
Abstract
Short-chain polyphosphate (polyP) is released from platelets upon platelet activation, but it is not clear if it contributes to thrombosis. PolyP has increased propensity to clot blood with increased polymer length and when localized onto particles, but it is unknown whether spatial localization of short-chain polyP can accelerate clotting of flowing blood. Here, numerical simulations predicted the effect of localization of polyP on clotting under flow, and this was tested in vitro using microfluidics. Synthetic polyP was more effective at triggering clotting of flowing blood plasma when localized on a surface than when solubilized in solution or when localized as nanoparticles, accelerating clotting at 10–200 fold lower concentrations, particularly at low to sub-physiological shear rates typical of where thrombosis occurs in large veins or valves. Thus, sub-micromolar concentrations of short-chain polyP can accelerate clotting of flowing blood plasma under flow at low to sub-physiological shear rates. However, a physiological mechanism for the localization of polyP to platelet or vascular surfaces remains unknown.
Similar content being viewed by others


Introduction
PolyP is an activator of blood coagulation through its ability to accelerate the activation of coagulation factors XII, XI, and V1,2, and by abrogating tissue factor pathway inhibitor (TFPI) function3,4. Long-chain polyP (hundreds to thousands of residues long) appears to be a much more potent activator of clotting, via activation of factor XII and the contact pathway, than short-chain polyp5,6,7. Short-chain polyP (60–100 phosphate residues long) is found in dense granules of human platelets and granules of mast cells (acidocalcisomes) and released upon their activation, while long-chain polyP occurs in microbes and some mammalian cells, such as in prostate cancer7,8,9,10. A characteristic of long-chain polyP is its ability to aggregate into particles, and this spatial localization may possibly contribute to its propensity to accelerate clotting11. It is less clear if there is a pathophysiological role for polyP released from human cells in thrombosis3. Short-chain endogenous polyP facilitates activation of FXII in vitro, albeit at supraphysiological concentrations12. It can also contribute to clotting in vitro under flow when tissue factor (TF) is present13. It is well-known that the local concentration of activators can profoundly influence their ability to initiate the clotting of blood14. Localization of polyP onto particles also accelerates coagulation under stagnant conditions15. Thus, we hypothesized that short-chain polyP may be a more effective activator when spatially localized onto surfaces, capable of accelerating clotting of flowing blood in vitro without participation of TF.
Initiation of blood coagulation is triggered when the local concentration of activators reaches a critical threshold, upon which the proteolytic cascade amplifies the local concentration of active enzymes to form a cross-linked fibrin mesh16,17. The spatial localization of activators to surfaces effectively increases their local concentration, allowing coagulation to be triggered with less total amount of activator18,19. Several activators have displayed this effect of spatial localization in microfluidic models of clotting, including TF, glass, and bacteria that activate prothrombin and factor X20. Flow influences coagulation in a variety of ways and enhances the effects of spatial localization21. Flow continuously strips clotting factors from catalytic surfaces, preventing activators from achieving a critical threshold and ultimately preventing clot initiation22,23. To accelerate clotting of flowing blood, greater amounts of activator need to be localized in order to achieve a higher local concentration24. In this study, we used numerical simulations and a microfluidic model of thrombosis to investigate whether the ability of localization to enhance clotting extends to short-chain polyP in vitro under flow. The shear rates used in this study range from low to sub-physiological (i.e. pathological) shear. These low shear rates mimic those typical of where thrombosis occurs in large veins and valves, such as in deep vein thrombosis (DVT) or those associated with airplane economy class syndrome25,26,27,28,29,30. This microfluidic model of thrombosis enabled clotting of plasma, or lack thereof, to be monitored over many hours in the absence of TF. In contrast to coagulation that occurs from acute injury to vessels, such as from puncture that exposes large amounts of TF, thrombosis may initiate over longer periods of time and can be potentiated by factor XII2,31,32,33,34,35. Our experiments were designed to determine if localization of physiologically-relevant concentrations of platelet-length polyP could contribute to coagulation in vitro at low to sub-physiological shear, but they do not validate whether or not localization of platelet-length polyP contributes to thrombosis in vivo.
Results
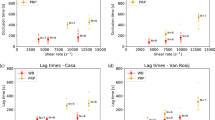
Numerical simulations predict the localization of polyP will increase its coagulability at low shear rates
To initially examine how localizing polyP onto surfaces affects thrombin generation, we used a two-dimensional numerical simulation that considered diffusion, convection, and the rates of 41 reactions of the coagulation cascade (Supplementary Tables S1–3). An established kinetic model for the coagulation cascade was used with the addition of polyP>1000 in three reactions that were previously characterized in kinetic assays1,4,5,7,20. PolyP was either spatially localized onto the surface of a cylindrical channel or dispersed throughout its volume. Shear rates were from 1 s−1 to 120 s−1, a range that encompasses sub-physiological shear rates (<~10 s−1) and shear rates in the inferior vena cava, venous valves, and large veins25,26,27,36. When polyP was localized onto the surface of the channel with a shear rate of 1 s−1, the local thrombin burst was 782-fold higher than when an equal amount of polyP was dispersed throughout the volume (1.83 × 10−8 M versus 2.34 × 10−11 M) (Fig. 1A). The amount of polyP in the simulations was 7.54 × 10−9 mol, which equates to 30 μM (with respect to phosphate monomer) when the total volume of the simulation was considered. The resulting thrombin burst was a consequence of the higher local concentration of polyP, which led to increased positive feedback from the coagulation cascade. Simulations showed that differences in thrombin generation persisted over various shear rates, up to 60 s−1 (Fig. 1B). However, at a set distance, the difference decreased as shear rate increased, because thrombin was rapidly transported down-stream.

Two-dimensional numerical simulations of the human blood coagulation cascade, comparing the generation of thrombin by polyP dispersed throughout a cylindrical channel versus polyP immobilized on the channel surface. The channel was 20 mm long with a radius of 2 mm. The overall number of polyP molecules was the same in all simulations (7.54 × 10−9 moles). (A) Plots show [thrombin], which is the sum of concentrations of thrombin and meizothrombin, for a two-dimensional longitudinal cut of the cylinder at 500 s into the simulation. (B) The fold difference in the maximum [thrombin] generated in the channel when polyP was surface-immobilized (SI-polyP) versus dispersed (D-polyP) at varying shear rates.
Surface-immobilized polyP accelerates clotting of flowing blood plasma
To determine if SI-polyP was able to accelerate clotting of flowing blood plasma, synthetic polyP400 was immobilized onto the walls of microfluidic channels (Fig. 2A). Half of each chamber was patterned with biotinylated lipids followed by an excess of streptavidin (Fig. 2B). Biotinylated-polyP400 was then flowed through the channel, becoming immobilized onto streptavidin. The surface concentration of SI-polyP400 was varied by diluting biotinylated-polyP400 in a solution of biotin-PEG before coating the channel. The concentration of polyP was determined by DAPI staining. Fluorescence intensities from known concentrations of stained D-polyP, which was soluble and dispersed throughout the channel, were used to generate a standard curve and used to calculate the surface concentration of SI-polyP (Fig. 2C). The surface concentration of SI-polyP was 300 nmol/m2, and could be decreased to 60 nmol/m2 by diluting with biotin-PEG. To test the ability of patterned polyP to induce clotting, platelet-poor human plasma was flowed through the chambers. Based on the simulation data, we tested the lowest shear (1 s−1) as it was predicted to have the largest effect on thrombin generation and therefor clotting. A range of shear rates are explored in later experiments. The plasma clotted selectively on areas with immobilized polyP400 (300 nmol/m2) in 50–70 min at a shear rate of 1 s−1. No clotting was observed over 5 hr in channels without polyP400 (Fig. 2D). All polyP concentrations are reported in terms of phosphate monomer.

(A) Schematic of biotinylated synthetic polyP (cyan) patterned onto the surface of half of a microfluidic channel, which induces production of thrombin and clotting (blue) of flowing blood plasma (grey). (B) Images of fluorescent-labeled agents flowing and patterned along one side of a microfluidic channel. Biotinylated lipids (tagged red) self-assembled on the channel wall. Non-biotinylated lipids (not tagged in these images) were simultaneously flowed and patterned on the other side of the chamber using laminar flow patterning. Then, streptavidin (tagged green) was flowed through and bound to the biotinylated lipids, followed by flowing biotinylated polyP labeled with DAPI (cyan), which bound streptavidin. A substrate (blue) for thrombin was activated, indicating initiation of clotting, selectively on patterned polyP400 (300 nmol/m2). Scale bar is 250 μm. (C) Quantifying of the amount of SI-polyP by measuring the fluorescence of DAPI bound to it. Channels with SI-polyP were compared to channels without polyP and to channels treated with polyP diluted with biotinylated PEG. Inset is a standard curve of known concentrations of solubilized D-polyP, which was used to calculate the surface concentration of SI-polyP in coated channels. (D) The clotting times of normal human plasma flowing through channels coated with polyP400 at a shear rate of 1 s−1. *p = < 0.01 compared to controls without polyP. Data indicate mean ± SEM, n = 3.
Measuring clot times simultaneously at various shear rates
A microfluidic system containing six regions with varying shear rates was used to measure clot times of flowing blood plasma (Fig. 3A). The range of shear rates was 1–110 s−1, which encompasses physiological shear rates which occur in the inferior vena cava, venous valves, and large veins; as well as, sub-physiological shear rates (<~10 s−1) that occur in pathological contexts25,26,27,28,36. These calculated shear rates were within 3–8% of the values obtained by measuring the flow velocity of micro particles by florescence microscopy. Clotting was monitored by visualizing the movement of fluorescent tracer beads specifically in the shear chambers, which became immobilized in clotted regions, and by a fluorogenic peptide substrate, which fluoresced when cleaved by thrombin during clotting (Fig. 3B). To characterize and determine the range of clot times of flowing blood plasma in the microfluidic system, coagulation factor VIIa (FVIIa) was used, and added to plasma at a range of concentrations (Fig. 3C). FVIIa does not circulate in plasma in appreciable amounts physiologically (~1% of total FVII circulates as FVIIa)37, but is administered during severe hemorrhage in some cases to aid in hemostasis at doses of 90 to 270 μg/kg, which roughly corresponds to 1 to 4 μg/mL in plasma38,39. In the device, plasma containing 16 μg/mL of FVIIa clotted in approximately 20–40 min, plasma containing 4 μg/mL of FVIIa clotted in approximately 60 min, and plasma containing 4 ng/mL did not clot within 6 hr. Intermediate clotting times occurred with concentrations of 400 ng/mL and 40 ng/mL and were dependent on shear rate. Clot formation always occurred from the channel wall, crudely mimicking how physiological thrombus formation occurs from the walls of blood vessels and is shear-dependent40.

(A) Schematic of the microfluidic system. Box (dashed lines) indicates the region where shear rates were varied and clot times were measured. (B) Fluorescence images showing that clotting was detected by the cessation of flow of tracer beads (pink) and by the cleavage of a substrate for thrombin (blue). Scale bar is 250 μm. (C) Assessing the range of clotting times in this flow system by adding various concentrations of FVIIa to the plasma. Data points indicate mean ± SEM, n = 3–4. Red circles indicate p = <0.05 between the data points, and blue circles indicate p = <0.01 between the data points.
Short-chain polyP accelerates clot formation faster when surface-localized than when dispersed in nanoparticles or in solution
PolyP160 was previously demonstrated to be a weak initiator of the contact pathway, but we examined the hypothesis that spatially localizing polyP160 onto a surface (SI-polyP160) would enhance its ability to contribute to clot formation compared to polyP160 dispersed as nanoparticles (NP-polyP160) or in solution (D-polyP160, Fig. 4A). With NP-polyP160 (1 μM, 250 nm diameter, Supplementary Fig. S1), clotting occurred in approximately 170 min and 200 min at a shear rate of 1 s−1 and 22 s−1 respectively. When a similar amount of polyP160 was localized onto the channel surface, clotting occurred significantly faster than both NP-polyP160 and D-polyP160. Clotting initiated from the parallel channel shear chamber walls, or in areas where the channel expanded from high to very low shear, and progressively grew outwards (Fig. 4B). Clotting with D-polyP160 (1 μM) was 4- to 2.8-fold slower than SI-polyP160 and 1.6- to 0-fold slower than NP-polyP160 at all shear rates. Overall, clotting occurred fastest with SI-polyP160 than dispersed polyP160 in either soluble or NP forms, even with 6–43 fold less SI-polyP160 in the channels.

(A) Clotting times of plasma by polyP160 at varying shear rates, comparing three states of polyP160: solubilized, self-assembled nanoparticles, and surface-immobilized. (B) Time-lapse images showing SI-polyP160 initiating clotting (detected by non-flowing beads) from the channel wall (dashed lines). Scale bar is 250 μm. (C) Comparing three states of polyP70: solubilized, surface-immobilized onto the microfluidic channels, and immobilized onto silica nanoparticles. Clotting tendencies of plasma containing silica nanoparticles coated with polyP70 (SNP-polyP70) compared to soluble and surface-immobilized polyP70 under shear in the microfluidic device. (D) Comparing two states of long-chain polyP: surface immobilized polyP400 and nanoparticles of self-assembled polyP>1000. (E) Schematic summarizing the relationship between spatial distribution of polyP and the acceleration of clotting in the above experiments. Data points indicate mean ± SEM, *p < 0.001, **p < 0.0001, n = 3–4. Statistical analysis represents comparisons between whole curves.
Platelet-length polyP can accelerate clotting when surface-localized
The concentration of polyP is approximately 1.1 mM in platelets, where it is stored in platelet dense granules, and can reach up to 2–7 μM in blood upon platelet activation41,42. To test whether synthetic polyP similar in length to those found in human platelets can clot flowing blood at physiological concentrations, polyP70 was tested (Fig. 4C). Soluble polyP70 (D-polyP70) at 400 nM did not accelerate clotting of flowing blood plasma at the shear rates tested. In contrast, an equivalent amount of SI-polyP70 substantially accelerated clotting, to 70 min and 160 min at shear rates of 1 s−1 and 110 s−1 respectively. The amount of SI-polyP70 used corresponded to a surface concentration of 24 nmol/m2 and a total concentration of around 400 nM in the volume of the channel. Initiation time of clotting by SI-polyP70 was dependent on FXII (Supplementary Fig. S2).
SI-polyP70 and D-polyP70 could not be directly compared to self-assembled nanoparticles of polyP70 (NP-polyP70), because the solubility of polyP70 is greater than longer chain polyP, and NP-polyP70 was not stable. Alternatively, we tested a second formulation of polyP nanoparticles, where polyP70 was coated on silica nanoparticles (SNP-polyP70)43. When SNP-polyP70 was added to plasma at varying shear, clotting occurred in approximately 70 min to 160 min at 200 μg/mL and 80 min to >360 min at 20 μg/mL. These masses of SNP-polyP70 corresponded to concentrations of polyP70 of 6 μM and 0.6 μM, respectively, but include both polyP70 and silica. Silica is also an activator of factor XII, so an equal comparison between SI-polyP70 and SNP-polyP70 cannot be made. Nevertheless, the clotting times of SI-polyP70 (400 nM) were significantly faster than 20 μg/mL of a SNP-polyP70, and were nearly identical to 200 μg/mL of a SNP-polyP70 even though there was a 15-fold lower concentration of polyP70.
Clotting by long-chain polyP is also enhanced by surface localization
To understand if the effect of surface localization extends to long-chain polyP, we tested a range of concentrations of long-chain polyP either surface localized (SI-polyP400) or dispersed as nanoparticles (NP-polyP>1000). PolyP>1000 naturally self-assembles, localizing into nanoparticles of 150 ± 30 nm in diameter in solutions containing Ca2+ at low millimolar concentrations11. It is a known activator of clotting under static conditions when dispersed throughout plasma3. We compared NP-polyP>1000 to SI-polyP400, rather than SI-polyP>1000, because surface patterning of polyP requires biotinylation of the polyP chains, and the biotinylation procedure caused degradation of long chain-lengths of polyP. When plasma was flowed over SI-polyP400, clotting occurred in approximately 60 min to 100 min at 7 μM and 140 min to 170 min at 1 μM at the shear rates examined (Fig. 4D). The clot times using NP-polyP>1000 demonstrated robust shear- and concentration-dependence at 2000, 200, 20, 7 and 1 μM. NP-PolyP>1000 was most potent at 2000 μM, initiating clotting at 60 min at 1 and 3 s−1, although requiring 285-fold more phosphate to match the propensity of SI-polyP400 to clot flowing plasma under the same conditions.
Discussion
Together, these data show that the spatial localization of synthetic polyP onto surfaces affects its ability to activate clotting under flow (Fig. 4E). Short-chain polyP polymers (polyP160 and polyP70) greatly accelerated clotting of flowing blood plasma at low to sub-physiological shear when surface-localized onto the walls of microfluidic chambers compared to when they are dispersed (nanoparticle or soluble forms). Soluble short-chain polyP only clotted stagnant blood (near-zero flow) in our experiments, and clotting did not occur within a span of hours even at sub-physiological shear rates. Localization of polyP onto the surface of channels showed the greatest activity overall. The concentration at which SI-polyP70 accelerated clotting in vitro is well-within the range of amounts of polyP released into plasma following platelet activation. Although it is not known if polyP localizes to cell surfaces or thrombi, or to the extent polyP contributes to physiological or pathophysiological coagulation, it is important to identify scenarios in which polyP could potentially elicit a role. These results propose that if polyP can surface-localize it may contribute to clotting at sub-physiological shear following platelet activation, but further in vitro and in vivo experiments are necessary to verify that this is a potential mechanism.
Remarkably, comparing SI-polyP70, SI-polyP160, and SI-polyP400, to each other shows that short-chain polyP could match the propensity of longer chain polyP to accelerate clotting under flow. SI-polyP70 accelerated clotting to a similar extent as SI-polyP160 and SI-polyP400 with a lower concentration of phosphate. Although clotting times were similar between them with respect to surface coverage of full-length polymers. This is likely because shorter chains have high surface coverage relative to the amount of monomer. Thus, clotting occurred faster with both increasing surface concentration of phosphate and increasing surface coverage.
The simulations predicted the trend observed in vitro. Localization creates high local concentrations of polyP, and in the numerical simulations this led to larger thrombin bursts due to increased positive feedback from the coagulation cascade. The simulations included polyP binding and inhibiting TFPI and accelerating activation of factors V and XI, which all occur in plasma. The mechanism is likely contact system mediated as under stagnant conditions FXII contributed to initiation of clotting by polyP, but we did not test this further in flow experiments. It was recently shown that short-chain polyP could complex with FXII in vitro to allostericly induce its activation at high polyP concentrations of 70–130 μM12. This polyP-induced activation of FXII was enhanced in the presence of zinc ions, which is known to bind robustly to both FXII and PolyP3,12. Short-chain polyP can also contribute to clotting independently of FXII when TF is present13. The results here, without TF, indicate that localization can further increase the propensity of short-chain polyP to clot blood plasma.
In these microfluidic experiments, shear rate and concentrations of either FVIIa or polyP influenced the clotting times over several hours. The shear rates mimicked the shear rates that are typical in large veins and valves; as well as, pathological shear which occurs it the context of thrombosis. The reported clotting times appear very long compared to clotting times in most in vitro, stagnant clotting assays, which occur in seconds to minutes. However, residence time of plasma in the microfluidic chambers was only ~10 sec, with plasma being continuously transported into and out of the chamber, and thus the rate of clotting cannot be directly compared to stagnant clotting assays. Long clot times were possible in this device, compared to most other flow systems, because platelet-poor plasma was recalcified on the device and because TF was not included44. The observed clotting times were much slower than what is typical in acute hemostasis and at high concentrations of TF, but they were within the time-frame that formation and growth of thrombi occurs inside veins and regions of low shear31,32,33. Thrombosis, in contrast to hemostasis, can involve progressive and gradual clot growth, where there is much less TF but increased contribution of factors XI and XII2,45. The clotting times measured in this microfluidic system are more representative of clotting times that would occur during thrombosis inside intact veins, rather than punctured vessels or stagnant clotting assays. In addition, the shear rates used in our microfluidic model include the rates which occur in large veins. Though platelets appear to contribute more to arterial thrombi than venous thrombi, they also contribute to venous thrombosis46,47. For example, antiplatelet drugs have also been beneficial in treating venous thrombosis47,48,49.
For several concentrations and chain-lengths of polyP, it was not possible to make equivalent comparisons between SI-polyP, NP-polyP, SNP-polyP, and D-polyP, because the chain-length and concentration are important determinants of whether polyP self-assembles into particles or remains soluble. The solubility of short-chain polyP is greater than long-chain polyP, but solubility also depends on the concentration of polyP and Ca2+. For example, polyP160 can be formulated to be soluble or to form NP-polyP by varying the concentration of polyP and Ca2+11. We used concentrations of polyP160 well below its limit of solubility. PolyP160 was first dissolved into water and then diluted into plasma. When added to citrated plasma, soluble polyP likely remained dissolved, as plasma has insufficient free divalent cations to facilitate nanoparticle formation11. Once plasma is recalcified, polyP likely remains protein-bound even in the presence of low millimolar amounts of ionic calcium, at least for the 35 sec that it is present in the microfluidic devices (Supplementary Fig. S3)11,50. In contrast, NP-polyP were formed by precipitating polyP160 in 5 mM Ca2+, generating nanoparticles that were stable for over 6 hr, as measured by dynamic light scattering. NP-polyP was diluted in the calcium-saline solution that mixed with blood plasma inside the microfluidic devices to keep the nanoparticles intact. The stability of NP-polyP in plasma is unknown; however, NP-polyP was initially prepared under supersaturated conditions, and the solubility of NP-polyP displays hysteretic behavior11. Thus, a large portion of NP-polyP, once formed, likely remained as NPs in the microfluidic devices. Although synthetic polyP was used in these experiments, natural polyP is also typically bound to calcium51.
In summary, this work shows that spatial localization of synthetic polyP, including short-chain polyP, increases its propensity to accelerate clotting of blood plasma at low to sub-physiological shear. The observed clotting times were much slower than what is typical in hemostasis, but they were within the time-frame that thrombosis occurs inside veins, particularly post-operative deep vein thrombi, which form over a period of days32,33,52. The experiments were designed solely to test if surface-localization of short-chain polyP accelerates clot formation under flow, at venous and sub-physiological shear rates. An important observation from this was that when localized, short-chain polyP could match the ability of long-chain polyP to accelerate clotting. The concentration required to accelerate clotting is markedly reduced when polyP is spatially localized onto surfaces, and to a lesser extent, into particles, even under flow and without TF. These biophysical insights provide a potential biophysical mechanism by which platelet-length polyP could contribute to thrombosis in regions of low shear, but further work is required to validate if this mechanism could indeed extend to in vivo scenarios. This effect of localization may potentially contribute to clotting at higher shear when TF is present13. Although these in vitro results, in an artificial flow system, support the notion that platelet-derived polyP could contribute to coagulation in vivo, the flow system used here does not include many factors that normally regulate clotting, such as platelets, platelet-derived polyP, red blood cells, immune cells, endothelium and other soluble factors. For these reasons, appropriate in vivo models are necessary to verify whether platelet-derived polyP and its spatial localization contributes to clot formation and thrombosis.
Materials and Methods
Numerical Simulations
Thrombin generation was modeled with the Transport of Diluted Species module of Comsol Multiphysics 4.4 by adding diffusion and convection to a previously reported kinetic model20. Changes to the model included the addition of three rate equations to describe the activity of polyP: 1) the binding and inhibition of TFPI; TFPI + polyP ↔ TFPI-polyP; kon = 4.0 × 105 M−1s−1, koff = 1.0 × 10−2 s−1, 2) the activation of factor V; V + Xa + polyP → Va + Xa + polyP; k = 8.0 × 1012 M−2s−1, 3) the activation of factor XI; XI + IIa + polyP → XIa + IIa + polyP; k = 8.8 × 109 M−2s−11,4,5,7,20. The diffusion coefficient for all soluble species was 5 × 10−11 m2/s and the velocity profile varied with the shear rate,  , where vz is the velocity in the axial direction at each radial coordinate r, R is the cylinder radius, and γw is the shear rate at the cylinder wall. The chemical species were flowed into a cylindrical geometry of radius 2 mm and length 20 mm. For each shear rate, [thrombin] was sampled after the incoming flow had displaced the channel volume 12.5 times. Both the experiments and simulations were performed in the same mass-transfer regime (Pe ≫ 1 and Gz > 3000).
, where vz is the velocity in the axial direction at each radial coordinate r, R is the cylinder radius, and γw is the shear rate at the cylinder wall. The chemical species were flowed into a cylindrical geometry of radius 2 mm and length 20 mm. For each shear rate, [thrombin] was sampled after the incoming flow had displaced the channel volume 12.5 times. Both the experiments and simulations were performed in the same mass-transfer regime (Pe ≫ 1 and Gz > 3000).
Preparing soluble polyP (D-polyP), self-assembled polyP nanoparticles (NP-polyP), polyP-coated silica nanoparticles (SNP-polyP), and surface-immobilized polyP (SI-polyP)
To make D-polyP, polyP was solubilized and diluted first in water and then added to citrated plasma (frozen citrated normal control plasma, Affinity Biologicals Inc.) prior to entering the microfluidic device. NP-polyP was generated as described previously11. Briefly, soluble polyP was added to a calcium solution (10 mM polyP, 5 mM CaCl2, 8 mM Tris, pH 6.0) followed by vortexing, during which polyP self-assembled into nanoparticles tightly bound to Ca2+ cations11. The formation and size of the nanoparticles were verified after adding them to this calcium solution by observing the scattering intensity and hydrodynamic diameter, as measured by dynamic light scattering (Zetasizer Nano ZSP, Malvern Instruments). NP-polyP formulations were added and diluted in calcium-saline solution, rather than the citrated-plasma, prior to entering the microfluidic device. The NP-polyP were stable for over 6 hr in these solutions. SNP-polyP70 were made by covalently attaching polyP70 onto silica nanoparticles as previously described43. Synthetic PolyP was generated by solubilization from Maddrell salts and biotinylated as previously described7,50. Synthetic polyP has been previously characterized, including its chain length, counterions and clotting activity7,11. Long-chain NP-polyP contained a heterogeneous preparation of very long, non-biotinylated polyP polymers ranging from around 200 mers to 1300 mers, referred to here as NP-PolyP>1000. Some experiments with SI-polyP employed heterogeneous long-chain biotinylated polyP consisting of chains 50 to 400 units in length, referred to here as biotin-polyP400. Some experiments employed fractionated material of narrower sizes (polyP70 and polyP160)7. All polyP concentrations are stated with respect to the concentration of phosphate monomer.
Preparing microfluidic devices with SI-polyP
Microfluidic devices were prepared from polydimethylsiloxane (PDMS) as previously described53. Channel dimensions are listed as follows (length × width, 125 μm height for all channels): 1.67 mm × 1000 μm (1 s−1), 3.33 mm × 500 μm (3 s−1), 5.83 mm × 286 μm (10 s−1), 8.33 mm × 200 μm (22 s−1), 12.50 mm × 133 μm (55 s−1), 16.67 mm, 100 μm (110 s−1). The devices were incubated in saline and kept under vacuum overnight to hydrate and remove air from the channels. Devices remained soaked in saline throughout the experiment to aid in coating the surfaces with lipids, and to reduce convective flow during experiments in the absence of flow. The devices were coated with phosphatidylcholine (PC) vesicles to prevent activation of clotting on the PDMS surface. In devices that were not coated with SI-polyP, vesicles were prepared with egg PC (Avanti Polar Lipids, Alabaster, USA) and fluorescent Texas Red 1,2-Dihexadecanoyl-sn-Glycero-3-Phosphoethanolamine (DHPE) (Invitrogen) in a 99.5:0.5 molar ratio. Lipids were extruded through a 100 nm membrane using a Lipex Thermobarrel Extruder (Northern Lipids, Burnaby, Canada). The vesicle solution (10 mg/mL in dH2O) were flowed through microfluidic channels at a rate of 1 μl/min for 15 min and rinsed out with saline. The coating of PC on the channels was stable for at least 10 hours (Supplementary Fig. S4). For devices where polyP was surface-immobilized, the channel was first coated with biotinylated lipids (1 μl/min, 15 min) and rinsed out with saline. To prepare biotinylated vesicles, 1-oleoyl-2-[12[biotinyl(aminododecanoyl)]-sn-glycero-3-phosphocholine (biotinylated-PC, Avanti Polar Lipids) was mixed with Egg PC and Texas Red DHPE in a molar ratio of 5.0:94.5:0.5 and extruded. Next, streptavidin (100 μg/mL) conjugated to Alexa Fluor® 488 (Molecular Probes, Inc.) was flowed through the device (1 μl/min, 40 min) and then rinsed with saline to wash away the unbound, excess streptavidin. Finally, a solution of biotin-polyP (50 μg/mL) and biotinylated-polyethylene glycol (biotin-PEG) (either 0 or 99 molar equivalents to biotin-polyP) was flowed through the device (1 μl/min, 40 min), binding to the patterned streptavidin followed by a saline rinse, which resulted in SI-polyP being selectively patterned on the walls of the microfluidic device shear chambers. Liposomes and saline were flowed into the device through a combination of inlet and outlet channels to achieve laminar flow patterning, such that the parallel streams of fluids were at low Reynolds number (≪1) and maintained sharp boundaries and excluded the possibility of turbulent flow54,55. This patterning allowed specific channel walls of the device to be coated, either all channels in the device, the channels in the shear chambers, or one wall of the chambers. To measure the amount of polyP, it was stained by flowing DAPI (40 μg/mL in 15 mM Tris acetate, 300 mM NaCl, 30 mM EDTA, and 0.02% NaN3) into the device. Thrombin generation during clotting was detected by adding 125 μg/mL of fluorescent peptide substrate for thrombin (Boc-Val-Pro-Arg-4-methylcoumaryl-7-amide, Peptide Institute Inc.) into normal plasma.
Flowing plasma and calcium into devices and measuring clotting
Flow rates were controlled using a syringe pump (Harvard Apparatus PHD 2000) by withdrawing solutions out of the outlets of the device at a rate of 1 μl/min. Shear rates in different channels were controlled by the width of each channel, while the residence time of plasma within the shear chambers were kept constant (~10 sec) by varying their respective lengths. Tubing connected to the outlets of the device were charged with 50 μl of Egg PC vesicles to prevent clotting from initiating in the tubing or syringes. A solution of sodium citrate (10 mM in dH2O) was initially pulled into both inlet channels to wash out the device and further charge the outlet tubing. Normal citrated human plasma (7 mM citrate) and calcium-saline solution (40 mM CaCl2 and 90 mM NaCl) were simultaneously pulled into the device and mixed at a ratio of 3:1 to recalcify the plasma, yielding a final free calcium concentration of 4–5 mM56. To measure clotting times, fluorescent beads (2.5 μg/mL, Fluoresbrite Plain YG 1.0 Micron Microsphere, Polysciences Inc.), and in some experiments 125 μg/mL fluorescent thrombin substrate, were mixed into the plasma and time-course imaging of each channel was performed using an epifluorescence microscope (Leica DMI6000B). The fluorescent beads did not influence clotting times (Supplementary Fig. S5). Clotting was determined by the immobilization of the fluorescent beads and in some experiments also by the generation of blue fluorescence upon cleavage of the thrombin substrate. In experiments where the effect of nanoparticle polyP (NP-polyP) on clotting was tested, the activators were mixed with the calcium solution prior to entering the device. For experiments with soluble polyP (D-polyP), polyP was added to the plasma instead to prevent nanoparticle formation. For experiments at zero shear, normal or congenital FXII-deficient plasma (Geroge King Bio-Medical, Inc.) and calcium were mixed together immediately before flowing them into the device and blocking all outlets to create stagnant plasma.
Additional Information
How to cite this article: Yeon, J. H. et al. Localization of Short-Chain Polyphosphate Enhances its Ability to Clot Flowing Blood Plasma. Sci. Rep. 7, 42119; doi: 10.1038/srep42119 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Muller, F. et al. Platelet Polyphosphates Are Proinflammatory and Procoagulant Mediators In Vivo . Cell 139, 1143–1156, doi: 10.1016/j.cell.2009.11.001 (2009).
Renne, T., Schmaier, A. H., Nickel, K. F., Blomback, M. & Maas, C. In vivo roles of factor XII. Blood 120, 4296–4303, doi: 10.1182/blood-2012-07-292094 (2012).
Morrissey, J. H., Choi, S. H. & Smith, S. A. Polyphosphate: an ancient molecule that links platelets, coagulation, and inflammation. Blood 119, 5972–5979, doi: 10.1182/blood-2012-03-306605 (2012).
Smith, S. A. et al. Polyphosphate modulates blood coagulation and fibrinolysis. Proc. Natl. Acad. Sci. USA 103, 903–908, doi: 10.1073/pnas.0507195103 (2006).
Choi, S. H., Smith, S. A. & Morrissey, J. H. Polyphosphate is a cofactor for the activation of factor XI by thrombin. Blood 118, 6963–6970, doi: 10.1182/blood-2011-07-368811 (2011).
Puy, C. et al. Factor XII promotes blood coagulation independent of factor XI in the presence of long-chain polyphosphates. J. Thromb. Haemost. 11, 1341–1352, doi: 10.1111/jth.12295 (2013).
Smith, S. A. et al. Polyphosphate exerts differential effects on blood clotting, depending on polymer size. Blood 116, 4353–4359, doi: 10.1182/blood-2010-01-266791 (2010).
Kumble, K. D. & Kornberg, A. Inorganic Polyphosphate in Mammalian-Cells and Tissues. J Biol Chem 270, 5818–5822 (1995).
Moreno-Sanchez, D., Hernandez-Ruiz, L., Ruiz, F. A. & Docampo, R. Polyphosphate Is a Novel Pro-inflammatory Regulator of Mast Cells and Is Located in Acidocalcisomes. J Biol Chem 287, 28435–28444, doi: 10.1074/jbc.M112.385823 (2012).
Nickel, K. F. et al. The polyphosphate-factor XII pathway drives coagulation in prostate cancer-associated thrombosis. Blood 126, 1379–1389, doi: 10.1182/blood-2015-01-622811 (2015).
Donovan, A. J., Kalkowski, J., Smith, S. A., Morrissey, J. H. & Liu, Y. Size-Controlled Synthesis of Granular Polyphosphate Nanoparticles at Physiologic Salt Concentrations for Blood Clotting. Biomacromolecules 15, 3976–3984, doi: 10.1021/bm501046t (2014).
Engel, R., Brain, C. M., Paget, J., Lionikiene, A. S. & Mutch, N. J. Single-chain factor XII exhibits activity when complexed to polyphosphate. J. Thromb. Haemost. 12, 1513–1522, doi: 10.1111/jth.12663 (2014).
Zhu, S., Travers, R. J., Morrissey, J. H. & Diamond, S. L. FXIa and platelet polyphosphate as therapeutic targets during human blood clotting on collagen/tissue factor surfaces under flow. Blood 126, 1494–1502, doi: 10.1182/blood-2015-04-641472 (2015).
Kastrup, C. J., Runyon, M. K., Lucchetta, E. M., Price, J. M. & Ismagilov, R. F. Using chemistry and microfluidics to understand the spatial dynamics of complex biological networks. Accounts Chem. Res. 41, 549–558, doi: 10.1021/ar700174g (2008).
Szymusiak, M. et al. Colloidal Confinement of Polyphosphate on Gold Nanoparticles Robustly Activates the Contact Pathway of Blood Coagulation. Bioconjug Chem 27, 102–109, doi: 10.1021/acs.bioconjchem.5b00524 (2016).
Okorie, U. M., Denney, W. S., Chatterjee, M. S., Neeves, K. B. & Diamond, S. L. Determination of surface tissue factor thresholds that trigger coagulation at venous and arterial shear rates: amplification of 100 fM circulating tissue factor requires flow. Blood 111, 3507–3513, doi: 10.1182/blood-2007-08-106229 (2008).
Balandina, A. N. et al. Positive Feedback Loops for Factor V and Factor VII Activation Supply Sensitivity to Local Surface Tissue Factor Density During Blood Coagulation. Biophys. J. 101, 1816–1824, doi: 10.1016/j.bpj.2011.08.034 (2011).
Kastrup, C. J., Runyon, M. K., Shen, F. & Ismagilov, R. F. Modular chemical mechanism predicts spatiotemporal dynamics of initiation in the complex network of hemostasis. Proc. Natl. Acad. Sci. USA 103, 15747–15752, doi: 10.1073/pnas.0605560103 (2006).
Kastrup, C. J. et al. Spatial localization of bacteria controls coagulation of human blood by ‘quorum acting’. Nat. Chem. Biol. 4, 742–750, doi: 10.1038/nchembio.124 (2008).
Chatterjee, M. S., Denney, W. S., Jing, H. Y. & Diamond, S. L. Systems Biology of Coagulation Initiation: Kinetics of Thrombin Generation in Resting and Activated Human Blood. PLoS Comput. Biol. 6, 24, doi: 10.1371/journal.pcbi.1000950 (2010).
Rana, K. & Neeves, K. B. Blood flow and mass transfer regulation of coagulation. Blood Rev, doi: 10.1016/j.blre.2016.04.004 (2016).
Fogelson, A. L., Hussain, Y. H. & Leiderman, K. Blood Clot Formation under Flow: The Importance of Factor XI Depends Strongly on Platelet Count. Biophys. J. 102, 10–18, doi: 10.1016/j.bpj.2011.10.048 (2012).
Haynes, L. M., Dubief, Y. C., Orfeo, T. & Mann, K. G. Dilutional Control of Prothrombin Activation at Physiologically Relevant Shear Rates. Biophys. J. 100, 765–773, doi: 10.1016/j.bpj.2010.12.3720 (2011).
Runyon, M. K., Kastrup, C. J., Johnson-Kerner, B. L., Van Ha, T. G. & Ismagilov, R. F. Effects of shear rate on propagation of blood clotting determined using microfluidics and numerical simulations. J. Am. Chem. Soc. 130, 3458–3464, doi: 10.1021/ja076301r (2008).
Cheng, C. P., Herfkens, R. J. & Taylor, C. A. Inferior vena caval hemodynamics quantified in vivo at rest and during cycling exercise using magnetic resonance imaging. Am J Physiol-Heart C 284, H1161–H1167, doi: 10.1152/ajpheart.00641.2002 (2003).
Karino, T. & Motomiya, M. Flow through a Venous Valve and Its Implication for Thrombus Formation. Thromb Res 36, 245–257, doi: 10.1016/0049-3848(84)90224-X (1984).
Papaioannou, T. G. & Stefanadis, C. Vascular wall shear stress: basic principles and methods. Hellenic journal of cardiology: HJC=Hellenike kardiologike epitheorese 46, 9–15 (2005).
Hathcock, J. J. Flow effects on coagulation and thrombosis. Arterioscler Thromb Vasc Biol 26, 1729–1737, doi: 10.1161/01.ATV.0000229658.76797.30 (2006).
Goel, M. S. & Diamond, S. L. Adhesion of normal erythrocytes at depressed venous shear rates to activated neutrophils, activated platelets, and fibrin polymerized from plasma. Blood 100, 3797–3803, doi: 10.1182/blood-2002-03-0712 (2002).
Gallus, A. S. Travel, venous thromboembolism, and thrombophilia. Seminars in thrombosis and hemostasis 31, 90–96, doi: 10.1055/s-2005-863810 (2005).
Casa, L. D. C., Deaton, D. H. & Ku, D. N. Role of high shear rate in thrombosis. J. Vasc. Surg. 61, 1068–1080, doi: 10.1016/j.jvs.2014.12.050 (2015).
Aghourian, M. N., Lemarie, C. A. & Blostein, M. D. In vivo monitoring of venous thrombosis in mice. J. Thromb. Haemost. 10, 447–452, doi: 10.1111/j.1538-7836.2011.04615.x (2012).
Wilson, J. S., Virag, L., Di Achille, P., Karsaj, I. & Humphrey, J. D. Biochemomechanics of Intraluminal Thrombus in Abdominal Aortic Aneurysms. J. Biomech. Eng.-Trans. ASME 135, 14, doi: 10.1115/1.4023437 (2013).
Stavrou, E. & Schmaier, A. H. Factor XII: What does it contribute to our understanding of the physiology and pathophysiology of hemostasis & thrombosis. Thromb Res 125, 210–215, doi: 10.1016/j.thromres.2009.11.028 (2010).
Yau, J. W. et al. Selective depletion of factor XI or factor XII with antisense oligonucleotides attenuates catheter thrombosis in rabbits. Blood 123, 2102–2107, doi: 10.1182/blood-2013-12-540872 (2014).
Sakariassen, K. S., Orning, L. & Turitto, V. T. The impact of blood shear rate on arterial thrombus formation. Future Sci. OA 1 (2015).
Morrissey, J. H., Macik, B. G., Neuenschwander, P. F. & Comp, P. C. Quantitation of Activated Factor-Vii Levels in Plasma Using a Tissue Factor Mutant Selectively Deficient in Promoting Factor-Vii Activation. Blood 81, 734–744 (1993).
Riddell, A., Abdul-Kadir, R., Pollard, D., Tuddenham, E. & Gomez, K. Monitoring low dose recombinant factor VIIa therapy in patients with severe factor XI deficiency undergoing surgery. Thromb. Haemost. 106, 521–527, doi: 10.1160/Th10-12-0816 (2011).
Hendriks, H. G. D. et al. An effective treatment of severe intractable bleeding after valve repair by one single dose of activated recombinant factor VII. Anesth. Analg. 93, 287–289, doi: 10.1097/00000539-200108000-00009 (2001).
Mackman, N. New insights into the mechanisms of venous thrombosis. J. Clin. Invest. 122, 2331–2336, doi: 10.1172/jci60229 (2012).
Ruiz, F. A., Lea, C. R., Oldfield, E. & Docampo, R. Human platelet dense granules contain polyphosphate and are similar to acidocalcisomes of bacteria and unicellular eukaryotes. J Biol Chem 279, 44250–44257, doi: 10.1074/jbc.M406261200 (2004).
Smith, S. A. et al. Inhibition of polyphosphate as a novel strategy for preventing thrombosis and inflammation. Blood 120, 5103–5110, doi: 10.1182/blood-2012-07-444935 (2012).
Kudela, D. et al. Clotting Activity of Polyphosphate-Functionalized Silica Nanoparticles. Angew. Chem.-Int. Edit. 54, 4018–4022, doi: 10.1002/anie.201409639 (2015).
Colace, T. V., Tormoen, G. W., McCarty, O. J. & Diamond, S. L. Microfluidics and coagulation biology. Annu Rev Biomed Eng 15, 283–303, doi: 10.1146/annurev-bioeng-071812-152406 (2013).
Geddings, J. E. & Mackman, N. New players in haemostasis and thrombosis. Thromb. Haemost. 111, 570–574, doi: 10.1160/th13-10-0812 (2014).
von Bruhl, M. L. et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo . J Exp Med 209, 819–835, doi: 10.1084/jem.20112322 (2012).
Becattini, C. & Agnelli, G. Aspirin for prevention and treatment of venous thromboembolism. Blood Rev 28, 103–108, doi: 10.1016/j.blre.2014.03.003 (2014).
Mekaj, Y. H., Daci, F. T. & Mekaj, A. Y. New insights into the mechanisms of action of aspirin and its use in the prevention and treatment of arterial and venous thromboembolism. Ther Clin Risk Manag 11, doi: 10.2147/Tcrm.S92222 (2015).
Collaborative Overview of Randomized Trials of Antiplatelet Therapy. 3. Reduction in Venous Thrombosis and Pulmonary-Embolism by Antiplatelet Prophylaxis among Surgical and Medical Patients. Brit Med J 308, 235–246 (1994).
Choi, S. H. et al. Phosphoramidate End Labeling of Inorganic Polyphosphates: Facile Manipulation of Polyphosphate for Investigating and Modulating Its Biological Activities. Biochemistry 49, 9935–9941, doi: 10.1021/bi1014437 (2010).
Jensen, T. E., Baxter, M., Rachlin, J. W. & Jani, V. Uptake of Heavy-Metals by Plectonema-Boryanum (Cyanophyceae) into Cellular-Components, Especially Polyphosphate Bodies - an X-Ray-Energy Dispersive Study. Environ Pollut A 27, 119–127, doi: 10.1016/0143-1471(82)90104-0 (1982).
Yamaguchi, T., Hasegawa, M., Niimi, R. & Sudo, A. Incidence and time course of asymptomatic deep vein thrombosis with fondaparinux in patients undergoing total joint arthroplasty. Thromb Res 126, E323–E326, doi: 10.1016/j.thromres.2010.03.018 (2010).
Whitesides, G. M., Ostuni, E., Takayama, S., Jiang, X. Y. & Ingber, D. E. Soft lithography in biology and biochemistry. Annu. Rev. Biomed. Eng. 3, 335–373, doi: 10.1146/annurev.bioeng.3.1.335 (2001).
Kenis, P. J. A., Ismagilov, R. F. & Whitesides, G. M. Microfabrication inside capillaries using multiphase laminar flow patterning. Science 285, 83–85, doi: 10.1126/science.285.5424.83 (1999).
Colace, T. V., Tormoen, G. W., McCarty, O. J. T. & Diamond, S. L. Microfluidics and Coagulation Biology. Annu Rev Biomed Eng 15, 283–303, doi: 10.1146/annurev-bioeng-071812-152406 (2013).
Mann, K. G., Whelihan, M. F., Butenas, S. & Orfeo, T. Citrate anticoagulation and the dynamics of thrombin generation. J. Thromb. Haemost. 5, 2055–2061, doi: 10.1111/j.1538-7836.2007.02710.x (2007).
Acknowledgements
This work was supported by Award WQ81XWH-11-2-0021 from the U.S. Army Medical Research and Material Command. The U.S. Army Medical Research Acquisition Activity, 820 Chandler Street, Fort Detrick, MD 21702-5014 is the awarding and administering acquisition office. The contents of this article do not necessarily reflect the position or the policy of the Government, and no official endorsement should be inferred. Aspects of the work were also funded by the by Natural Sciences and Engineering Research Council of Canada (418652–2012) the Canadian Institutes of Health Research (MOP-119426 and MSH-130166), Canadian Foundation for Innovation (31928) and the British Columbia Knowledge Development Fund. We thank Rustem F. Ismagilov for his helpful suggestions in conceptualizing and planning of experiments.
Author information
Authors and Affiliations
Contributions
J.H.Y and N.M. contributed equally to this work. J.H.Y., N.M., T.S.S., K.Y.T.C., J.R.B., S.A.S., A.J.D., and D.K., performed experiments and analyzed data; J.H.Y., N.M., T.S.S., C.J.K., J.H.M., Y.L., and G.D.S. conceptualized and planned experiments; J.H.Y., N.M., and C.J.K. wrote the manuscript; and all authors reviewed and edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
S.A.S., D.K., G.D.S., and J.H.M. are co-inventors on pending patent applications covering potential medical uses of SNP-polyP. A.J.D. and Y.L. are co-inventors on a pending patent application related to the therapeutic usage and delivery of NP-polyP. D.K., G.D.S., and J.H.M. declare competing financial interests in this work. The remaining authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Yeon, J., Mazinani, N., Schlappi, T. et al. Localization of Short-Chain Polyphosphate Enhances its Ability to Clot Flowing Blood Plasma. Sci Rep 7, 42119 (2017). https://doi.org/10.1038/srep42119
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep42119
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
