive effects of SCE on overall stages of wound healing, our results indicate that individual constituents of SCE exert their beneficial activity on different stages of wound healing process by regulating inflammatory, proliferative and remodelling phases.
In conclusion, our results suggest that SCE and its constituents induce cutaneous wound healing in SD rats, enhance keratinocyte motility and mRNA expression of type I and III collagen and inhibit PGE2 production and inflammatory mediator expression. SCE enhanced re-epithelialisation in wounds at an early stage of wound healing. Many wound healing drugs, including chemical molecules and growth factors, often have serious side effects58. However, SCE can be used to treat cutaneous wounds without any side effects. Therefore, SCE and its constituents may be beneficial for the treatment of wounds.
Materials and Methods
Reagents and materials
Foetal bovine serum (FBS), penicillin and Dulbecco’s modified Eagle medium (DMEM) were purchased from Hyclone (South Logan, UT, USA). Antibodies targeting β-catenin, phospho-p44/42 MAPK (ERK1/2), p44/42 MAPK (ERK1/2), phospho-Akt (Tyr525/526), Akt, phospho-Smad2 (Ser465/467)/Smad3 (Ser423/425), Smad2/3, and β-actin were purchased from Cell Signaling Technology Inc. (Danvers, MA, USA). Antibodies targeting filaggrin, loricrin, and involucrin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). LPS (Escherichia coli, serotype 0111:B4) and all other chemicals were purchased from Sigma Chemical (St. Louis, MO, USA). The plasmid (β-catenin) was transfected using Lipofectamine Plus (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s guidelines. Centella asiatica extract (CAE), as a positive control, was obtained from Korea Chong Kun Dang Pharma. Co. Ltd., in Korea.
Plant material
S. chamaejasme was collected in Gachuurt, Ulaanbaatar, Mongolia and was identified by Dr C. Sanchir, Institute of Botany, Mongolian Academy of Sciences. A voucher specimen was placed in the Flora and Plant Systematic Laboratory, Institute of Botany, Mongolian Academy of Sciences.
Extraction and isolation of compounds
Dried aerial parts of S. chamaejasme plants (5 kg) were extracted with 95% ethanol for 10 days at room temperature and filtered through filter paper. The filtrates were evaporated in a vacuum to yield the ethanol extract (203 g). This ethanol extract was suspended in distilled water and partitioned with n-hexane, ethyl acetate and n-butanol. The n-butanol fraction (5.2 g) was chromatographed on a RP-18 column, eluting with a H2O-MeOH gradient system (60:40 → 50:50), to give five fractions (fractions 1–5). Compound 1 (daphnin) was purified from fraction 3 (980 mg) using Sephadex LH-20 (MeOH) and Silica gel [CH2Cl2-MeOH-Water (3:1:0.1 → 1:1:0.1)] column chromatography. From fraction 5 (200 mg), compound 2 (daphnetin-8-O-glucoside) was isolated with Sephadex LH-20 (MeOH) column chromatography.
The ethyl acetate fraction (22 g) was chromatographed on Silica gel using CH2Cl2-MeOH (50:1 → 0:100) to give 13 fractions (fractions 1–13). From fraction 4 (1.2 g), compound 3 (daphnetin) was re-chromatographed on a RP-18 column, eluting with a H2O-MeOH gradient system (60:40 → 50:50) and Sephadex LH-20 (MeOH). Compounds 4 (rutarensin) and 5 (isoquercitrin) were purified from fraction 13 (2.3 g) using Silica gel [CH2Cl2-MeOH-Water (5:1:0.1 → 1:1:0.1)] and Sephadex LH-20 (MeOH) column chromatography. From fraction 8 (240 mg), compound 6 (chamaechromone) was re-chromatographed on a RP-18 column, eluting with a H2O-MeOH gradient system (50:50 isocratic) and Silica gel [CH2Cl2-MeOH-Water (8:1:0.1 → 4:1:0.1)]. Compound 7 (daphnoretin) was isolated from fraction 3 on Silica gel, eluting with Hexane-EtOAc (2:1 → 1:1), and recrystallised with CHCl3-MeOH to obtain white crystals.
The chemical structures of compounds 1–7 (Fig. 1A) were determined by 1H and 13C nuclear magnetic resonance data and compared with reported data59,60,61,62,63,64,65.
High-performance liquid chromatography (HPLC) analysis
The high-performance liquid chromatography (HPLC) system consisted of an Agilent infinity series 1260 liquid chromatography system with a G1311B quaternary pump, G1329B autosampler, G1316A column oven and G1315D DAD detector, connected to Agilent ChemStation software (Agilent, Waldbronn, Germany). The separation was conducted with a YMC-PACK ODS column (4.6 mm × 250 mm, 5 μm). The gradient profile was as follows: 0–5 min, initial mobile phase of 0.1% formic acid prepared in acetonitrile and 0.1% formic acid prepared in water (10:90); 5–45 min, linear gradient of 70:30; and 45–50 min, isocratic 70:30. The flow rate was 1.0 ml/min, and detection was at 254 nm. The sample concentration was 10 mg/min in MeOH, and the injection volume was 10 μl. The HPLC chromatogram of SCE is shown in Fig. 1B.
Cell culture
The human keratinocyte cell line (HaCaT), human foreskin fibroblast cell line (Hs68) and mouse macrophage cell line (RAW 264.7) were obtained from the American Type Culture Collection (Manassas, VA, USA) via the Korean Cell Line Bank (Seoul, Korea), and were routinely cultured in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin. Cells were grown in a humidified atmosphere with 5% CO2 at 37 °C.
Cell viability assay
Cell viability was assayed using a 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay (Sigma-Aldrich, St Louis, MO, USA). HaCaT, Hs68 and RAW 264.7 cells were grown in 24-well plates (1 × 105 cells/ml) for 24 h. After treatment with various concentrations of SCE and its constituents for 6 h, cells were washed and incubated with MTT (0.5 mg/ml) at 37 °C for 4 h. Cells were then washed, and the insoluble formazan products were dissolved in 200 μl of DMSO. Absorbance was measured at 550 nm by spectrophotometry (Bio-Tek Instruments, Winooski, VT, USA).
Cell migration assay
HaCaT cells were seeded into 24-well plates (1 × 105 cells/well). After 24 h of incubation, the monolayers were scratched with sterile pipette tips and treated with or without SCE (5 or 10 μg/ml) and its constituents (20 μM). After 48 h, the cells were fixed in 4% formalin for 20 min and stained with 2% crystal violet. The wound closure rate was evaluated using an Eclipse TE2000U inverted microscope with twin CCD cameras (magnification, ×200; Nikon, Tokyo, Japan; n = 3).
Reporter assay
Luciferase transactivation was measured as described previously66. TOPflash and FOPflash assays were performed in HaCaT cells. Cells were seeded in 24-well plates (6 × 104 cells/well). After 24 h, cells were co-transfected with luciferase reporter constructs (TOPflash or FOPFlash) and the pRL-CMV-Renilla reporter plasmid using Fugene 6 (Roche Applied Science, Indianapolis, IN, USA). After 24 h, SCE (5 or 10 μg/ml) and its constituents (10 or 20 μM) were added to the HaCaT cells for another 24 h. The cells were then lysed with lysis reagent (Promega, Madison, WI, USA), and the luciferase assay substrate (Promega) was added using a dual-luciferase assay system according to the manufacturer’s instructions. Transcriptional activity values were measured using a luminometer (Bio-Tek Instruments).
Enzyme-linked immunosorbent assay (ELISA) and NO assay
RAW 264.7 cells were incubated with SCE (50, 100 or 200 μg/ml) and its constituents (20 or 40 μM) for 1 h and stimulated with LPS (1 μg/ml) for 24 h. Cell culture medium was collected, and the secretion of PGE2 was determined using enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions. Nitrite (indicator of NO production) was measured in culture medium by adding Griess reagent. The culture medium was mixed with Griess reagent for 20 min, and then the absorbance at 540 nm was measured by spectrophotometry (Bio-Tek Instruments).
Hs68 cells were incubated with serum-free DMEM containing SCE (1, 5 or 10 μg/ml) and its constituents (20 μM). Cell culture medium was collected after 24 h, type I procollagen production were quantified using a procollagen type I C-peptide enzyme immunoassay kit (MK101; Takara, Shiga, Japan).
Animal study
Seven-week-old male Sprague-Dawley (SD) rats (Central Laboratory Animal Inc., Seoul, Korea) were housed in a controlled environment (25 ± 2 °C, 55% ± 5% relative humidity, 12 h light-dark cycle) and allowed to acclimatise for 1 week. All mouse experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the Korea Institute of Science and Technology in Seoul, Korea (2015–017). One day after dorsal hair removal, full-thickness incision wounds were made. SCE (1%, 2% or 3%) was topically applied to the wounds every day (n = 10), and wound sizes were measured three times per week. At the end of experiments, animals were anaesthetised with a Zoletil-Rompun mixture and skin lesions were removed and stored at −80 °C until analysis.
Histological analysis
Skin lesions were embedded in paraffin, sectioned (4 mm thick), deparaffinised in xylene and rehydrated in a gradient of alcohol solutions. The sections were stained with haematoxylin-eosin (H&E). The H&E-stained slides were visualised using an Eclipse TE2000U inverted microscope with twin CCD cameras (magnification, ×200; Nikon).
Abramov’s histological scoring system was used to score epithelisation, fibrosis, angiogenesis and collagen levels67,68. This system assigns a score of 0–3 for each parameter. Inflammation (the number of macrophages) was scored as follows: 0–25 = 1, 26–50 = 2 and >51 = 3. Fibroplasia was graded as follows: 0 (none to minimal fibroblasts), 1 (a few fibroblasts), 2 (more fibroblasts) and 3 (predominantly fibroblasts). Angiogenesis was graded as follows: 0 (none), 1 (up to five vessels per high-power field [HPF]), 2 (6–10 vessels per HPF) and 3 (more than ten vessels per HPF). Epithelisation was graded as follows: 0 (none), 1 (partial), 2 (complete, but immature or thin) and 3 (complete and mature).
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was isolated from the skin lesion using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions and quantified by spectrophotometry at 260 nm. An equal amount of total RNA was used to synthesise cDNA with reverse transcriptase premix (Elpis-Biotech. Inc., Taejeon, Korea). Reverse transcription was performed at 42 °C for 55 min and was terminated by incubation at 94 °C for 5 min. PCR amplification of the cDNA products (3 μl) was performed with PCR premix (Elpis-Biotech) and the primer pairs (Bioneer, Daejeon, Korea) shown in Table 1. Prior to PCR amplification, primers were denatured at 94 °C for 5 min. PCR was performed on a GeneAmp PCR System 2700 (Applied Biosystems, Foster City, CA, USA). PCR products were separated by 1.5% agarose gel electrophoresis and visualised with ethidium bromide. β-Actin was used as an internal control for normalising data.
Western blot analysis
HaCaT and Hs68 cells were lysed in RIPA buffer containing protease inhibitors and then incubated on ice for 10 min. The protein concentrations of the lysates were normalised by the Bradford method (Bio-Rad Laboratories, Hercules, CA, USA). Equal amounts of protein (20 μg) were resolved by 10% SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (Whatman GmbH, Dassel, Germany). The membrane was further incubated with specific primary antibodies for 16 h at 4 °C, followed by appropriate secondary antibodies coupled to horseradish peroxidase (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 2 h. Proteins were developed with ECL Western detection reagents (Amersham Biosciences, Little Chalfont, UK) and visualised by enhanced chemiluminescence (GE Healthcare, Hatfield, UK).
Statistical analysis
Each experiment was performed at least three times. All data are expressed as mean ± standard deviation. Differences between groups were analysed using a one-way analysis of variance followed by Scheffe’s test (SPSS 17.0, Chicago, IL, USA). ##p < 0.01, *p < 0.05 and **p < 0.01 were considered statistically significant.
Additional Information
How to cite this article: Kim, M. et al. Stellera chamaejasme and its constituents induce cutaneous wound healing and anti-inflammatory activities. Sci. Rep. 7, 42490; doi: 10.1038/srep42490 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Singer, A. J. & Clark, R. A. Cutaneous wound healing. The New England Journal of Medicine 341, 738–746 (1999).
Mendonça, R. J. & Coutinho-Netto, J. Cellular aspects of wound healing. Anais Brasileiros de Dermatologia 84, 257–262 (2009).
Zhang, D. L. et al. Effect of Wnt signaling pathway on wound healing. Biochemical and Biophysical Research Communications 378, 149– 151 (2009).
Weber, T. J. et al. Basic fibroblast growth factor regulates persistent ERK oscillations in pre-malignant but not malignant JB6 cells. Journal of Investigative Dermatology 130, 1444–1456 (2010).
Squarize, C. H., Castilho, R. M., Bugge, T. H. & Gutkind, J. S. Accelerated wound healing by mTOR activation in genetically defined mouse models. PLoS One 5, e10643 (2010).
Moon, R. T., Kohn, A. D., De Ferrari, G. V. & Kaykas, A. WNT and beta-catenin signalling: diseases and therapies. Nature Reviews Genetics 5, 691–701 (2004).
Widelitz, R. B. Wnt signaling in skin organogenesis. Organogenesis 4, 123–133 (2008).
Blanpain, C. & Fuchs, E. Epidermal homeostasis: a balancing act of stem cells in the skin. Nature Reviews Molecular Cell Biology 10, 207–217 (2009).
Grumolato, L. et al. Canonical and noncanonical Wnts use a common mechanism to activate completely unrelated coreceptors. Genes & Development 24, 2517–2530 (2010).
Cheon, S. et al. Prolonged beta-catenin stabilization and tcf-dependent transcriptional activation in hyperplastic cutaneous wounds. Laboratory Investigation 85, 416–425 (2005).
Cheon, S. S. et al. beta-Catenin stabilization dysregulates mesenchymal cell proliferation, motility, and in-vasiveness and causes aggressive fibromatosis and hyperplastic cutaneous wounds. Proceedings of the National Academy of Sciences USA 99, 6973–6978 (2002).
Kern, F., Niault, T. & Baccarini, M. Ras and Raf pathways in epidermis development and carcinogenesis. British Journal of Cancer 104, 229–234 (2011).
Peng, X. D. et al. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes & Development 17, 1352–1365 (2003).
Watson, A., Morris, V. L. & Chan, B. M. Coordinated integrin and growth factor regulation of primary keratinocyte migration mediated through extracellular signal regulated kinase and phosphoinositide 3-kinase. Archives of Dermatological Research 301, 307–317 (2009).
Abe, M. et al. Evidence that PI3K, Rac, Rho, and Rho kinase are involved in basic fibroblast growth factor-stimulated fibroblast-Collagen matrix contraction. Journal of Cellular Biochemistry 102, 1290–1299 (2007).
Xu, K. & Yu, F. S. Impaired epithelial wound healing and EGFR signaling pathways in the corneas of diabetic rats. Investigative Ophthalmology & Visual Science 52, 3301–3308 (2011).
Massague, J. & Wotton, D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J 19, 1745–1754 (2000).
Quaglino, D. J. et al. Transforming growth factor-beta stimulates wound healing and modulates extracellular matrix gene expression in pig skin: incisional wound model. Journal of Investigative Dermatology 97, 34–42 (1991).
Lowenstein, C. J. & Snyder, H. Nitric oxide, a novel biologic messenger. Cell 70, 705–707 (1992).
Smith, W. L., Garavito, R. M. & Dewitt, D. L. Prostaglandin endoperoxide H synthase (cyclooxygenase)-1 and -2. The Journal of Biological Chemistry 271, 33157–33160 (1996).
Hammond, R. A. et al. Endotoxin induction of nitric oxide synthase and cyclooxygenase-2 in equine alveolar macrophages. American Journal of Veterinary Research 60, 426–431 (1999).
Bauer, J. A., Rao, W. & Smith, D. J. Evaluation of linear polyethyleneimine/nitric oxide adduct on wound repair: therapy versus toxicity. Wound Repair and Regeneration 6, 569–577 (1998).
Mulligan, M. S., Moncada, S. & Ward, P. A. Protective effects of inhibitors of nitric oxide synthase in immune complex-induced vasculitis. British Journal of Pharmacology 107, 1159–1162 (1992).
Mack Strong, V. E. et al. NS-398 treatment after trauma modifies NFkappaB activation and improves survival. Journal of Surgical Research 98, 40–46 (2001).
Masatake, N. et al. A new coumarin glucosides from Daphne arisanensis . Chemical and Pharmaceutical Bulletin 39, 2422–2424 (1991).
Zhao, M. et al. A review of the most economically important poisonous plants to the livestock industry on temperate grasslands of China. Journal of Applied Toxicology 33, 9–17 (2013).
Asada, Y. et al. Stelleralides A-C, novel potent anti-HIV daphnane-type diterpenoids from Stellera chamaejasme L. Organic Letters 13, 2904–2907 (2011).
Yang, G. & Chen, D. Biflavanones, flavonoids, and coumarins from the roots of Stellera chamaejasme and their antiviral effect on hepatitis B virus. Chemistry & Biodiversity 5, 1419–1424 (2008).
Wang, M., Jia, Z. P., Ma, J. & Wang, B. The antitumor effects of total-flavonoid from Stellera chamaejasme . Zhongguo Zhong Yao Za Zhi 30, 603–606 (2005).
Shi. G. L. et al. Acaricidal activities of extracts of Stellera chamaejasme against Tetranychus viennensis (Acari: Tetranychidae). Journal of Economic Entomology 97, 1912–1926 (2004).
Xu, Z. H., Qin, G. W., Li, X. Y. & Xu, R. S. New biflavanones and bioactive compounds from Stellera chamaejasme L . Yao Xue Xue Bao 36, 669–671 (2001).
Tang, X. & Hou, T. Isolation and identification of 2-isopropyl-5-methylphenol from Stellera chamaejasme and its insecticidal activity against Aphis craccivora and Pieris rapae. Natural Product Research 25, 381–386 (2011).
Darby, I. A. & Hewitson, T. D. Fibroblast differentiation in wound healing and fibrosis. International Review of Cytology 257, 143–179 (2007).
Kawai, T. et al. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 11, 115–122 (1999).
Samavati, L. et al. STAT3 tyrosine phosphorylation is critical for interleukin 1 beta and interleukin-6 production in response to lipopolysaccharide and live bacteria. Molecular Immunology 46, 1867–1877 (2009).
Gentleman, E. et al. Mechanical characterization of collagen fibers and scaffolds for tissue engineering. Biomaterials 24, 3805–3813 (2003).
Santoro, M. M. & Gaudino, G. Cellular and molecular facets of keratinocyte reepithelization during wound healing. Experimental Cell Research 304, 274–286 (2005).
Ouji, Y., Yoshikawa, M., Shiroi, A. & Ishizaka, S. Wnt-10b promotes differentiation of skin epithelial cells in vitro . Biochemical and Biophysical Research Communications 342, 28–35 (2006).
Sato, M. Upregulation of the Wnt/beta-catenin pathway induced by transforming growth factor-beta in hypertrophic scars and keloids. Acta Dermato-Venereologica 86, 300–307 (2006).
Mutsaers, S. E., Bishop, J. E., McGrouther, G. & Laurent, G. J. Mechanisms of tissue repair: from wound healing to fibrosis. The International Journal of Biochemistry & Cell Biology 29, 5–17 (1997).
O’Kane, S. & Ferguson, M. W. Transforming growth factor beta s and wound healing. The International Journal of Biochemistry & Cell Biology 29, 63–78 (1997).
Guo, X. & Wang, X. F. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Research 19, 71–88 (2009).
Cheon, S. S., Nadesan, P., Poon, R. & Alman, B. A. Growth factors regulate beta-catenin-mediated TCF-dependent transcriptional activation in fibroblasts during the proliferative phase of wound healing. Experimental Cell Research 293, 267–274 (2004).
Carthy, J. M., Garmaroudi, F. S., Luo, Z. & McManus, B. M. Wnt3a induces myofibroblast differentiation by upregulating TGF-b signaling through SMAD2 in a b-catenin-dependent manner. PLoS One 6, e19809 (2011).
DeGeorge, G. L., Heck, D. E. & Laskin, J. D. Arginine meta-bolism in keratinocytes and macrophages during nitric oxide biosynthesis: multiple modes of action of nitric oxide synthase inhibitors. Biochemical Pharmacology 54, 103–112 (1997).
Rubbo, H., Tarpey, M. & Freeman, B. A. Nitric oxide and reactive oxygen species in vascular injury. Biochemical Society Symposium 61, 33–45 (1995).
Schaffer, M. R., Tantry, U., Thornton, F. J. & Barbul, A. Inhibition of nitric oxide synthesis in wounds: pharma-cology and effect on accumulation of collagen in wounds in mice. European Journal of Surgery 165, 262–267 (1999).
Pat Patel, R. et al. Regulation of cytosolic COX-2 and prostaglandin E 2 production by nitric oxide in activated murine macrophages. Journal of Immunology 162, 4191–4197 (1999).
Pentland, A. P. & Needleman, P. Modulation of keratinocyte proliferation in vitro by endogenous prostaglan-din synthesis. Journal of Clinical Investigation 77, 246–251 (1986).
Form, D. M. & Auerbach, R. PGE2 and angiogenesis. Proceedings of the Society for Experimental Biology and Medicine 172, 214–218 (1983).
Futagami, A. et al. Wound healing involves induction of cyclo-oxygenase-2 expression in rat skin. Laboratory Investigation 82, 1503–1513 (2002).
Lin, W. W. & Karin, M. A. Cytokine-mediated link between innate immunity, inflammation, and cancer. Journal of Clinical Investigation 117, 1175–1183 (2007).
Lawrence, T., Willoughby, D. A. & Gilroy, D. W. Anti-inflammatory lipid mediators and insights into the resolu-tion of inflammation. Nature Reviews Immunology 2, 787–795 (2002).
Arora, D., Kumar, M. & Dubey, S. D. Centella asiatica – a review of its medicinal uses and pharmacological effects. Journal of Natural Remedies 2, 143–149 (2002).
Gohil, K. J., Patel, J. A. & Gajjar, A. K. Pharmacological review on Centella asiatica: a potential herbal cure-all. Indian Journal of Pharmaceutical Sciences 72, 546–556 (2010).
Hashim, P. et al. Triterpene composition and bioactivities of Centella asiatica . Molecules 16, 1310–1322 (2011).
James, J. T. & Dubery, I. A. Pentacyclic triterpenoids from the medicinal herb, Centella asiatica (L) urban. Molecules 14, 3922–3941 (2009).
Fu, X. Cheng, B. & Sheng, Z. Growth factors and wound healing: review and prospect in recent ten years. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi 18, 508–512 (2004).
Tang, X., Chen, S. & Wang, L. Purification and identification of carvacrol from the root of Stellera chamaejasme and research on its insecticidal activity. Natural Product Research 25, 320–325 (2011).
Wei, Z. et al. Coumarins from the bark of Daphne marginata . Chemistry of Natural Compounds 43, 317–318 (2007).
Si-Cheng L. et al. Identification and characterization of human UDP-glucuronosyltransferases responsible for the in vitro glucuronidation of daphnetin. Drug Metab Dispos 38, 973–980 (2010).
Guanghou, S. & Leong, L. P. An improved method for the analysis of major antioxidants of Hibiscus esculentus Linn. J Chromatogr A 1048, 17–24 (2004).
Bernhard, K., Andreas, N. & Hildebert, W. Triumbellin, A tricoumarin rhamnopyranoside from Daphne mezereum . Phytochemsitry 29, 3633–3637 (1990).
Yuchi, Z. et al. Efficient combination of circulating ultrasound-assisted extraction and centrifugal partition chromatography for extraction and on-line separation of chemical constituents from Stellera chamaejasme L. Phytochemical Analysis 26, 301–309 (2015).
Wing-Shan, H. et al. Antiviral acitivity of daphnorentin isolated from Wikstroemia indica . Phytother Res 24, 657–661 (2010).
Yoo, J. H. et al. α- and γ-Mangostin inhibit the proliferation of colon cancer cells via β-catenin gene regulation in Wnt/cGMP signaling. Food Chemistry 129, 1669–1566 (2011).
Abramov, Y. et al. Histologic characterization of vaginal vs. abdominal surgical wound healing in a rabbit model. Wound Repair and Regeneration 15, 80–86 (2007).
Greenhalgh, D. G., Sprugel, K. H., Murray, M. J. & Ross, R. PDGF and FGF stimulate wound healing in the genetically diabetic mouse. American Journal of Pathology 136, 1235–1246 (1990).
Acknowledgements
This work was supported by the Center Project for Korea-Mongolia Science and Technology Cooperation sponsored by the Ministry of Education, Science and Technology (2U04650) and the National Research Council of Science & Technology (NST) grant by the Korea government (MSIP) (No. CRC-15-01-KIST).
Author information
Authors and Affiliations
Contributions
M. Kim and C.W. Nho designed the studies. M. Kim, H.J. Lee, J. Yun, and A. Randy performed the experiments. M. Kim, H.J. Lee, S.R. Oh, and C.W. Nho analyzed the data. M. Kim and H.J. Lee wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Kim, M., Lee, H., Randy, A. et al. Stellera chamaejasme and its constituents induce cutaneous wound healing and anti-inflammatory activities. Sci Rep 7, 42490 (2017). https://doi.org/10.1038/srep42490
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep42490
This article is cited by
-
An advanced wood preservative based on the extract of Stellera chamaejasme root with high antifungal activity
Advanced Composites and Hybrid Materials (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
