
Abstract
Inherited retinal dystrophies (IRD) comprise a wide group of clinically and genetically complex diseases that progressively affect the retina. Over recent years, the development of next-generation sequencing (NGS) methods has transformed our ability to diagnose heterogeneous diseases. In this work, we have evaluated the implementation of whole exome sequencing (WES) for the molecular diagnosis of IRD. Using Ion ProtonTM system, we simultaneously analyzed 212 genes that are responsible for more than 25 syndromic and non-syndromic IRD. This approach was used to evaluate 59 unrelated families, with the pathogenic variant(s) successfully identified in 71.18% of cases. Interestingly, the mutation detection rate varied substantially depending on the IRD subtype. Overall, we found 63 different mutations (21 novel) in 29 distinct genes, and performed in vivo functional studies to determine the deleterious impact of variants identified in MERTK, CDH23, and RPGRIP1. In addition, we provide evidences that support CDHR1 as a gene responsible for autosomal recessive retinitis pigmentosa with early macular affectation, and present data regarding the disease mechanism of this gene. Altogether, these results demonstrate that targeted WES of all IRD genes is a reliable, hypothesis-free approach, and a cost- and time-effective strategy for the routine genetic diagnosis of retinal dystrophies.
Similar content being viewed by others


Introduction
Inherited retinal dystrophies (IRD) are a highly heterogeneous group of diseases characterized by the degeneration of photoreceptors and retinal pigment epithelium (RPE) cells. They affect approximately 1 in 3,000 people, and represent the major cause of incurable familial blindness, with more than 2 million people affected worldwide1. In general, IRD are classified according to the type of cells in the retina that are primarily affected —cones or rods—, the age of onset of the first symptoms, and the progression of degeneration over the years2. Accordingly, the spectrum of IRD comprises diseases that predominantly affect (i) the central retina, such as Stargardt disease (STGD), cone dystrophy (CD), cone–rod dystrophy (CRD), and achromatopsia (ACHR); (ii) the peripheral retina, such as retinitis pigmentosa (RP); or (iii) both, such as Leber congenital amaurosis (LCA). However, some patients do not display specific clinical representations associated with a particular IRD, but exhibit overlapping phenotypes that are consistent with more than one particular dystrophy. Moreover, intrafamilial variability and incomplete penetrance are not uncommon, so assigning a definitive and precise clinical diagnosis can be difficult. In most cases, the retina is the only affected tissue (non-syndromic forms), while in some patients, other tissues can also be involved (syndromic forms)3. The latter includes Usher syndrome (US), Bardet-Biedl syndrome (BBS), and Joubert syndrome (JS), among others. In some families, discrimination between a non-syndromic or syndromic IRD is not always simple, as the existence of affected tissues other than the retina can be coincidental4. In this regard, genetic analysis becomes essential, as it is the only current tool that has the potential to provide a reliable and conclusive diagnosis. Nevertheless, providing a molecular diagnosis for these pathologies can be challenging due to the large number of candidate genes (>200) and mutations (>4,000) described, the absence of major genes or mutations, multiple inheritance patterns (autosomal dominant, autosomal recessive, and X-linked), the possible existence of modifier alleles, and the presence of non-causative variants5.
Previously, conventional genetic diagnosis of IRD patients was based on the use of arrayed primer extension reactions (APEX) and Sanger sequencing method, which allow for the examination of known mutations or specific exons and gene targets6. However, these techniques guarantee the identification of the causative mutations in only 10–20% of cases7. More recently, next-generation sequencing (NGS) technologies have revolutionized genetic diagnosis of IRD. These methods facilitate the simultaneous screening of a large number of genes, are at least 1,000 times faster than conventional sequencing, and are much less expensive per sequence8. Within this context, some authors have opted to develop their own disease-specific gene panels that include a list of genes responsible for one particular IRD subtype9,10,11,12,13,14, but they can only be applied when a clinical interpretation can be made with relative assurance15.
Different studies suggest that 35% of IRD cases remain unsolved at the genetic level2; however, it is thought that the most common IRD genes have already been identified. Therefore, undiagnosed patients may carry mutations either in syndromic genes (usually not analyzed in non-syndromic patients), in genes associated with other IRD subtypes (not included in disease-specific panels), or in as-yet-unknown IRD genes4,16. This fact suggests that the best approach for the genetic diagnosis of these pathologies may be the simultaneous analysis of known genes responsible for both syndromic and non-syndromic forms of all IRD types. This could be through targeted/custom non-disease-specific panels or through more broad-based NGS strategies such as whole exome sequencing (WES) or whole genome sequencing (WGS). Within this context, new associations between already-known IRD genes and particular phenotypes would be established. Indeed, during the last few years, many IRD genes have been reclassified using these approaches12,17,18,19,20,21. In the present study, we used WES to examine 212 genes responsible for more than 25 syndromic and non-syndromic retinal dystrophies in a cohort of 59 unrelated families affected by a wide range of IRD phenotypes.
Results
Clinical diagnosis of IRD patients
Almost 89% of the cases included in our cohort had a well-defined clinical diagnosis (52/59). Most of the phenotypes were non-syndromic (53/59), with RP being the most common of the IRD (26 isolated cases and six US cases), followed by CD and STGD (six and five cases, respectively) (Table 1). In seven cases (11.86%), a specific clinical diagnosis could not be assigned, as the displayed phenotype was compatible with two, or even three, different retinal dystrophies (unclear phenotype).
WES approach for the genetic diagnosis of IRD patients
Ion AmpliseqTM Exome technology and Ion ProtonTM platform allowed for the capturing, amplification and sequencing of more than 97% of the coding regions of >19,000 genes. The generated data was filtered to obtain coverage information and variants of 212 genes included in our IRD panel (the full list of genes is given in Supplementary Table S1). In particular, 5,294 amplicons covered 98.4% of the coding regions and flanking exon/intron boundaries (around 50 bp) of the IRD genes. After the analysis, 96.9% of target regions had a coverage of >20x, 90% had >40x, 56.4% had >100x, and 12.8% had >200x. Only 3% of the amplicons had coverage of <20x (Supplementary Fig. S1).
On average, depth coverage of IRD genes was 121x, with values that ranged from 52x (CHM) to 217x (NRL). Most of the genes (73.5%, 147/212) displayed an average depth of >100x. A schematic representation of the percentage of nucleotides covered at different depth ranges, as well as the mean value for each gene, is shown in Fig. 1.

The percentage of nucleotides with 0x, 1x–14x, 15x–34x, or ≥35x depth coverage per gene is shown. Black lines represent the average depth in each case.
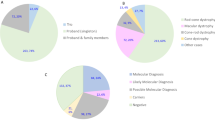
For each sample, a mean of 1,050 variants were identified in the 212 genes, including single nucleotide variants and small indels. After discarding common polymorphisms (MAF > 0.01), putative false-positives (depth < 15x), and non-deleterious variants (synonymous substitutions, UTR variants), a mean of 14 variants remained, for which we used different prediction tools to evaluate their putative pathogenic impact. In addition, cosegregation studies were performed when relatives’ samples were available. In the present study, a total of 63 different causative mutations were identified in 29 distinct genes (Table 2). ABCA4, USH2A, and PDE6A were the most frequent causative genes in our cohort (the frequency distribution of each gene is shown in Fig. 2a). Of the 63 mutations, 21 were identified for the first time (33.3%). A deleterious function of new missense variants was attributed when at least three of the four prediction programs assigned a damaging or disease-causing effect (see Supplementary Table S2). In family Fi15/09, the missense variant c.3G > A affected the first methionine of PROM1; therefore, it was assumed to be deleterious, regardless of the bioinformatic predictions. The pathogenic effect of novel variants located in introns was estimated using five splice site predictors. Only those variants estimated to weak canonical splice sites, or those that create new donor or acceptor sites, were considered pathogenic (Supplementary Table S3). The identified mutations were mainly missense (43%), followed by small deletions (24%), nonsense (17%), splicing (9%), small insertions (5%), and gross deletions (2%) (Fig. 2b).

(a) Distribution and frequencies of IRD genes. ABCA4, USH2A and PDE6A were the most prevalent genes. (b) Types of mutations identified and their frequencies. (c) Comparison of the mutation detection rate of different IRD subtypes. Abbreviations: ACHR, achromatopsia; CD, cone dystrophy; CRD, cone–rod dystrophy; GA, gyrate atrophy; LCA, Leber congenital amaurosis; RP, retinitis pigmentosa; STGD, Stargardt disease; US, Usher syndrome.
The causative mutation (or mutations) was identified in 71.18% of the families (42/59). The mutation detection rate varied considerably depending on the retinal dystrophy subtype. In some diseases, such US and LCA, it reached 100%; however, in CD and CRD, it did not exceed 35% (Fig. 2c). Regarding RP, pathogenic mutations were identified in 18/26 cases (69.2%). Interestingly, phenotypic unclear cases reached a mutation detection rate of 71.4% (5/7). Finally, the genetic diagnosis was inconclusive in 17 families, representing 28.81% of the cohort, although in four of these, we identified one putative pathogenic variant in a recessive gene (see Supplementary Table S4).
CDHR1 as a candidate for retinitis pigmentosa with early macular affectation
Spanish family Fi15/19 contained three affected members from two different branches (two siblings, III:1 and III:2, and their cousin, III:4) who developed progressive nyctalopia and a reduction in peripheral visual field during the second decade of life (Fig. 3a). Patient III:4 presented a best-corrected visual acuity (BCVA) of 20/32 and 20/40 at the ages of 25 and 35 years, respectively. BCVA decreased rapidly between the fourth and fifth decade of life, progressing to light perception at the age of 45. Fundus examination of the affected members of the family revealed typical signs of RP, including marked attenuation of the retinal blood vessels, waxy pallor of the optic discs, and pigmented bone spicules in the periphery (Fig. 3b). Retinal autofluorescence showed diffuse RPE disturbances, both in the periphery and the macula. The macula showed a mottled hypoautofluorescence, similar to that seen in the very early stages of CRD, but without the characteristic RPE atrophy seen in the later stages (Fig. 3c). Electroretinography (ERG) showed abolished responses in scotopic conditions. The a-wave was also undetectable in photopic ERG, though there was a clearly reduced, but detectable, b-wave (Fig. 3d). The clinical and electrophysiological findings showed some overlapping traits between CRD and RP. However, the initial symptoms were clearly dominated by nyctalopia and peripheral visual field loss, while central vision decreased later, although to a much greater extent than found for most cases of RP. Altogether, these findings support a diagnosis of RP with early macular degeneration for this particular family.

(a) Cosegregation analysis of CDHR1 variants identified in family Fi15/19. (b and c) Fundus eye photographs and autofluorescence images of the affected member III:2. (d) Electroretinographic recordings under scotopic (0 dB) and photopic (0 dB) conditions from both eyes of patient III:4. A b-wave could be detected in photopic ERG (amplitude and latency values are shown). Photopic 30-Hz flicker (0 dB) was also recorded. (e) RT-PCR analysis of CDHR1 blood mRNA of affected patients (III:1 and III:2), carriers (III:3, III:6, III:8, and IV:1), and one control individual (WT). Patients and carriers showed a dramatic decrease in the CDHR1 normal transcript. III:1, III:2, and III:3, who carry c.1554-2A > C, also produced a lower mass band corresponding to an mRNA that skips exon 15, whereas members III:6, III:8 and IV:1, carriers of c.1485 + 2T > C, presented a 769 bp band that directly links exon 12 and 14 of the gene.
Genetic analysis using the IRD panel showed that patient III:4 carried a frameshift mutation (c.1868_1869insA, p.Asn623Lysfs*53) in compound heterozygosity with a splicing variant (c.1485 + 2T > C) in the CDHR1 gene. The first variant was considered to be new as it was not found in any public or private database, whereas the splicing variant was previously described in patients affected with a diffuse retinal dystrophy involving both cones and rods22. Cosegregation analysis of family Fi15/19 revealed that the variants came from different alleles, that no healthy sibling carried both variants, and that affected members III:1 and III:2 only carried the c.1868_1869insA variant. At this point, all coding regions of CDHR1 were analyzed by Sanger sequencing in patient III:2, finding a previously unknown putative pathogenic splicing variant, c.1554-2A > C (Fig. 3a). All the splicing predictors defined this new variant as clearly pathogenic (Supplementary Table S3).
In order to confirm the pathogenicity of these three CDHR1 variants in vivo, RT-PCR analysis was performed on RNA blood samples from two affected members (III:1 and III:2), four carriers (III:3, III:6, III:8, and IV:1), and one non-related control individual (wild-type [WT]). The results revealed that affected patients and most carriers presented lower levels of the CDHR1 canonical isoform compared with the WT (934 bp band in Fig. 3e). Interestingly, family members III:1, III:2 and III:3 (carriers of the c.1554-2A > C variant) also showed a 705 bp band that skipped exon 15 of the gene. This generated transcript created several premature termination codons (PTC) and apparently did not undergo nonsense-mediated mRNA decay (NMD), maybe due to the position of the first PTC, located in the second-to-last exon. On the other hand, members III:6, III:8 and IV:1 (carriers of the c.1485 + 2T > C variant) produced a 769 bp band, which directly linked exons 12 and 14, and generated an in-frame transcript that likely translates to a CDHR1 protein that lacks 55 amino acids of two cadherin domains. Concerning the frameshift mutation (c.1868_1869insA), it was detected at the cDNA level of the carriers, suggesting that this variant also avoids NMD.
Novel functional studies of IRD mutations
Retinitis pigmentosa
Pedigree Fi15/20 is a Spanish consanguineous family with two siblings severely affected by RP. The analysis of the IRD panel in patient III:2 revealed a novel homozygous missense mutation in MERTK, c.1961G > T (p.Gly654Val). This variant cosegregated with the disease (Fig. 4a–i) and was predicted as deleterious by different missense prediction algorithms (Supplementary Table S2). Interestingly, the variant affected the first nucleotide of exon 15 (a highly conserved guanine), which highlighted a putative effect on the splicing mechanism. In accordance with the splicing predictors, this variant abolishes the recognition of the intron 14 acceptor site (Supplementary Table S3). To assess whether the c.1961G > T mutation resulted in an altered splicing pattern, we performed a comparative RT-PCR analysis of MERTK in RNA from white blood cells of the affected siblings (III:2 and III:4), one carrier (IV:1), and one non-related control individual (WT), using primers located within exons 11 and 18 of the gene (see Supplementary Table S5 for primer sequences). The WT sample produced a single 857 bp band, as was expected from analysis of a correctly spliced transcript, whereas both patients and the carrier showed not only the WT band, but also another lower band of 738 bp, which lacked exon 15 and directly fused exons 14 and 16 (Fig. 4a–ii). This aberrant transcript contained PTC and was found abundantly in the patients, indicating that it probably avoids the NMD degradation mechanism. The level of WT transcript was quantified in each sample using real-time RT-PCR, showing that the carrier individual produced around 38% WT transcript, whereas the patients only expressed 5–10%, compared with the control sample (Fig. 4a–iii). However, it is worth mentioning that this small amount of correct mRNA still carried the missense mutation, which is predicted to be deleterious.

(a-i) Pedigree of consanguineous family Fi15/20, showing the cosegregation analysis of variant c.1961G > T of MERTK. (a-ii) Electrophoresis gel of the RT-PCR products obtained from affected patients (III:2 and III:4), carrier (IV:1), and WT blood samples. Patients produced high levels of an aberrantly spliced transcript, whereas the WT produced only the expected 857 bp band. (a-iii) Quantitative analysis of MERTK levels by real-time RT-PCR. The WT sample was set at 100%. (b-i) Cosegregation analysis of the two new CDH23 variants identified in family Fi15/36. (b-ii) RT-PCR assay using blood samples from the affected patient (III:2) and an unrelated WT individual. Two different bands were obtained in the patient, the expected one and another skipping exon 51. (b-iii) Chromatogram of the 2,566 bp band obtained from cDNA of patient III:2. (c-i) Fi15/12 family pedigree and RPGRIP1 variants cosegregation. (c-ii) RT-PCR analysis revealed intrafamilial differences in the expression of RPGRIP1 gene. (c-iii) Quantification of RPGRIP1 canonical transcript by real-time RT-PCR. The noncarrier family member III:3 was used as a control, and her sample was set at 100%.
Usher syndrome
The analysis of individual III:2, of the Spanish family Fi15/36, who was diagnosed with US, identified two novel mutations in CDH23: a deletion of a single nucleotide that creates a frameshift mutation (c.5546del, p.Pro1849Leufs*4), and a splicing variant (c.7482 + 1G > A) (Fig. 4b–i). In order to assess the effects of the two variants on CDH23 expression, RT-PCR analysis of blood samples from the patient and a non-related WT individual was performed using primers located in exons 39 and 54. In contrast with the WT sample, two different transcripts were obtained from the affected patient: one corresponding to the WT band (2,566 bp), and another (2,446 bp) skipping exon 51 (Fig. 4b–ii). This finding suggests that the c.7482 + 1G > A splicing variant weakens the canonical donor splice site of exon/intron 51. The direct link between exons 50 and 52 generated an in-frame transcript that likely produces a CDH23 protein that lacks 40 amino acids of a highly conserved sequence of the extracellular domain. Concerning the c.5546del frameshift mutation, Sanger sequencing of the obtained WT band from the patient’s cDNA suggested that the aberrant transcript did not undergo NMD degradation, as the chromatogram showed double peaks starting from the variant position (Fig. 4b–iii).
Leber congenital amaurosis
Family Fi15/12, which contained one male affected with LCA, was previously genetically analyzed by de Castro-Miro et al. using a cosegregation chip based on SNP genotyping, followed by Sanger sequencing of the candidate genes23. This analysis identified two new variants in RPGRIP1, a deletion of two nucleotides that created a frameshift mutation (c.895_896del, p. Glu299Serfs*21), and an intronic variant with unknown pathogenic effect (c.2367 + 23del) (Fig. 4c–i). However, the genetic diagnosis was finally inconclusive, as the pathogenic impact of the intronic variant could not be proved. At this point, with consideration of the family’s interest in obtaining a reliable molecular diagnosis, the affected patient was analyzed using the IRD panel. Nevertheless, no clearly pathogenic mutations were detected other than the two RPGRIP1 variants. Within this context, we aimed to evaluate the pathogenic impact of the c.2367 + 23del variant, which did not show any substantial pathogenic impact according to the in silico predictions (see Supplementary Table S3). The RPGRIP1 gene displays high transcriptional complexity, with two different promoters (the canonical one and another internal promoter located before exon 10) and several alternative exons. Previous studies in animal models have revealed that many of the RPGRIP1 isoforms are likely to have tissue-specific expression, and that both promoters are active in the retina, whereas only the one with the transcription start site in exon 10 is active in other tissues such as the lung, heart, or testis24,25. Thus, we first assessed the expression of RPGRIP1 isoforms in human blood by using several pairs of primers, with cDNA amplification obtained when using primers located after exon 10, suggesting that the internal promoter is the only one active in this tissue. Taking into account these results, RT-PCR analysis using primers located at exons 14 and 17 was performed in the affected patient (III:2), his carrier parents (II:3 and II:4), the carrier brother (III:1) and the noncarrier sister (III:3). (see Supplementary Table S5 for primer sequences). All family members expressed two different bands, the WT band (665 bp) and a lower one (170 bp) that lacked exons 15 and 16 (Fig. 4c–ii). Intrafamilial variable expression of these two transcripts was observed. At this point, we aimed to quantify by real-time RT-PCR the amount of WT transcript in each case, using a Taqman probe located between exons 14 and 15 (Fig. 4c–iii). Interestingly, those family members who carried the intronic variant exhibited reduced expression of the WT isoform ranging from 37–75%. This suggests that the c.2367 + 23del variant strongly impairs the recognition of RPGRIP1 canonical splice sites, favoring the production of the transcript that skips exons 15 and 16. The latter would likely generate an in-frame sequence that lacks 492 nucleotides (164 amino acids) that code for the C2 protein domain, which is responsible for RPGRIP1 interaction with other proteins in the ciliary transition zone. In this regard, several RPGRIP1 deleterious mutations have been shown to disrupt this interaction26,27.
Discussion
Some authors claim that custom targeted-NGS of specific panels is the best strategy for genetic screening of IRD, and that WES is useful for uncovering new candidate genes involved in these diseases only when known genes have already been ruled out28. In this report, we propose that WES is an effective tool not only for the identification of new genes, but also for routine IRD molecular diagnosis. Within this context, we used Ion ProtonTM system to sequence coding regions of > 19,000 genes and specifically analyze a panel of 212 IRD candidates in a cohort of 59 genetically and clinically unselected families. The pathogenic variants were detected in 71.18% of cases (42/59), reaching a mutation detection rate higher than most previous studies, where either custom targeted-NGS or WES was used (Table 3)9,10,11,13,19,29,30,31,32,33,34,35,36,37,38,39. The improvement displayed in our study may be explained by a number of different factors: i) more accurate clinical characterization, which assures that all patients included in the cohort show features compatible with retinal dystrophies caused by a genetic alteration; ii) superior gene panel design, ensuring inclusion of all potential candidates; iii) a more optimal data analysis pipeline (for example, 15x as the acceptable coverage threshold); and iv) better coverage of the genes of interest, especially those that contain mutational hotspots or prevalent variants.
WES offers several advantages compared to targeted-NGS, especially in genetic heterogeneous diseases such as IRD, for which novel disease genes are continually being discovered. In this regard, targeted-NGS is limited in its flexibility for including new disease candidates, whereas the WES strategy allows for rapid enlargement of the panel. Moreover, for unsolved cases, once all known genes have been ruled out by WES, the same generated data can be used for the identification of new candidates located throughout the entire exome. However, it is worth mentioning that targeted-NGS retains some advantages as it can achieve a higher depth of coverage in the regions of interest than that possible with WES37. In our case, the mean depth of the genes included in the panel was 121x, whereas in most targeted-NGS studies, this value ranged between 250x and 1,330x (Table 3)9,10,13,19,31,36,37. A greater depth of coverage not only reduces the detection of false-positives, but also allows for the analysis of copy number variations (CNV)4,6. However, in complex sequences such as exon ORF15 of RPGR, custom targeted-NGS does not achieve better coverage than that possible with WES. Our strategy allowed for the analysis of 76% of the whole RPGR gene and 41% of exon ORF15, with these values being similar to previous target-NGS studies9. Within this context, we were able to identify by WES a frameshift mutation in exon ORF15 in one patient of the cohort (Fi15/41).
In our study, CNV detection was not possible due to the average depth obtained; thus, we assume that gross rearrangements may be responsible for a proportion of the genetically undiagnosed patients (28.82%). Additionally, the failure to identify mutations in these patients may be due to the causative variants being located in coding regions poorly covered by WES, in non-coding regions or regulatory sequences, or in novel genes that have not yet been associated with IRD. Interestingly, the percentage of unsolved patients was significantly higher in CD and CRD in comparison to other conditions (see Fig. 2c), suggesting that these dystrophies have been studied to a lesser extent, and that more novel genes are yet to be discovered for these particular phenotypes.
The phenotypic overlap of retinal dystrophies greatly diminishes the ability to arrive at an accurate clinical diagnosis. In fact, some authors estimate that IRD patients visit an average of seven ophthalmologists before the final diagnosis is made40. In our cohort, a significant group of patients (11.86%) showed clinical features that were compatible with different retinal dystrophies, prompting us to design a non-disease-specific panel that encompassed all syndromic and non-syndromic IRD genes. Once the analysis was performed, the pathogenic mutation was successfully identified in almost 72% of the families with an unclear phenotype, providing evidence that our strategy is effective not only for patients with a precise and particular clinical diagnosis, but also for cases with uncertain phenotypic features. Moreover, this non-hypothesis-driven approach allowed us to propose the already known IRD gene CDHR1 as a good RP candidate. This gene was previously described in families mainly affected by CRD41,42,43, or by a retinal dystrophy that involves both rods and cones at the same time22,44. In our study, family Fi15/19 was found to carry mutations in CDHR1, and displayed the main clinical traits corresponding to autosomal recessive RP, with a remarkable premature degeneration of cones. These particular traits are similar to those observed in some patients that carry mutations in PROM1, a gene also involved in RP and CRD45. In fact, PROM1 and CDHR1 proteins co-localize at the base of the developing outer segment of photoreceptors, with both participating in disc morphogenesis46. This suggests that their alterations could lead to similar clinical phenotypes. Here, we propose CDHR1 as a good candidate in molecular genetic studies of patients showing either CRD or RP symptoms, and add two new mutations to the molecular spectrum of this gene. Previously, only seven different CDHR1 variants have been described, most of them resulting in premature stop codons or splicing alterations that should lead to NMD. In this work, we report for the first time in vivo functional assays for CDHR1 mutations that demonstrate that pathogenic mechanisms other than NMD may be responsible for the deleterious effect of truncating/splicing variants.
In addition to basic molecular genotyping, in certain cases, functional studies are needed in order to provide a conclusive genetic diagnosis. This was the case for family Fi15/12, who carry an intronic deletion in RPGRIP1 (c.2367 + 23del) that, according to in silico prediction programs, has a slight effect on splicing. However, here we provide in vivo functional evidence that this variant has a clear impact on the RPGRIP1 splicing mechanisms. Similarly, a recent study demonstrated the pathogenic effect of a prevalent ABCA4 intronic mutation, c.5461-10T > C, which was also predicted to be neutral by bioinformatic tools47. Together, these studies highlight the importance of functional analysis, with splicing predictors being frequently difficult to interpret and sometimes unreliable, especially for variants located outside of the intronic canonical signals. In addition, in vivo studies of mutations present in heterogeneous diseases, such as those investigated in the present analysis, are also useful for the establishment of genotype–phenotype correlations. This is the case for CDH23, where previous work has shown that truncated peptides or loss of numerous amino acid residues in the CDH23 protein results in US, whereas missense mutations cause non-syndromic deafness48,49. Such evidence is in accordance with the in vivo results that we obtained from the analysis of family Fi15/36, who is affected by US, and carries two new mutations altering the open reading frame of CDH23.
In conclusion, we analyzed a large cohort of clinically heterogeneous IRD patients using WES, and report a diagnostic yield greater than 70%. Altogether, our results indicate that WES using Ion ProtonTM system is a valuable strategy for application to IRD.
Methods
Patients
A cohort of 59 clinically and genetically unselected IRD families was included in the present study. None of the families was previously screened for gene mutations, except pedigree Fi15/12 that presented previous genetic data23, which was inconclusive and therefore not considered in this work. Most patients originated from Spain (n = 46), while the others were from the Arabian Peninsula (n = 11), Venezuela (n = 1), and Costa Rica (n = 1). Clinical diagnoses were established at the Institut de Microcirurgia Ocular (Barcelona, Spain) and were based on standard ophthalmic evaluations (best corrected visual acuity, retinography, fundus autofluorescence, optical coherence tomography, electroretinography, and visual field). Peripheral blood (in EDTA tubes) or saliva samples were obtained from patients and their relatives. Automated extraction of genomic DNA was performed by using the KingFisher Duo purification system (Thermo Fisher Scientific, Waltham, MA). All procedures used in this study were in accordance with the Declaration of Helsinki. Ethics approval was received from the Ethics Committee of Institut de Microcirurgia Ocular (160321_96). All patients and their relatives were fully informed of the purpose and procedures of this study, and written consent was obtained from each individual.
Gene panel design, whole exome sequencing and data processing
A total of 212 genes previously associated with inherited retinal disorders were included in our IRD panel. Genes were selected according to the information available in RetNet (https://sph.uth.edu/retnet/) and Pubmed databases (http://www.ncbi.nlm.nih.gov/pubmed/). These genes were known to be responsible for 13 non-syndromic IRD forms (CD, CRD, LCA, RP, ACHR, STGD, congenital stationary night blindness, gyrate atrophy, familial exudative vitreoretinopathy, choroideremia, Sorsby’s dystrophy, Norrie disease, and retinoschisis) and more than 10 syndromic diseases (US, BBS, JS, Alström syndrome, Senior–Løken syndrome, Stickler syndrome, Alport syndrome, Alagille syndrome, Wagner disease, and oculoauricular syndrome, among others).
WES was performed in patients from 59 unrelated families using libraries designed and constructed using the Ion AmpliSeqTM Exome technology (ThermoFisher Scientific). Generated amplicons were genotyped with the Ion ProtonTM platform (Life Technologies), following the manufacturer’s instructions. Sequences were aligned against the reference genome (GRCh37/hg19) by using TMAP Alignment (Thermo Fisher Scientific). WES was performed in collaboration with a private company (NIMGenetics, Madrid, Spain), which provided the BAM, BAI, and FASTQ files, as well as VCF and TSV files that contained a compilation of all of the variants detected using the Ion Reporter software (Thermo Fisher Scientific). Moreover, the company supplied a TSV file that specifically included the variants identified in the 212 genes (after running a custom pipeline), and a document that detailed the coverage data of the panel, specifying the number of reads of each amplicon. The coverage information obtained from the 59 samples was used to measure the average depth and coverage percentage of each gene.
Determination of pathogenic variants
Variants that were detected in genes included in the IRD panel were filtered according to coverage (≥15x), minor allele frequency (≤0.01), and deleterious potential. All resulting variants were contrasted with the mutation databases, HGMD (http://www.hgmd.cf.ac.uk/ac/index.php) and Uniprot (http://www.uniprot.org/). The pathogenicity of missense changes was evaluated using the following in silico predictors: SIFT, MutationTaster, PolyPhen-2, and Align GV-GD. When new variants potentially affected the splicing mechanism, the splice site score values of the wild-type and the mutated sequence were predicted online using SpliceSite, MaxEntScan, NNSPLICE, GeneSplicer and Human Splicing Finder. Moreover, nucleotide conservation was evaluated using the PhastCons and PhyloP programs.
Sanger sequencing was performed to confirm all of the putative pathogenic variants obtained after WES genotyping, and mutation segregation analysis was carried out when relatives’ samples were available.
In partially solved recessive cases, with only one pathogenic variant detected in an IRD gene, the coverage data of this gene was carefully evaluated, and those regions poorly covered (<15x) were Sanger sequenced. If the second pathogenic allele was not found, and gross deletion or duplication rearrangements were previously described in the gene of interest, a copy number variation analysis was also carried out. In particular, in family Fi15/40, a deletion/duplication analysis for GPR98 was performed using a custom designed gene centric microarray (Baylor Miraca, Houston, TX). In family Fi15/44 Supplementary Table S4, multiplex ligation-dependent probe amplification (MLPA) analysis was used to evaluate the presence of rearrangements of USH2A (SALSA MLPA probemixes P361 and P362).
RNA expression
Total RNA from particular patients and their relatives was obtained from 500 μl of blood stabilized with 1.3 ml of RNAlater by using the RiboPure-Blood purification kit (Thermo Fisher Scientific), according to the manufacturer’s instructions. First, cDNA chains were obtained by reverse transcription (RT) using the Transcriptor High Fidelity cDNA Synthesis Kit (Roche Diagnostics, Indianapolis, IN). Specific amplification of transcripts was obtained for MERTK, RPGRIP1, CDH23 or CDHR1 genes. GAPDH was used as a control for normalization. Primer sequences are given in Supplementary Table S5.
Quantitative real-time RT-PCR was performed using TaqmanTM Gene Expression Assays (Applied Biosystems, Carlsbad, CA) from 20–30 ng of cDNA per well. Relative gene expression was assayed in triplicate and compared with wild-type samples, which served as the set point. All real-time RT-PCR reactions were performed on a QuantStudio 3 instrument (Applied Biosystems), following the manufacturer’s instructions. Housekeeping genes β2 M, GAPDH and ACTB were used for normalization. Relative quantification was assessed by using the 2−ΔΔCT method. Taqman Gene Expression Assays references were Hs00187842_m1 (β2 M), Hs99999905_m1 (GAPDH), Hs01060665_g1 (ACTB), Hs01031970_m1 (MERTK) and Hs00971456_g1 (RPGRIP1).
Additional Information
How to cite this article: Riera, M. et al. Whole exome sequencing using Ion Proton system enables reliable genetic diagnosis of inherited retinal dystrophies. Sci. Rep. 7, 42078; doi: 10.1038/srep42078 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Sahel, J. A., Marazova, K. & Audo, I. Clinical characteristics and current therapies for inherited retinal degenerations. Cold Spring Harbor perspectives in medicine 5, a017111, doi: 10.1101/cshperspect.a017111 (2015).
den Hollander, A. I., Black, A., Bennett, J. & Cremers, F. P. Lighting a candle in the dark: advances in genetics and gene therapy of recessive retinal dystrophies. The Journal of clinical investigation 120, 3042–3053, doi: 10.1172/JCI42258 (2010).
Werdich, X. Q., Place, E. M. & Pierce, E. A. Systemic diseases associated with retinal dystrophies. Seminars in ophthalmology 29, 319–328, doi: 10.3109/08820538.2014.959202 (2014).
Chiang, J. P. et al. Progress and prospects of next-generation sequencing testing for inherited retinal dystrophy. Expert review of molecular diagnostics 15, 1269–1275, doi: 10.1586/14737159.2015.1081057 (2015).
Ran, X. et al. ‘RetinoGenetics’: a comprehensive mutation database for genes related to inherited retinal degeneration. Database: the journal of biological databases and curation 2014, doi: 10.1093/database/bau047 (2014).
Nash, B. M., Wright, D. C., Grigg, J. R., Bennetts, B. & Jamieson, R. V. Retinal dystrophies, genomic applications in diagnosis and prospects for therapy. Translational pediatrics 4, 139–163, doi: 10.3978/j.issn.2224-4336.2015.04.03 (2015).
Avila-Fernandez, A. et al. Mutation analysis of 272 Spanish families affected by autosomal recessive retinitis pigmentosa using a genotyping microarray. Molecular vision 16, 2550–2558 (2010).
Daiger, S. P., Sullivan, L. S. & Bowne, S. J. Genes and mutations causing retinitis pigmentosa. Clinical genetics 84, 132–141, doi: 10.1111/cge.12203 (2013).
Perez-Carro, R. et al. Panel-based NGS Reveals Novel Pathogenic Mutations in Autosomal Recessive Retinitis Pigmentosa. Scientific reports 6, 19531, doi: 10.1038/srep19531 (2016).
Aparisi, M. J. et al. Targeted next generation sequencing for molecular diagnosis of Usher syndrome. Orphanet journal of rare diseases 9, 168, doi: 10.1186/s13023-014-0168-7 (2014).
Eisenberger, T. et al. Increasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: the example of retinal dystrophies. PloS one 8, e78496, doi: 10.1371/journal.pone.0078496 (2013).
Fernandez-San Jose, P. et al. Targeted Next-Generation Sequencing Improves the Diagnosis of Autosomal Dominant Retinitis Pigmentosa in Spanish Patients. Investigative ophthalmology & visual science 56, 2173–2182, doi: 10.1167/iovs.14-16178 (2015).
Glockle, N. et al. Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. European journal of human genetics: EJHG 22, 99–104, doi: 10.1038/ejhg.2013.72 (2014).
Yoon, C. K. et al. The diagnostic application of targeted re-sequencing in Korean patients with retinitis pigmentosa. BMC genomics 16, 515, doi: 10.1186/s12864-015-1723-x (2015).
Jin, X., Qu, L. H., Meng, X. H., Xu, H. W. & Yin, Z. Q. Detecting genetic variations in hereditary retinal dystrophies with next-generation sequencing technology. Molecular vision 20, 553–560 (2014).
Chiang, J. P. & Trzupek, K. The current status of molecular diagnosis of inherited retinal dystrophies. Current opinion in ophthalmology 26, 346–351, doi: 10.1097/ICU.0000000000000185 (2015).
Hull, S. et al. Nonsyndromic Retinal Dystrophy due to Bi-Allelic Mutations in the Ciliary Transport Gene IFT140. Investigative ophthalmology & visual science 57, 1053–1062, doi: 10.1167/iovs.15-17976 (2016).
Avila-Fernandez, A. et al. Whole-exome sequencing reveals ZNF408 as a new gene associated with autosomal recessive retinitis pigmentosa with vitreal alterations. Human molecular genetics 24, 4037–4048, doi: 10.1093/hmg/ddv140 (2015).
Boulanger-Scemama, E. et al. Next-generation sequencing applied to a large French cone and cone-rod dystrophy cohort: mutation spectrum and new genotype-phenotype correlation. Orphanet journal of rare diseases 10, 85, doi: 10.1186/s13023-015-0300-3 (2015).
Wang, F. et al. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: identification of a novel genotype-phenotype correlation and clinical refinements. Human genetics 133, 331–345, doi: 10.1007/s00439-013-1381-5 (2014).
Estrada-Cuzcano, A. et al. BBS1 mutations in a wide spectrum of phenotypes ranging from nonsyndromic retinitis pigmentosa to Bardet-Biedl syndrome. Archives of ophthalmology 130, 1425–1432, doi: 10.1001/archophthalmol.2012.2434 (2012).
Nikopoulos, K. et al. Identification of two novel mutations in CDHR1 in consanguineous Spanish families with autosomal recessive retinal dystrophy. Scientific reports 5, 13902, doi: 10.1038/srep13902 (2015).
de Castro-Miro, M. et al. Combined genetic and high-throughput strategies for molecular diagnosis of inherited retinal dystrophies. PloS one 9, e88410, doi: 10.1371/journal.pone.0088410 (2014).
Kuznetsova, T., Zangerl, B., Goldstein, O., Acland, G. M. & Aguirre, G. D. Structural organization and expression pattern of the canine RPGRIP1 isoforms in retinal tissue. Investigative ophthalmology & visual science 52, 2989–2998, doi: 10.1167/iovs.10-6094 (2011).
Lu, X. & Ferreira, P. A. Identification of novel murine- and human-specific RPGRIP1 splice variants with distinct expression profiles and subcellular localization. Investigative ophthalmology & visual science 46, 1882–1890, doi: 10.1167/iovs.04-1286 (2005).
Roepman, R. et al. Interaction of nephrocystin-4 and RPGRIP1 is disrupted by nephronophthisis or Leber congenital amaurosis-associated mutations. Proceedings of the National Academy of Sciences of the United States of America 102, 18520–18525, doi: 10.1073/pnas.0505774102 (2005).
Arts, H. H. et al. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nature genetics 39, 882–888, doi: 10.1038/ng2069 (2007).
Almoguera, B. et al. Application of Whole Exome Sequencing in Six Families with an Initial Diagnosis of Autosomal Dominant Retinitis Pigmentosa: Lessons Learned. PloS one 10, e0133624, doi: 10.1371/journal.pone.0133624 (2015).
Xin, W. et al. Identification of Genetic Defects in 33 Probands with Stargardt Disease by WES-Based Bioinformatics Gene Panel Analysis. PloS one 10, e0132635, doi: 10.1371/journal.pone.0132635 (2015).
Beryozkin, A. et al. Whole Exome Sequencing Reveals Mutations in Known Retinal Disease Genes in 33 out of 68 Israeli Families with Inherited Retinopathies. Scientific reports 5, 13187, doi: 10.1038/srep13187 (2015).
Bravo-Gil, N. et al. Improving the management of Inherited Retinal Dystrophies by targeted sequencing of a population-specific gene panel. Scientific reports 6, 23910, doi: 10.1038/srep23910 (2016).
Patel, N. et al. Expanding the clinical, allelic, and locus heterogeneity of retinal dystrophies. Genetics in medicine: official journal of the American College of Medical Genetics, doi: 10.1038/gim.2015.127 (2015).
Zhao, L. et al. Next-generation sequencing-based molecular diagnosis of 82 retinitis pigmentosa probands from Northern Ireland. Human genetics 134, 217–230, doi: 10.1007/s00439-014-1512-7 (2015).
Huang, X. F. et al. Genotype-phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Genetics in medicine: official journal of the American College of Medical Genetics 17, 271–278, doi: 10.1038/gim.2014.138 (2015).
O’Sullivan, J. et al. A paradigm shift in the delivery of services for diagnosis of inherited retinal disease. Journal of medical genetics 49, 322–326, doi: 10.1136/jmedgenet-2012-100847 (2012).
Weisschuh, N. et al. Mutation Detection in Patients with Retinal Dystrophies Using Targeted Next Generation Sequencing. PloS one 11, e0145951, doi: 10.1371/journal.pone.0145951 (2016).
Oishi, M. et al. Comprehensive molecular diagnosis of a large cohort of Japanese retinitis pigmentosa and Usher syndrome patients by next-generation sequencing. Investigative ophthalmology & visual science 55, 7369–7375, doi: 10.1167/iovs.14-15458 (2014).
Neveling, K. et al. Next-generation genetic testing for retinitis pigmentosa. Human mutation 33, 963–972, doi: 10.1002/humu.22045 (2012).
Shanks, M. E. et al. Next-generation sequencing (NGS) as a diagnostic tool for retinal degeneration reveals a much higher detection rate in early-onset disease. European journal of human genetics: EJHG 21, 274–280, doi: 10.1038/ejhg.2012.172 (2013).
Koenekoop, R. K., Lopez, I., den Hollander, A. I., Allikmets, R. & Cremers, F. P. Genetic testing for retinal dystrophies and dysfunctions: benefits, dilemmas and solutions. Clinical & experimental ophthalmology 35, 473–485, doi: 10.1111/j.1442-9071.2007.01534.x (2007).
Ostergaard, E., Batbayli, M., Duno, M., Vilhelmsen, K. & Rosenberg, T. Mutations in PCDH21 cause autosomal recessive cone-rod dystrophy. Journal of medical genetics 47, 665–669, doi: 10.1136/jmg.2009.069120 (2010).
Ba-Abbad, R. et al. Clinical characteristics of early retinal disease due to CDHR1 mutation. Molecular vision 19, 2250–2259 (2013).
Cohen, B. et al. A novel splice site mutation of CDHR1 in a consanguineous Israeli Christian Arab family segregating autosomal recessive cone-rod dystrophy. Molecular vision 18, 2915–2921 (2012).
Henderson, R. H. et al. Biallelic mutation of protocadherin-21 (PCDH21) causes retinal degeneration in humans. Molecular vision 16, 46–52 (2010).
Permanyer, J. et al. Autosomal recessive retinitis pigmentosa with early macular affectation caused by premature truncation in PROM1. Investigative ophthalmology & visual science 51, 2656–2663, doi: 10.1167/iovs.09-4857 (2010).
Yang, Z. et al. Mutant prominin 1 found in patients with macular degeneration disrupts photoreceptor disk morphogenesis in mice. The Journal of clinical investigation 118, 2908–2916, doi: 10.1172/JCI35891 (2008).
Sangermano, R. et al. Photoreceptor Progenitor mRNA Analysis Reveals Exon Skipping Resulting from the ABCA4 c.5461-10T->C Mutation in Stargardt Disease. Ophthalmology 123, 1375–1385, doi: 10.1016/j.ophtha.2016.01.053 (2016).
Becirovic, E. et al. Usher syndrome type 1 due to missense mutations on both CDH23 alleles: investigation of mRNA splicing. Human mutation 29, 452, doi: 10.1002/humu.9526 (2008).
von Brederlow, B. et al. Identification and in vitro expression of novel CDH23 mutations of patients with Usher syndrome type 1D. Human mutation 19, 268–273, doi: 10.1002/humu.10049 (2002).
Xiao, X. et al. A recurrent mutation in GUCY2D associated with autosomal dominant cone dystrophy in a Chinese family. Molecular vision 17, 3271–3278 (2011).
Kelsell, R. E. et al. Mutations in the retinal guanylate cyclase (RETGC-1) gene in dominant cone-rod dystrophy. Human molecular genetics 7, 1179–1184 (1998).
McKie, A. B. et al. Mutations in the pre-mRNA splicing factor gene PRPC8 in autosomal dominant retinitis pigmentosa (RP13). Human molecular genetics 10, 1555–1562 (2001).
Kohl, S. et al. Total colourblindness is caused by mutations in the gene encoding the alpha-subunit of the cone photoreceptor cGMP-gated cation channel. Nature genetics 19, 257–259, doi: 10.1038/935 (1998).
Pras, E. et al. Cone-rod dystrophy and a frameshift mutation in the PROM1 gene. Molecular vision 15, 1709–1716 (2009).
Mashima, Y. et al. Nonsense-codon mutations of the ornithine aminotransferase gene with decreased levels of mutant mRNA in gyrate atrophy. American journal of human genetics 51, 81–91 (1992).
Perrault, I. et al. Spectrum of NPHP6/CEP290 mutations in Leber congenital amaurosis and delineation of the associated phenotype. Human mutation 28, 416, doi: 10.1002/humu.9485 (2007).
Lotery, A. J. et al. Mutations in the CRB1 gene cause Leber congenital amaurosis. Archives of ophthalmology 119, 415–420 (2001).
den Hollander, A. I. et al. Mutations in a human homologue of Drosophila crumbs cause retinitis pigmentosa (RP12). Nature genetics 23, 217–221, doi: 10.1038/13848 (1999).
Sundin, O. H. et al. Genetic basis of total colourblindness among the Pingelapese islanders. Nature genetics 25, 289–293, doi: 10.1038/77162 (2000).
Khan, M. I. et al. The molecular basis of retinal dystrophies in pakistan. Genes 5, 176–195, doi: 10.3390/genes5010176 (2014).
Barragan, I. et al. Mutation spectrum of EYS in Spanish patients with autosomal recessive retinitis pigmentosa. Human mutation 31, E1772–1800 (2010).
Lenassi, E. et al. A detailed clinical and molecular survey of subjects with nonsyndromic USH2A retinopathy reveals an allelic hierarchy of disease-causing variants. European journal of human genetics: EJHG 23, 1318–1327, doi: 10.1038/ejhg.2014.283 (2015).
Baux, D. et al. Molecular and in silico analyses of the full-length isoform of usherin identify new pathogenic alleles in Usher type II patients. Human mutation 28, 781–789, doi: 10.1002/humu.20513 (2007).
Corton, M. et al. Exome sequencing of index patients with retinal dystrophies as a tool for molecular diagnosis. PloS one 8, e65574, doi: 10.1371/journal.pone.0065574 (2013).
Lewis, R. A. et al. Genotype/Phenotype analysis of a photoreceptor-specific ATP-binding cassette transporter gene, ABCR, in Stargardt disease. American journal of human genetics 64, 422–434, doi: 10.1086/302251 (1999).
Paloma, E., Martinez-Mir, A., Vilageliu, L., Gonzalez-Duarte, R. & Balcells, S. Spectrum of ABCA4 (ABCR) gene mutations in Spanish patients with autosomal recessive macular dystrophies. Human mutation 17, 504–510, doi: 10.1002/humu.1133 (2001).
Lotery, A. J. et al. Mutation analysis of 3 genes in patients with Leber congenital amaurosis. Archives of ophthalmology 118, 538–543 (2000).
Danciger, M. et al. Mutations in the PDE6B gene in autosomal recessive retinitis pigmentosa. Genomics 30, 1–7, doi: 10.1006/geno.1995.0001 (1995).
Dryja, T. P., Rucinski, D. E., Chen, S. H. & Berson, E. L. Frequency of mutations in the gene encoding the alpha subunit of rod cGMP-phosphodiesterase in autosomal recessive retinitis pigmentosa. Investigative ophthalmology & visual science 40, 1859–1865 (1999).
Corton, M. et al. High frequency of CRB1 mutations as cause of Early-Onset Retinal Dystrophies in the Spanish population. Orphanet journal of rare diseases 8, 20, doi: 10.1186/1750-1172-8-20 (2013).
Baum, L. et al. ABCA4 sequence variants in Chinese patients with age-related macular degeneration or Stargardt’s disease. Ophthalmologica. Journal international d’ophtalmologie. International journal of ophthalmology. Zeitschrift fur Augenheilkunde 217, 111–114, doi: 68553 (2003).
Jaakson, K. et al. Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Human mutation 22, 395–403, doi: 10.1002/humu.10263 (2003).
Fujinami, K. et al. The clinical effect of homozygous ABCA4 alleles in 18 patients. Ophthalmology 120, 2324–2331, doi: 10.1016/j.ophtha.2013.04.016 (2013).
Allikmets, R. et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nature genetics 15, 236–246, doi: 10.1038/ng0397-236 (1997).
Maugeri, A. et al. The 2588G–>C mutation in the ABCR gene is a mild frequent founder mutation in the Western European population and allows the classification of ABCR mutations in patients with Stargardt disease. American journal of human genetics 64, 1024–1035 (1999).
Simonelli, F. et al. New ABCR mutations and clinical phenotype in Italian patients with Stargardt disease. Investigative ophthalmology & visual science 41, 892–897 (2000).
Janecke, A. R. et al. Twelve novel myosin VIIA mutations in 34 patients with Usher syndrome type I: confirmation of genetic heterogeneity. Human mutation 13, 133–140, doi: 10.1002/(SICI)1098-1004(1999)13:2<133::AID-HUMU5>3.0.CO;2-U (1999).
Roux, A. F. et al. Survey of the frequency of USH1 gene mutations in a cohort of Usher patients shows the importance of cadherin 23 and protocadherin 15 genes and establishes a detection rate of above 90%. Journal of medical genetics 43, 763–768, doi: 10.1136/jmg.2006.041954 (2006).
Garcia-Garcia, G. et al. Mutational screening of the USH2A gene in Spanish USH patients reveals 23 novel pathogenic mutations. Orphanet journal of rare diseases 6, 65, doi: 10.1186/1750-1172-6-65 (2011).
Aller, E. et al. Identification of 14 novel mutations in the long isoform of USH2A in Spanish patients with Usher syndrome type II. Journal of medical genetics 43, e55, doi: 10.1136/jmg.2006.041764 (2006).
Breuer, D. K. et al. A comprehensive mutation analysis of RP2 and RPGR in a North American cohort of families with X-linked retinitis pigmentosa. American journal of human genetics 70, 1545–1554, doi: 10.1086/340848 (2002).
Mears, A. J. et al. Protein-truncation mutations in the RP2 gene in a North American cohort of families with X-linked retinitis pigmentosa. American journal of human genetics 64, 897–900, doi: 10.1086/302298 (1999).
Tiwari, A. et al. Identification of Novel and Recurrent Disease-Causing Mutations in Retinal Dystrophies Using Whole Exome Sequencing (WES): Benefits and Limitations. PloS one 11, e0158692, doi: 10.1371/journal.pone.0158692 (2016).
Acknowledgements
The authors thank Bernard Faure for his contribution and the nurses from Departament de Fotografia Oftàlmica of Institut de Microcirurgia Ocular (IMO) for technical assistance.
Author information
Authors and Affiliations
Contributions
M.R., R.N. and E.P. designed the study and experiments. R.N., A.B.-J. and B.C. diagnosed all patients at clinical level and collected phenotypic data. M.R., S.R.-N., P.M. and E.P. performed NGS, Sanger sequencing and cosegregation analyses. M.R. and S.R.-N. carried out the in vivo functional assays. M.R., R.N. and E.P. wrote the manuscript and prepared all the figures. All authors approved the final version to be published.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Riera, M., Navarro, R., Ruiz-Nogales, S. et al. Whole exome sequencing using Ion Proton system enables reliable genetic diagnosis of inherited retinal dystrophies. Sci Rep 7, 42078 (2017). https://doi.org/10.1038/srep42078
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep42078
This article is cited by
-
Probability of high-risk genetic matching with oocyte and semen donors: complete gene analysis or genotyping test?
Journal of Assisted Reproduction and Genetics (2022)
-
Species classifier choice is a key consideration when analysing low-complexity food microbiome data
Microbiome (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
