
Abstract
To date, there is no periadventitial drug delivery method available in the clinic to prevent restenotic failure of open vascular reconstructions. Resveratrol is a promising anti-restenotic natural drug but subject to low bioavailability when systemically administered. In order to reconcile these two prominent issues, we tested effects of periadventitial delivery of resveratrol on all three major pro-restenotic pathologies including intimal hyperplasia (IH), endothelium impairment, and vessel shrinkage. In a rat carotid injury model, periadventitial delivery of resveratrol either via Pluronic gel (2-week), or polymer sheath (3-month), effectively reduced IH without causing endothelium impairment and vessel shrinkage. In an in vitro model, primary smooth muscle cells (SMCs) were stimulated with elevated transforming growth factor (TGFβ) and its signaling protein Smad3, known contributors to IH. TGFβ/Smad3 up-regulated Kruppel-like factor (KLF5) protein, and SMC de-differentiation which was reversed by KLF5 siRNA. Furthermore, TGFβ/Smad3-stimulated KLF5 production and SMC de-differentiation were blocked by resveratrol via its inhibition of the Akt-mTOR pathway. Concordantly, resveratrol attenuated Akt phosphorylation in injured arteries. Taken together, periadventitial delivery of resveratrol produces durable inhibition of all three pro-restenotic pathologies — a rare feat among existing anti-restenotic methods. Our study suggests a potential anti-restenotic modality of resveratrol application suitable for open surgery.
Similar content being viewed by others


Introduction
Post-operative vessel re-narrowing (or restenosis) occurs in a great number of patients undergoing vascular reconstructions because of intimal hyperplasia (IH) and/or constrictive vessel remodeling1. Currently the clinically used anti-restenotic drugs are rapamycin (or analogs) and paclitaxel. While these drugs delivered by endovascular stents are effective in retarding IH, there is evidence that they cause stent-edge constrictive remodeling2. Moreover, they delay the recovery of damaged endothelium, predisposing patients to in-stent thrombosis and sudden death3,4. Therefore, drugs/delivery methods that can mitigate IH without causing endothelium impairment and constrictive remodeling make attractive candidates for next-generation anti-restenotic therapy5. Resveratrol, a polyphenol enriched in grapes, has been shown to be somewhat beneficial in animal models of atherosclerosis and restenosis in several reports6,7,8,9,10,11. However, the efficacy is relatively low in these studies, presumably due to the use of systemic delivery approaches (intraperitoneal, intravenous, intra-gavage, or dietary). It is well known that the extremely low bioavailability of resveratrol poses a major limitation for its translation into clinical use12,13,14. Thus, local delivery methods, which hold promise for prolonging bioactivity of this drug, are being actively pursued14.
In contrast to systemic delivery, periadventitial administration prevents drug from directly entering circulation and hence its rapid metabolic degradation15,16. More importantly, a periadventitial approach is uniquely suited for over 300,000 patients (each year in the US alone) who undergo open surgeries including bypass, endarterectomy, and dialysis access17. Of particular note, currently there are no drug delivery methods available for these patients to prevent restenosis15. Drug-eluting stents are used for the management of restenosis following angioplasty, but this method cannot be applied to open surgeries18. Hence in this specific context, to evaluate the potential of resveratrol as a candidate anti-restenotic therapeutic for open surgery, we employed a periadventitial15, local (as opposed to systemic) delivery approach to determine its full-spectrum impact on the major pathological processes contributing to restenosis, including IH, endothelium damage, as well as constrictive remodeling. Periadventitial delivery of resveratrol has not been previously assessed as an alternative approach to improve its anti-restenotic efficacy.
Given the therapeutic potential of resveratrol for prevention of restenosis, it is important to understand its functional mechanisms. In previous studies using primary vascular SMCs treated with various stimuli, resveratrol was shown to inhibit cell proliferation and migration, both IH-promoting behaviors5,19. Most recently, Thompson et al. found that resveratrol inhibited platelet derived growth factor (PDGF-BB)-stimulated SMC de-differentiation20. It has been recognized that the phenotypic plasticity of vascular SMCs is key to understanding the pathological mechanisms of IH19. Following surgical damage of the endothelium and elastic laminas, SMCs in the media become exposed to a range of stimuli, particularly potent being PDGF and transforming growth factor (TGFβ)19,21. Activated SMCs then undergo de-differentiation, as manifested by reduced expression of contractile proteins22 including calponin, smooth muscle myosin heavy chain (SM-MHC), and smooth muscle actin (α-SMA)20. They migrate into the intima, proliferate, and form a highly cellular neointima layer intruding into the lumen (restenosis)23. Thus SMC de-differentiation is fundamental for the SMC transformation to pathogenic phenotypes (e.g. migration and proliferation)24. In this regard it is both important and advantageous to use resveratrol as a pharmacological probe to interrogate the molecular mechanisms of SMC de-differentiation and abrogation thereof (or re-differentiation). Although previous studies reveal that the anti-proliferative function of resveratrol in SMCs is related to anti-oxidant and anti-inflammatory effects10,11, the mechanism underlying resveratrol inhibition of SMC de-differentiation is relatively little understood. Thompson et al. found that at low and high concentrations resveratrol promotes SMC contractile phenotype by activating sirtuin1 and AMPK, respectively20. However, it remains unknown whether resveratrol re-differentiates SMCs by modulating transcription factors, which play a master role for IH-promoting SMC de-differentiation25.
The best-known transcription factors involved in SMC phenotype switching are KLF4 and KLF526. Whereas the role of KLF4 is somewhat controversial, KLF5 has been consistently shown to promote SMC de-differentiation, migration and proliferation in vitro, all phenotypes contributing to IH. While KLF5 siRNA knockdown reduces, KLF5 overexpression enhances IH27. Up-regulation of KLF5 is found in human and rat restenotic lesions, and higher KLF5 levels are correlated with higher incidence of restenosis and cardiac allograft vasculopathy in human patients28. As such, KLF5 is an ideal transcription factor for our mechanistic study of SMC de-differentiation. We have previously found that TGFβ and its signaling protein Smad3 are both up-regulated in neointimal SMCs and contribute to IH21. Most recently, we further observed that TGFβ/Smad3 effected potent stimulation for SMC de-differentiation in cultured SMCs22. However, it is not known whether TGFβ/Smad3-stimulated SMC de-differentiation is related to KLF5.
In this study, we first evaluated the periadventitial delivery mode of resveratrol as a potentially viable method for restenosis prevention after open surgery, by systematically analyzing its effects on all three major pro-restenotic pathologies using a rat balloon injury model of restenosis. We found that the periadventitial approach had a markedly improved inhibitory effect on IH versus systemic delivery in previous reports. Remarkably, the recovery of damaged endothelium was not impaired but rather enhanced, and constrictive remodeling did not occur. Then using an established in vitro system we investigated the mechanism by which resveratrol re-differentiates rat aortic SMCs that are de-differentiated by elevated TGFβ/Smad3 signaling. We focused our mechanistic studies on SMC de-differentiation because it is fundamentally important for SMC pathogenic phenotype transformation and IH yet the molecular mechanism remains relatively poorly understood22,25. Moreover, it is of our particular interest to further investigate a novel finding from our latest report, i.e. TGFβ/Smad3 stimulates SMC de-differentiation22. Thus, it was based upon this specific context that we determined the mechanism of re-differentiation of TGFβ/Smad3-activated SMCs by resveratrol. We found that TGFβ/Smad3 treatment de-differentiated SMCs via up-regulation of KLF5 protein, and resveratrol could re-differentiate those cells by blocking KLF5 up-regulation. Ultimately, our results in this study suggest that when periadventitially delivered, resveratrol is a favorable anti-restenotic agent with significant potential clinic utility.
Methods
Animals
All animal procedures conformed to the NIH guide for the ethical care and use of laboratory animals. Animal protocols were approved by the Institutional Animal Care and Use Committee of University of Wisconsin-Madison. All surgeries were performed under isoflurane anesthesia (inhalation at 2 ml/min flow rate), and every effort was made to minimize animal suffering. Animals were euthanized in a chamber gradually filled with CO2.
Rat carotid artery balloon angioplasty model
Carotid artery balloon angioplasty was performed in male Sprague-Dawley rats (Charles River, 300–350 g) as previously described29. Briefly, rats were anesthetized by inhalation of 2.5% isoflurane (throughout this study). The left common carotid artery was exposed through a midline cervical incision. A 2 F Fogarty catheter (Edwards Lifesciences) was inserted into the common carotid artery via an arteriotomy in the external carotid artery. To produce arterial injury, the balloon was inflated and withdrew to the carotid bifurcation and this action was repeated three times. The external carotid artery was then permanently ligated, and blood flow was resumed.
Periadventitial local delivery of resveratrol to injured arteries
For short-term periadventitial resveratrol delivery, we applied a widely used Pluronic gel method as described in our previous report5,29. Briefly, to ensure complete solubility, 500 μg of resveratrol (Sigma-Aldrich) from a DMSO stock was first dissolved in 30 μl of 10% DMSO and then mixed with 270 μl of 25% F-127 Pluronic gel (Sigma-Aldrich) that was kept on ice. Immediately after balloon angioplasty in the left common carotid artery, the gel containing resveratrol was applied to the outside of the injured segment of the carotid artery and solidified right away. In the control group, equal volume of vehicle (30 μl of 20% DMSO) mixed with Pluronic gel was applied. Since Pluronic gel dissolves within 3 days16, for long-term periadventitial delivery we used a poly(ε-caprolactone) (PCL) sheath approach that was established by our groups and able to produce steady release for at least 2 months. Briefly, resveratrol-loaded PCL sheaths (100 μg per sheath) were prepared using our solvent casting method18. Control sheaths were prepared using the same procedures but with no resveratrol added. Immediately after balloon injury, a PCL sheath was longitudinally placed onto the injured artery segment. It was wrapped in such a way that only ~90% of the outer surface of the vessel was covered so as to avoid constriction of normal blood flow. The neck incision was then closed using a suture and animals were kept on a 37 °C warm pad for recovery. To avoid resveratrol photoisomerization, aliquots of the stock solution were stored in a sealed Eppendorf tube wrapped in aluminum foil, and experiments involving resveratrol were kept from constant light exposure.
Mass spectrometry analysis of resveratrol administered into the arterial wall
In order to detect the molecules of resveratrol (and/or its metabolites) that penetrated into the arterial wall, we used a mass spectrometry (MS)-based strategy following our published method30,31.
Sample preparation
We collected (1 day after surgery) the injured rat carotid arteries that were treated with resveratrol-containing periadventitial Pluronic gel, and immediately snap-froze the samples in liquid N2. For resveratrol extraction, 500 μl MeOH/H2O (80:20, V/V) was added to ground tissue homogenates, incubated for 30 min at 60 °C on shaking, and then centrifuged for 5 min at 10,000 rcf. The supernatant was transferred to a new Eppendorf tube and dried with Speed-Vac. The tissue extract was finally dissolved in 100 μl MeOH/H2O (80:20, V/V) and a 5 ul aliquot was injected for MS analysis.
Mass spectrometry
The MS analysis was performed using an ultra-performance liquid chromatography system equipped with a Q-Exactive Hybrid Quadrupole-Orbitrap mass spectrometer in positive ion mode. A reversed phase C18 column was employed. Mobile phases: 0.1% FA/H2O (A) and 0.1% FA/ACN (B). LC gradient: 0–5 min, 0–50% B; 5–7 min, 50–100% B; 7–8 min, 100 % B; 8–9 min, 100–0% B; 9–12 min, 0% B. The flow rate was 0.3 ml/min and column temperature was 30 °C. The injection volume for each run was 2 μl. MS experiments were performed with a resolution of 70,000 (at m/z 400), a mass range of m/z 90–1000, a maximum injection time of 100 ms, and 2 microscans per scan. The spray voltage was 3.5 kV and the sheath gas flow was 48 psi. A standard sample of resveratrol was used for reference. A series of resveratrol standard solutions were prepared at concentrations of 1 ng/μl, 0.5 ng/μl, 0.1 ng/μl and 0.01 ng/μl. Each 5 μl standard solution was injected into the mass spectrometer.
Morphometric analysis of intimal hyperplasia, vessel remodeling, and restenosis
Two weeks (Pluronic gel delivery) or three months (PCL sheath delivery) after balloon angioplasty, common carotid arteries were collected from anesthetized animals (under 2.5% isoflurane) following perfusion fixation at a physiological pressure of 100 mmHg29, the animals were then euthanized in a CO2 chamber. Paraffin sections (5 μm thick) were excised at equally spaced intervals and eosin or van Gieson-stained for morphometric analysis, as described in our previous reports29. Planimetric parameters as follows were measured on the sections and calculated using Image J: the area inside external elastic lamina (EEL area) or internal elastic lamina (IEL area), lumen area, intima area (=IEL area − lumen area), and media area (=EEL area − IEL area). Measurements were performed by a student blinded to the experimental conditions using 3–5 sections from each of 4–6 rats treated with either vehicle (DMSO) or resveratrol. The data from all sections were pooled to generate the mean for each animal. The means from all the animals in each treatment group were then averaged, and the standard error of the mean (SEM) was calculated. The vessel size is defined by EEL length; intimal hyperplasia is quantified as a ratio of intima area versus media area.
Immunohistochemistry for assessment of phospho-Akt or caspase-3 positive cells in the rat arterial wall
Immunostaining was performed on rat carotid artery sections following our published methods5,29. Briefly, the sections were first incubated with each of the primary antibodies: anti- caspase-3 (Cell Signaling, Cat#9661), 1:500 dilution, incubated for 1 h; anti-p-AKT (Cell Signaling, Cat#4060, clone D9E), 1:40 dilution, incubated for overnight at 4 °C. The sections were then incubated with the ImmPRESS HRP-conjugated goat-anti-rabbit secondary antibody (Vector Laboratories, 1:200), followed by visualization with 3, 3-diaminobenzidine (DAB). The slides were counterstained with hematoxylin. The number of positively stained cells was counted on 8-bit binary images converted by Image J from the pictures of immunostained sections and normalized by the high power microscopic field (HPF). Cell counting was performed by a student blinded to experimental conditions. In each experimental group (DMSO control or resveratrol treatment), at least 5 sections from each of 4–6 animals were used. The data from all sections were pooled to generate the mean for each animal. The means from all animals in each experimental group were then averaged, and the standard error of the mean (SEM) was calculated.
Assessment of post-angioplasty re-endothelialization
Re-endothelialization in balloon-injured arteries was evaluated on day 14 after angioplasty by CD31 immunostaining using the method described above5. Briefly, a goat anti-CD31 primary antibody (R&D Sytems,1:150) was incubated with the sections for 1 h followed by an incubation with a biotinylated rabbit-anti-goat secondary antibody for 30 min. Immunostaining of CD31 was then visualized by using streptavidin-HRP and DAB. For quantification of re-endothelialization, we used Image J to measure the percentage of CD31-stained versus total peri-luminal perimeter5,16.
In vitro stimulation of primary SMCs and treatment with resveratrol
Rat aortic vascular SMCs were isolated from thoracoabdominal aortas of male Sprague–Dawley rats and maintained at 37 °C and 5% CO2 in low glucose Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, Carlsbad, CA) supplemented with 10% fetal bovine solution (FBS)5. Cells were used at passages 5–6 for all experiments. Cell viability was >95% as indicated by Trypan Blue exclusion assay. For stimulation of SMCs with elevated TGFβ/Smad3 signaling, adenoviral vectors expressing Smad3 (AdSmad3) and GFP (AdGFP control) were constructed as previously described22,32. SMCs were infected for 4 h with AdSmad3 (or AdGFP) (3 × 104 particles/cell) in DMEM containing 2% FBS, and recovered for 20 h with 10% FBS, and then starved with 0.5% FBS for 24 h followed by treatment with human recombinant TGFβ1 (5 ng/ml, R&D Systems, Minneapolis, MN) or equivalent amount of solvent (final 4 μM HCl and 1 μg/ml BSA) for the time length indicated in figure legends. Cells were then harvested for various assays. For treatment with small molecular inhibitors, cells were pre-incubated for 2 h with an inhibitor at desired concentrations or with equal volume of DMSO (vehicle control) before TGFβ1 was added. To illustrate SMC morphology, Calcein (2 μM, Fisher Scientific) was incubated with the cell culture for 30 min at 37 °C, and cells were washed 3× with PBS prior to fluorescence microscopy followed by cell area calculation using Image J.
siRNA knockdown of KLF5
SMCs were infected with AdGFP or AdSmad3 as described above. Prior to treatment with TGFβ1, knockdown of KLF5 was performed using siRNA oligos according to manufacturer’s protocols. Briefly, SMCs of 60–70% confluence were transfected with 20 nM KLF5-specific siRNA (Silencer siRNA, s136575) or scrambled control siRNA (Silencer siRNA, AM4611) (Ambion, Carlsbad, CA) for 6 h using Lipofectamine RNAiMAX (13778, Invitrogen, Carlsbad, CA) in OptiMEM I medium containing 0.5% FBS but no antibiotics.
Quantitative real-time PCR (qRT-PCR)
mRNA was isolated from collected cells using Trizol (Qiagen, Valencia, CA) following the manufacturer’s instructions. Purified mRNA (1 μg) was used for the first-strand cDNA synthesis using iScript cDNA synthesis kit (Bio-Rad) and quantitative RT-PCR was performed using the 7500 Fast Real-Time PCR System (Applied Biosystems, Carlsbad, CA). Each cDNA template was amplified in triplicates using SYBR Green PCR Master Mix (Applied Biosystems, Carlsbad, CA) with primers for rat CD34 or rat KLF5.
Western blotting for evaluation of protein levels
SMCs were lysed in RIPA buffer containing protease inhibitors (50 mM Tris, 150 mM NaCl, 1% Nonidet P-40, 0.1% sodium dodecyl sulfate, and 10 μg/ml aprotinin). Protein concentrations of cell lysates were determined using a Bio-Rad DC™ Protein Assay kit. Approximately 15~30 μg of proteins from each sample were separated on 4–20% Mini-PROTEAN TGX precast gels (Bio-Rad) and transferred to a PVDF membrane. Proteins of interest were detected by immunoblotting using the following primary antibodies and dilution ratios: anti-calponin (1:1000) from Santa Cruz (sc-58707), anti-SM-MHC (1:1000) from Santa Cruz (sc-6956), anti-KLF5 (1:250) from Sigma-Aldrich, anti-pmTOR (1:1000) from Cell Signaling (5536 S), anti-total mTOR (1:1000) from Cell Signaling (2972), anti-pS6K (1:1000) from Cell Signaling (9204 S), anti-total S6K (1:1000) from Cell Signaling (2708 S), anti-pAkt (1:1000) from Cell Signaling (9271 S), anti-total Akt (1:1000) from Cell Signaling (4691 S), anti-pSmad3 (1:1000) from Invitrogen (44246 G), anti-total Smad3 (1:1000) from Invitrogen (511500) and mouse anti-β-actin from Sigma-Aldrich. After incubation of the blots with HRP-conjugated secondary antibodies (1:3000 for goat anti-rabbit or 1:10000 for goat anti-mouse, Bio-Rad), specific protein bands on the blots were visualized by applying enhanced chemiluminescence reagents (Pierce) and then recorded with a LAS-4000 Mini imager (GE, Piscataway NJ). Band intensity was quantified using ImageJ.
Statistical analysis
Data are presented as means ± SEM. Means were compared between two groups using two-tailed unpaired Student’s t-test. For multiple comparisons the analysis of variance (ANOVA) test followed by Bonferroni’s correction was applied. A value of P < 0.05 was considered significant. All P-values are two-sided. Statistics were performed using the GraphPad prism 6.0 Software (GraphPad Software, Inc., La Jolla, CA, USA).
Results
Periadventitial delivery of resveratrol eliminates neointima without causing constrictive remodeling two weeks after balloon injury of rat carotid arteries
In several reports resveratrol was shown to suppress IH but with limited efficacy (~30–60% inhibition)6,7,8,9,10,11. It is notable that in these studies resveratrol was administered via systemic approaches, which might have led to its quick destruction by metabolic systems. To circumvent this problem, here we adopted a periadventitial delivery approach so that resveratrol could evade circulation and rapid systemic clearance.
We performed rat carotid balloon angioplasty, a widely accepted model of IH18, and then applied resveratrol in Pluronic gel around the injured artery. We confirmed that the resveratrol compound was able to penetrate into the arterial wall, as indicated by mass spectrometry analysis using artery tissue homogenates (Figure S1). At 24 h after periadventitial application, resveratrol was present in the injured rat carotid arteries in two major forms, non-metabolized resveratrol and its monoglucuronide conjugate (Figure S2), with a ratio of the latter versus the former at 28.4% ± 17.3 (standard error of the mean, n = 3 rats). This data indicates that majority of resveratrol molecules in the vessel wall were non-metabolized one day after periadventitial administration.
We then evaluated the effect of resveratrol on IH. At day 14 after balloon injury and resveratrol administration, carotid arteries were collected and cross sections prepared. Morphometric analyses (Fig. 1) indicate that treatment with resveratrol resulted in a pronounced 86% decrease of IH (intima/media ratio) compared to vehicle (DMSO) control. The EEL length, which measures the overall vessel size (or remodeling), was unaltered. The ultimate outcome of resveratrol treatment is an approximately 2 fold increase of lumen area. These results demonstrate that periadventitial delivery of resveratrol is highly efficacious versus previously reported systemic delivery (~30–60% inhibition of IH)6,7,8,9,10,11.

(A) Representative stained sections from arteries treated with vehicle (DMSO) and resveratrol respectively and collected on day 14 after injury. Arrows indicate external elastic lamina (EEL); arrowheads mark internal elastic lamina (IEL). (B–D) Quantified data of IH (ratio of intima/media area), lumen area, and arterial remodeling (EEL length), respectively. Each bar represents a mean (±SEM) of sections from 5 animals, *P < 0.05 compared to vehicle control.
Periadventitial delivery of resveratrol enhances re-endothelialization two weeks after balloon injury of rat carotid arteries
Current clinical anti-restenotic managements (stents releasing rapalogs or paclitaxel) impair endothelium recovery following surgical damage4. As such these methods are associated with a high risk of in-stent thrombosis, a condition associated with ~50% mortality3. We therefore evaluated whether resveratrol delays post angioplasty re-endothelialization, by immunostaining CD31, a commonly used marker for vascular endothelium. Remarkably, statistical analysis of CD31 staining indicates that compared to vehicle control resveratrol treatment did not impair endothelium at two weeks after periadventitial administration, but rather, increased re-endothelialization by 2 fold (Fig. 2A). This result reveals an interesting protective effect of periadventitially delivered resveratrol on the vascular endothelium in vivo. Furthermore, a lack of apoptotic cells, which would otherwise be positively stained for cleaved caspase-3, indicates a healthy vessel after resveratrol treatment (Fig. 2B). The caspase-3 immunostaining method was validated by a positive control sample.

Rat carotid arteries were treated with either vehicle or resveratrol and collected on day 14 after injury. Arrows indicate external elastic lamina (EEL); arrowheads mark internal elastic lamina (IEL). (A) Immunostaining of CD31. Quantification: mean ± SEM; n = 3–5 animals; *P < 0.05 compared to vehicle control. (B) Immunostaining of cleaved caspase-3. Scale bar = 20 μm. A placenta sample serves as positive control.
Periadventitial delivery of resveratrol mitigates neointima without causing constrictive remodeling three months after balloon injury of rat carotid arteries
Studies testing long-term effect of drug delivery have been rare presumably owing to a lack of suitable drug-releasing platforms as well as the difficulty of carrying out such experiments. We have recently developed a biodegradable PCL sheath that enables steady release of a hydrophobic compound (e.g. rapamycin) for at least 2 months18. Taking advantage of this technology, we produced resveratrol-loaded PCL sheaths and deployed them around injured segments of rat carotid arteries. After 3 months arteries were collected. Morphometric analyses indicate that IH was reduced by 42% in resveratrol-treated arteries compared to those treated with control PCL sheaths without resveratrol (Fig. 3). There was no significant change of EEL length. As a result, the lumen area was increased by 162%. These results indicate that given an appropriate periadventitial delivery method resveratrol can produce prolonged inhibitory effect on IH without causing constrictive remodeling.

(A) Representative van Gieson-stained sections from arteries treated respectively with control PCL sheaths (no resveratrol) and resveratrol-loaded sheaths and collected at 3 months after injury. Arrows indicate external elastic lamina (EEL); arrowheads mark internal elastic lamina (IEL). (B–D) Quantified data of IH (ratio of intima/media area), lumen area, and arterial remodeling (EEL length), respectively. Each bar represents a mean (±SEM) of sections from 4–5 animals, *P < 0.05 compared to vehicle control.
Resveratrol reverses TGFβ/Smad3-stimulated de-differentiation of cultured SMCs
Following surgical damage of the integrity of the endothelium and vascular wall, SMCs are exposed to various stimuli (e.g. growth factors) and undergo de-differentiation losing their signature contractile phenotype. This SMC phenotype change is critical for neointima formation19,25. Our group has previously shown that TGFβ and its signaling effector Smad3 are up-regulated following arterial injury and this signaling axis is an important stimulator of neointima formation21. Recently we observed that elevated TGFβ/Smad3 stimulate SMC de-differentiation22. Whether resveratrol inhibits TGFβ/Smad3-stimulated SMC de-differentiation is not known.
To mimic post-injury TGFβ/Smad3 up-regulation, we first expressed Smad3 (or GFP control) in rat primary aortic SMCs using adenovirus, and then added recombinant TGFβ1 (5 ng/ml) to activate Smad3 signaling22,33. Resveratrol (or DMSO control) was pre-incubated with the SMC culture for 2 h prior to TGFβ1 stimulation. We used resveratrol at 50 μM, an effective concentration without triggering apoptosis, as reported by Brito et al. using rat primary SMCs34. We defined SMC de-differentiation by changes in cellular morphology and in the expression of calponin and SM-MHC, the two most stringent vascular SMC markers20,25. As shown in Fig. 4, while TGFβ/Smad3 treatment markedly down-regulated calponin and SM-MHC as compared to AdGFP control, resveratrol completely reversed this change. Accordingly, while under TGFβ/Smad3 stimulation SMCs lost their characteristic spindle shape and transformed to an expanded morphology (quantified as increase of cell-occupied area) indicative of de-differentiation, treatment with resveratrol restored SMC spindle-shape morphology. Since it is known that rapamycin inhibits SMC de-differentiation19, we used rapamycin as a positive control and observed an effect similar to that of resveratrol. Together these results demonstrate that resveratrol can re-differentiate SMCs that are de-differentiated due to elevated TGFβ/Smad3 signaling.

Rat aortic SMCs were infected with AdGFP (control) or AdSmad3 followed by pre-incubation with vehicle (DMSO) or resveratrol (50 μM) or rapamycin (200 nM) for 2 h, and then treated with solvent (for AdGFP control) or TGFβ1 (5 ng/mL) for 24 h. Cells were then used for Western blotting of calponin (A) or SM-MHC (B), or staining with Calcein for cell area measurement (C). Quantification: mean ± SEM of three independent experiments; normalization to β-actin; *P < 0.05 compared to AdGFP control; #P < 0.05 compared to AdSmad3 + TGFβ1 without pre-incubation with a drug.
Resveratrol abrogates TGFβ/Smad3-stimulated KLF5 protein up-regulation in cultured SMCs
We then further investigated the molecular mechanism underlying the resveratrol inhibitory effect on TGFβ/Smad3-stumulated SMC de-differentiation. SMC de-differentiation is known to be a transcriptionally regulated process but the molecular mechanisms remain to be better understood25. We first investigated KLF4 and KLF5, two prominent transcription factors involved in SMC de-differentiation28,35. We did not detect a significant change of KLF4 protein following TGFβ/Smad3 stimulation (data not shown). Interestingly however, through Western blotting, we found that either TGFβ or AdSmad3 alone increased KLF5 protein, and the combination of these two resulted in a 4-fold increase of KLF5 versus AdGFP control (Fig. 5A). We did not find a significant change of KLF5 mRNA by RT-PCR (Fig. 5B), which agrees with the Affymetrix result derived from our recent study22. These results reveal a novel finding that TGFβ/Smad3 up-regulate KLF5 protein (but not mRNA). Remarkably, resveratrol reversed this up-regulation by reducing KLF5 protein to the basal level (AdGFP control).

Rat aortic SMCs were infected with AdGFP (control) or AdSmad3 followed by pre-incubation with vehicle (DMSO) or resveratrol (50 μM) for 2 h, and then treated with solvent or TGFβ1 (5 ng/mL) for 24 h. Cells were then collected for Western blotting analysis (A) or RT-PCR (B). Microarray data were retrieved from our recent report22. Quantification: mean ± SEM of three independent experiments; normalization to β-actin; **P < 0.05 compared to any of the first 3 conditions; #P < 0.05 compared to AdSmad3 + TGFβ1 without pre-incubation with resveratrol. To determine the effect of KLF5 knockdown on TGFβ/Smad3-stimulated SMC de-differentiation, rat aortic SMCs were infected with AdGFP (control) or AdSmad3 and then transfected with scrambled or KLF5-specific siRNA for 6 h followed by treatment with solvent (control) or TGFβ1 (5 ng/mL) for 24 h. Cells were then used for Western blotting of KLF5 (C), calponin (D) or SM-MHC (E), or staining with Calcein for cell area measurement (F). Quantification: mean ± SEM of three independent experiments; normalization to β-actin; *P < 0.05 compared to AdSmad3 + TGFβ1 with scrambled siRNA (the middle bar).
In order to determine a specific role of KLF5 in mediating TGFβ/Smad3-stimulated SMC de-differentiation, we used siRNA to knock down KLF5 in SMCs. As shown in Fig. 5C, in comparison to scrambled siRNA, KLF5-specific siRNA effectively reduced TGFβ/Smad3-stimulated KLF5 protein production. Consequently, KLF5 siRNA substantially restored protein levels of SMC markers calponin and SM-MHC that were down-regulated by TGFβ/Smad3 treatment (Fig. 5D and E). SMC re-differentiation due to specific KLF5 knockdown was accordantly reflected from SMC morphological changes; i.e. TGFβ/Smad3-transformed SMCs were reverted to a differentiated spindle shape by KLF5 siRNA (Fig. 5F). Thus, these results indicate that TGFβ/Smad3 initiate SMC de-differentiation in a KLF5-dependent manner.
Resveratrol blocks TGFβ/Smad3-stimulated mTOR activation in cultured SMCs
For identification of upstream regulators responsible for KLF5 up-regulation, we noticed that similar to resveratrol, rapamycin was also able to re-differentiate TGFβ/Smad3-activated SMCs back to a contractile phenotype (Fig. 4). We thus determined the effect of rapamycin on KLF5 expression, and found that rapamycin at 200 nM (an effective concentration used in our previous report36) reversed TGFβ/Smad3-stimulated KLF5 protein up-regulation (Fig. 6A). As rapamycin is a specific mTOR inhibitor19, it is conceivable that elevated TGFβ/Smad3 signaling may activate the mTOR pathway in SMCs, which has never been previously reported. Indeed, we found that while TGFβ and Smad3 each increased p-mTOR, treatment with both stimulated mTOR phosphorylation nearly 4 fold (Fig. 6B). Interestingly, resveratrol substantially mitigated TGFβ/Smad3-stimulated mTOR phosphorylation. The assay was validated by the effect of rapamycin in blocking TGFβ/Smad3-stimulated mTOR phosphorylation. Furthermore, the same pattern of TGFβ/Smad3 stimulation and resveratrol inhibition was also observed with phosphorylation of S6K (Fig. 6C), the downstream effector and a frequently used surrogate of mTOR activation. This result confirms the TGFβ/Smad3 stimulatory effect on mTOR activation and its blockade by resveratrol.

Rat aortic SMCs were infected with AdGFP (control) or AdSmad3 followed by pre-incubation with vehicle (DMSO), resveratrol (50 μM), rapamycin (200 nM), or LY249004 (20 μM) for 2 h, and then treated with solvent or TGFβ1 (5 ng/mL) for 24 h. Cells were then collected for Western blotting analysis of KLF5 (A), p-mTOR (B) or p-S6K (C). Quantification: mean ± SEM of three independent experiments; normalization to β-actin or to total protein; *P < 0.05 compared to any of the first 3 conditions; #P < 0.05 compared to AdSmad3 + TGFβ1 without pre-incubation with a drug.
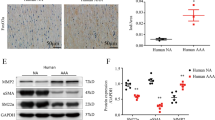
Resveratrol inhibits TGFβ/Smad3-stimulated Akt phosphorylation with no effect on Smad3 phosphorylation in cultured SMCs
We previously observed that TGFβ/Smad3 treatment enhanced Akt phosphorylation in rat aortic SMCs32. In a recent study, it was found that in rat SMCs oxidized LDL activated the PI3K-Akt-mTOR pathway, which was inhibited by resveratrol34. In addition, in our current study we found that LY249004, a commonly used inhibitor of the PI3K-Akt pathway, was able to block TGFβ/Smad3-stimulated KLF5 up-regulation (Fig. 6A). These observations together raise an interesting question as to whether TGFβ/Smad3 activated the Akt-mTOR pathway which in turn acted as the upstream regulator of KLF5 protein up-regulation and hence the resveratrol target. Therefore, after establishing that resveratrol inhibits TGFβ/Smad3-stimulated mTOR activation and KLF5 up-regulation, we determined the effect of resveratrol on Akt activation. We found that while agreeing with our previous report32 TGFβ/Smad3 increased phospho-Akt ~2 fold, resveratrol abolished this stimulation (Fig. 7A). In contrast, rapamycin did not show an effect on Akt phosphorylation, placing mTOR downstream of Akt activation. We then further determined whether resveratrol inhibited TGFβ/Smad3-stimulated Akt activation by blocking Smad3 phosphorylation. As shown in Fig. 7B, a lack of resveratrol effect on TGFβ-stimulated Smad3 phosphorylation ruled out this possibility. In sum, these results together tracked down the target of resveratrol inhibitory effect on TGFβ/Smad3-stimulated KLF5 protein up-regulation and SMC de-differentiation, to the Akt-mTOR pathway which was downstream of TGFβ/Smad3 in our experimental system.

(A,B) Rat aortic SMCs were infected with AdGFP (control) or AdSmad3 followed by pre-incubation with vehicle (DMSO), resveratrol (50 μM), or rapamycin (200 nM) for 2 h, and then treated with solvent or TGFβ1 (5 ng/mL). For Western blotting analyses of p-Akt (A) and p-Smad3 (B), cells were collected after TGFβ1 treatment for 2 h and 0.5 h, respectively. Quantification: mean ± SEM of three independent experiments; normalization to total Akt (t-Akt) or to total Smad3 (t-Smad3); *P < 0.05 compared to each of the first 2 conditions; **P < 0.05 compared to AdSmad3 only; #P < 0.05 compared to AdSmad3 + TGFβ1 without pre-incubation with a drug; n.s., not significant. (C). Rat carotid arteries were treated with either vehicle or resveratrol and collected on day 14 after angioplasty. Shown on the left are representative sections of p-Akt Immunostaining. Arrows indicate external elastic lamina (EEL); arrowheads mark internal elastic lamina (IEL). Scale bar = 20 μm. Negative staining: IgG instead of a primary antibody was used. Quantification: mean ± SEM of 4–5 animals; *P < 0.05 compared to vehicle control; HPF, high power field.
After tracking down the resveratrol functional target to Akt phosphorylation, we finally evaluated this effect in balloon-injured arterial wall, where the TGFβ/Smad3 signaling is known to be elevated21. Using the artery sections from the periadventitial delivery experiments, we found that while p-Akt staining was enhanced in the injured arteries compared to uninjured control at 14 days after surgery, periadventitial treatment with resveratrol significantly reduced Akt phosphorylation (Fig. 7C). As the effect of resveratrol on p-Akt has not been previously examined in neointima-producing injured arteries, our data provide new information in regard to in vivo targets of resveratrol for potential pharmacological interventions to inhibit IH.
Discussion
Restenosis remains a major cause of failure of reconstructive vascular interventions. The failure rate is particularly high among the patients undergoing open surgical procedures including bypass, endarterectomy, and dialysis access, as there is no translated anti-restenotic method for these patients18. With this clinical need in mind, we carried out research with two chief objectives: 1) To evaluate periadventitial delivery as a viable approach to optimize the effects of resveratrol on all three major pro-restenotic pathologies. We observed that periadventitial delivery of resveratrol produced a markedly improved neointima-inhibiting effect versus previously reported systemic administration (86% versus ~30–60%6,7,8,9,10,11). Importantly, perivascular delivery of resveratrol did not retard, but rather accelerated post-surgery endothelial recovery and did not cause constrictive remodeling. 2) To investigate the molecular mechanism of resveratrol-promoted re-differentiation of SMCs using an in vitro model of TGFβ/Smad3 stimulation. We found that while TGFβ/Smad3 stimulated mTOR activation and up-regulated KLF5 protein — a transcription factor known to promote SMC de-differentiation and IH28, resveratrol abolished KLF5 up-regulation and re-differentiated SMCs by targeting the Akt-mTOR pathway downstream of TGFβ/Smad3 (Fig. 8). These findings suggest that resveratrol represents a promising candidate drug for an anti-restenotic perivascular delivery paradigm that is uniquely suited for open surgeries.

A schematic of resveratrol-targeted pathway in rat vascular smooth muscle cells.
Three major pathologies contribute to restenosis: SMC hyperplasia in the intima (IH) due to SMC pathogenic phenotype transformation, impairment of re-endothelialization which exacerbates IH, and constrictive remodeling that shrinks the vessel diameter and lumen. An unfavorable effect on any of these three could compromise the long-term efficacy of an anti-restenotic therapy5. Current clinical methods of rapamycin or paclitaxel-eluting stents not only cause constrictive remodeling at stent edge2 but also impinge on the endothelium leading to thrombosis4. These inherent defects are increasingly recognized necessitating vigorous research on alternative therapeutics/paradigms. Recently, resveratrol has shown an anti-IH effect in several preclinical studies6,7,8,9,10,11. However, the overall outcome is not satisfactory. We noted that in these studies resveratrol was administered exclusively via systemic approaches, which are not optimal because natural compounds such as resveratrol are extremely susceptible to metabolic clearance when systemically delivered13. Even though resveratrol coated on a stent reduced stenosis in rat carotid artery by 60% 4 weeks after stenting, resveratrol on the stent is subject to loss to the circulation and its longer-term effect is not known37. We thus adopted a periadventitial approach to circumvent this problem. Moreover, our studies differ from the existing publications also in that we determined the resveratrol effects on all three major pro-restenotic pathologies as well as apoptosis in the vessel wall whereas none of the previous reports offered a full-spectrum analysis. Significantly, our data indicate that periadventitial delivery of resveratrol produced highly desirable effects on IH prevention and endothelium protection, and also favorably, no constrictive remodeling. Furthermore, another unique progress made in our study is that we also tested a 3-month long-term resveratrol treatment of injured rat carotid arteries using a PCL polymer sheath18, which produced a lasting inhibitory effect on IH without causing constrictive remodeling. Over a long term (months) of drug delivery multiple factors including the degradation products of the drug carrier would elicit inflammatory or mechanical stress on the vessel wall leading to constrictive remodeling. In this regard, the positive outcome from our 3-month periadventitial resveratrol treatment is promising and lays a basis for future optimization.
Improvement of resveratrol efficacy by using a periadventitial approach versus systemic delivery could be rationalized by the following. 1) Since resveratrol applied outside the artery does not directly enter the circulation, its bioavailability could be protected15. Resveratrol would be otherwise subjected to quick degradation in various organs if systemically delivered12,14. 2) Periadventitial local delivery may provide relatively high focal drug concentrations in pathogenic SMCs in the vessel wall. In support of these assertions, at 24 h after periadventitial application we still detected non-metabolized resveratrol as the major form in the vessel wall (Figure S2), whereas resveratrol in the circulation (serum) can be completely metabolized in less than 15 min14,38. Moreover, in a new report by Tolva et al.14, local delivery of a resveratrol-containing compound into injured rabbit arteries with a balloon catheter effectively preserved active forms of resveratrol in the arterial wall and reduced the intima/media area ratio by ~70% compared to sham control. 3) In addition, accelerated post-surgery endothelial recovery may also contribute to improved efficacy of periadventitial delivery of resveratrol. Unlike SMCs, ECs are highly susceptible to drug toxicity4,36. In a recent report using systemic delivery, whereas a low concentration of resveratrol promotes re-endothelialization after angioplasty, a high resveratrol concentration failed to produce this effect6. Indeed, high concentrations of resveratrol have been shown to induce apoptosis in several cell types34. Of note, periadventitial release could generate a gradient15,16 of resveratrol with higher concentrations in the adventitia and lower concentrations in the endothelium, and may have thus protected endothelial recovery. A protective effect of appropriate concentrations of resveratrol on ECs has been previously demonstrated in vitro6,39. While ECs are highly sensitive to oxidative and inflammatory damage, resveratrol produces antioxidant and anti-inflammatory effects11,34, or stimulates eNOS expression6, which is generally thought to be beneficial to ECs. Taken together, our in vivo results suggest that periadventitial delivery of resveratrol represents a viable strategy to optimally produce multi-beneficial effects of this promising anti-restenotic drug.
The functional mechanisms of resveratrol are complex. This complexity stems from the fact that resveratrol targets multiple proteins and pathways. To date four direct-binding targets of resveratrol have been discovered. Earlier sirtuin-1 was identified as a resveratrol target40. It was then reported that resveratrol is a PDE4 inhibitor which activates AMPK by lowering intracellular AMP concentrations41. Most recently, resveratrol was found to also directly bind to estrogen receptors42 and tyrosine transfer RNA synthetase43. Each of these resveratrol-binding sites is associated with numerous pathways, upstream and downstream. Therefore, it is impractical to delineate the resveratrol-associated mechanism in SMCs by investigating all the possible pathways. On the other hand, a growing body of evidence indicates that the therapeutic action of resveratrol is highly context-dependent. It may function by distinct mechanisms depending on cell type, the nature and duration of stimulation, and cell or disease state12. In order to minimize those confounding variables, we focused the resveratrol mechanistic studies specifically on vascular SMC de-differentiation using elevated TGFβ/Smad3 signaling as a stimulant, which is a known important contributor to IH21.
Built on our recent finding that TGFβ/Smad3 promote SMC de-differentiation22,24, herein we further uncovered that KLF5 plays a key role in this event. Up-regulation of KLF5 in SMCs by TGFβ/Smad3 has not been previously reported. Demonstrating a specific role of the TGFβ/Smad3 signaling axis in KLF5 regulation, TGFβ and AdSmad3 treatment each increased KLF5 protein and their combination further substantially enhanced KLF5 up-regulation. In addition, in our experimental system we did not observe a significant change of KLF5 protein levels after PDGF-BB treatment (data not shown), further supporting a specific role of TGFβ/Smad3 in KLF5 up-regulation. An ensuing question was whether TGFβ/Smad3 regulated KLF5 mRNA or protein, since Smad3 is a transcription factor that regulates the transcription of hundreds of genes. Surprisingly, our data did not show a significant difference in KLF5 mRNA levels between TGFβ/Smad3 stimulation and GFP control. We were thus able to narrow down the TGFβ/Smad3 regulation of KLF5 to the protein level. Inasmuch as both the TGFβ/Smad3 axis and KLF5 are proven important contributors to IH21,27, our finding of KLF5 regulation by TGFβ/Smad3 opens a new conduit for better understanding of neointimal pathogenesis and its mitigation.
Using resveratrol and rapamycin as powerful pharmacological tools, we further discovered that activation of mTOR is responsible for TGFβ/Smad3-stimulated up-regulation of KLF5. While resveratrol treatment abrogated TGFβ/Smad3-stimulated activation of mTOR and its downstream effector S6K, it also reversed TGFβ/Smad3-stimulated KLF5 up-regulation, suggesting mTOR as an upstream positive regulator of KLF5. Further confirming the role of mTOR in KLF5 regulation, treatment with rapamycin eliminated the increase of KLF5 protein caused by TGFβ/Smad3 stimulation. The up-regulation of KLF5 protein by activated mTOR observed herewith is a novel finding that has never been previously reported, neither in SMCs nor in other cell types. It is established that the central function of activated mTOR is promotion of protein translation, consistent with our data that KLF5 protein rather than mRNA was increased following TGFβ/Smad3-stimulated mTOR activation. Importantly, while KLF5 is a transcription factor known to promote SMC de-differentiation28, rapamycin inhibits SMC de-differentiation by targeting mTORC119. Furthermore, Liu et al. noted that while rapamycin in SMCs up-regulated TET2, a pro-differentiation epigenetic factor, siRNA knockdown of TET2 increased de-differentiating factors including KLF519. Although in their paper there is no data available for a direct effect of rapamycin on KLF5 protein levels, the stimulatory effect of rapamycin on TET2 and the inhibitory action of TET2 on KLF5 combined support a real possibility of an inhibitory effect of rapamycin on KLF5, as observed herewith. It is worth noting however, TET2 siRNA increased KLF5 mRNA but in our study TGFβ/Smad3 stimulated the production of KLF5 protein but not mRNA. It is therefore premature to propose that TGFβ/Smad3-stimulated mTOR activation enhanced KLF5 protein production via TET2 down-regulation. Rather, in our experimental setting, activated mTOR likely up-regulates KLF5 protein by a distinct mechanism, which is our next research subject.
Our finding of positive regulation of KLF5 protein by mTOR is significant especially considering that mTOR and KLF5 both are crucial regulators in SMC pathophysiology and vascular disease. mTOR is a master regulator with influence on almost every cellular activity. In complex with protein partners, this atypical kinase senses and integrates diverse environmental cues including growth factors, and couples these signals to promotion of protein translation and lipid synthesis. A critical role of mTOR in recurrent vascular disease is best demonstrated by the clinical use of rapamycin as an anti-restenotic drug (on stents) that potently blocks SMC proliferation and migration19. Moreover, as reported by Liu et al., rapamycin also inhibits SMC de-differentiation, and this function is likely related to KLF4 and KLF519.
With regard to a role in SMC de-differentiation, KLF4 has been at the center of interests as well as debates, partly because it is one of the four Yamanaka factors essential for reprogramming differentiated cells to pluripotency26. Whereas some studies show that KLF4 is a potent stimulator of SMC de-differentiation25,35, there are also reports indicating an opposite KLF4 function44. Further confounding evidence was from in vivo studies. As demonstrated by at least two independent groups, knockout or knockdown of KLF4 did not mitigate but rather exacerbated IH after arterial injury35. Unlike KLF4, KLF5 has been shown to promote SMC de-differentiation, proliferation and migration in vitro consistently by different research groups27,28. As for the mechanism, a plausible explanation is that KLF5 competes with myocardin for binding with the transcription factor SRF thus suppressing pro-differentiation genes in SMCs28. Importantly, while overexpression of KLF5 enhanced, knockdown of KLF5 reduced IH in a rat balloon angioplasty model27. Moreover, application of miR145, which down-regulated its target protein KLF5, reduced IH as well45. Taken together these solid studies and our own results, the mTOR/KLF5 axis appears to be an important player in SMC phenotype transformation, in particular, in TGFβ/Smad3-initiated SMC de-differentiation. Significantly, resveratrol provides an effective inhibitor alternative to rapamycin in curbing IH-promoting SMC pathophysiology.
In pursuit of the regulators upstream of the mTOR/KLF5 axis and downstream of TGFβ/Smad3, we focused our attention on the well-established Akt-mTOR pathway. While we previously showed that TGFβ/Smad3 stimulated Akt phosphorylation via direct protein interactions32, Brito et al. reported that resveratrol inhibited the Akt-mTOR pathway in SMCs in vitro34. It is important to note that although our results agree with Brito et al. on the resveratrol inhibition of the Akt-mTOR pathway in SMCs, our studies differ in at least three areas: (1) We focused on SMC de-differentiation which was not addressed by Brito et al. (2). Whereas Brito et al. used ox-LDL to stimulate SMC proliferation34, we used TGFβ/Smad3 to de-differentiate SMCs. (3) Most importantly, our in vitro experiments identified mTOR/KLF5 as a novel regulatory axis mediating TGFβ/Smad3-stimulated SMC de-differentiation. Nonetheless, both studies support the important role of resveratrol as an inhibitor of the Akt-mTOR pathway. Furthermore, an inhibitory effect of resveratrol on TGFβ/Smad3-stimulated Akt activation and a lack of resveratrol effect on Smad3 activation together confine the target of resveratrol to the downstream of Smad3 and upstream of Akt (Fig. 7C). Although there is evidence that resveratrol targets PI3K, it remains unclear whether resveratrol binds directly to this kinase34.
It was recently reported that at a concentration ≥25 μM, resveratrol activates AMPKα (phosphorylation at T172) and inhibits Akt in cardiac myocytes46 as well as in human20 and rat34 vascular SMCs. Moreover, Thompson et al. reported that resveratrol inhibits human SMC de-differentiation via AMPK activation20. We therefore asked the question whether in our study resveratrol influenced TGFβ/Smad3-sitmulated signaling via AMPKα phosphorylation in rat SMCs. Our Western blot assays indicate that stimulation of rat SMCs with TGFβ/Smad3 did not alter levels of phosphorylated AMPKα(T172) at either of the six time points from 15 min to 24 h (data not shown). In accordance to our observations, Brito et al. reported that ox-LDL, another stimulant, did not change phospho-AMPKα(T172) either, and they concluded that resveratrol inhibited the Akt-mTOR pathway independent of AMPK activation34. In addition, a report using fibroblasts indicates that resveratrol inhibits mTOR signaling in an AMPK- independent manner47. Taken together, our results suggest a mechanism underlying the effective resveratrol inhibition of TGFβ/Smad3-stimulated SMC de-differentiation, i.e., resveratrol abrogates KLF5 protein up-regulation by blocking the activation of the Akt-mTOR pathway, as summarized in Fig. 8.
Conclusions
Through systematic and longitudinal in vivo analysis we find that periadventitial delivery of resveratrol produces highly desirable anti-restenotic outcomes. Two weeks after balloon injury of rat carotid arteries, neointima was nearly eliminated, endothelium recovery was accelerated, and constrictive remodeling was avoided. This therapeutic effect is significant especially considering a paucity of publications reporting anti-restenotic methods (or agents) with favorable effects on mitigating all three major pro-restenotic pathologies. Furthermore, 3 months of periadventitial treatment with resveratrol delivered by a polymer sheath produced lasting suppression of neointima growth without a side effect of constrictive remodeling. Given that restenosis in human patients is a chronic process, while still rare, demonstration of long-term efficacy of endothelium-protective anti-restenotic methods is highly desirable. This is particularly true for resveratrol, a promising anti-restenotic drug but fretting researchers for its extremely low bioavailability14. Furthermore, our in vitro studies using resveratrol as a pharmacological probe identified a novel regulation; i.e., while elevated TGFβ/Smad3 signaling de-differentiate SMCs by up-regulating transcription factor KLF5, resveratrol blocks KLF5 up-regulation and re-differentiates these activated SMCs by disrupting the Akt-mTOR pathway. Inasmuch as there is no clinical method available to date for the prevention of restenosis following open surgery, periadventitial delivery of resveratrol would potentially lead to an effective paradigm to meet this urgent clinical need, in thousands of patients who are subject to frequent failure of open surgical interventions.
Additional Information
How to cite this article: Zhu, Y. et al. Restenosis Inhibition and Re-differentiation of TGFβ/Smad3-activated Smooth Muscle Cells by Resveratrol. Sci. Rep. 7, 41916; doi: 10.1038/srep41916 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Goel, S. A., Guo, L. W., Liu, B. & Kent, K. C. Mechanisms of post-intervention arterial remodelling. Cardiovascular research 96, 363–371 (2012).
Ichimoto, E. et al. Mechanism of edge restenosis after sirolimus-eluting stent implantation. J Invasive Cardiol 24, 55–57 (2012).
Inoue, T. et al. Vascular inflammation and repair: implications for re-endothelialization, restenosis, and stent thrombosis. JACC. Cardiovascular interventions 4, 1057–1066 (2011).
Holy, E. W. et al. PI3K/p110alpha inhibition selectively interferes with arterial thrombosis and neointima formation, but not re-endothelialization: potential implications for drug-eluting stent design. European heart journal 35, 808–820 (2014).
Guo, L. W. et al. Halofuginone stimulates adaptive remodeling and preserves re-endothelialization in balloon-injured rat carotid arteries. Circ Cardiovasc Interv 7, 594–601 (2014).
J, G. et al. Effects of resveratrol on endothelial progenitor cells and their contributions to reendothelialization in intima-injured rats. Journal of cardiovascular pharmacology 47, 711–721 (2006).
Orozco-Sevilla, V. et al. Epigallocatechin-3-gallate is a potent phytochemical inhibitor of intimal hyperplasia in the wire-injured carotid artery. Journal of vascular surgery 58, 1360–1365 (2013).
Choi, K. H. et al. Phosphoinositide 3-kinase is a novel target of piceatannol for inhibiting PDGF-BB-induced proliferation and migration in human aortic smooth muscle cells. Cardiovascular research 85, 836–844 (2010).
Khandelwal, A. R. et al. Resveratrol and quercetin interact to inhibit neointimal hyperplasia in mice with a carotid injury. The Journal of nutrition 142, 1487–1494 (2012).
Zhang, J. et al. Resveratrol attenuates oxidative stress induced by balloon injury in the rat carotid artery through actions on the ERK1/2 and NF-kappa B pathway. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology 31, 230–241 (2013).
Kim, J. W. et al. Inhibition of neointimal formation by trans-resveratrol: role of phosphatidyl inositol 3-kinase-dependent Nrf2 activation in heme oxygenase-1 induction. Molecular nutrition & food research 54, 1497–1505 (2010).
Smoliga, J. M., Baur, J. A. & Hausenblas, H. A. Resveratrol and health–a comprehensive review of human clinical trials. Molecular nutrition & food research 55, 1129–1141 (2011).
Cottart, C. H., Nivet-Antoine, V., Laguillier-Morizot, C. & Beaudeux, J. L. Resveratrol bioavailability and toxicity in humans. Molecular nutrition & food research 54, 7–16 (2010).
Tolva, V. et al. A successful experimental model for intimal hyperplasia prevention using a resveratrol-delivering balloon. Journal of vascular surgery 63, 788–794 (2016).
Chaudhary, M. A. et al. Periadventitial drug delivery for the prevention of intimal hyperplasia following open surgery. Journal of controlled release: official journal of the Controlled Release Society 233, 174–180 (2016).
Shi, X. et al. Periadventitial application of rapamycin-loaded nanoparticles produces sustained inhibition of vascular restenosis. PloS one 9, e89227 (2014).
Jim, J., Owens, P. L., Sanchez, L. A. & Rubin, B. G. Population-based analysis of inpatient vascular procedures and predicting future workload and implications for training. Journal of vascular surgery 55, 1394–1399, discussion 1399–1400 (2012).
Yu, X. et al. A rapamycin-releasing perivascular polymeric sheath produces highly effective inhibition of intimal hyperplasia. Journal of controlled release: official journal of the Controlled Release Society 191, 47–53 (2014).
Liu, R. et al. Ten-eleven translocation-2 (TET2) is a master regulator of smooth muscle cell plasticity. Circulation 128, 2047–2057 (2013).
Thompson, A. M., Martin, K. A. & Rzucidlo, E. M. Resveratrol induces vascular smooth muscle cell differentiation through stimulation of SirT1 and AMPK. PloS one 9, e85495 (2014).
Tsai, S. et al. TGF-beta through Smad3 signaling stimulates vascular smooth muscle cell proliferation and neointimal formation. American journal of physiology. Heart and circulatory physiology 297, H540–549 (2009).
Shi, X. et al. TGF-beta/Smad3 stimulates stem cell/developmental gene expression and vascular smooth muscle cell de-differentiation. PloS one 9, e93995 (2014).
Nemenoff, R. A. et al. SDF-1alpha induction in mature smooth muscle cells by inactivation of PTEN is a critical mediator of exacerbated injury-induced neointima formation. Arteriosclerosis, thrombosis, and vascular biology 31, 1300–1308 (2011).
Shi, X. et al. Local CXCR4 Upregulation in the Injured Arterial Wall Contributes to Intimal Hyperplasia. Stem Cells 34, 2744–2757 (2016).
Alexander, M. R. & Owens, G. K. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu Rev Physiol 74, 13–40 (2012).
Shankman, L. S. et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nature medicine 21, 628–637 (2015).
Suzuki, T. et al. Kruppel-like factor 5 shows proliferation-specific roles in vascular remodeling, direct stimulation of cell growth, and inhibition of apoptosis. The Journal of biological chemistry 284, 9549–9557 (2009).
Dong, J. T. & Chen, C. Essential role of KLF5 transcription factor in cell proliferation and differentiation and its implications for human diseases. Cellular and molecular life sciences: CMLS 66, 2691–2706 (2009).
Wang, B. et al. BET Bromodomain Blockade Mitigates Intimal Hyperplasia in Rat Carotid Arteries. EBioMedicine 2, 1650–1661 (2015).
Hao, L. et al. In-Depth Characterization and Validation of Human Urine Metabolomes Reveal Novel Metabolic Signatures of Lower Urinary Tract Symptoms. Scientific reports 6, 30869 (2016).
Hao, L., Zhong, X., Greer, T., Ye, H. & Li, L. Relative quantification of amine-containing metabolites using isobaric N, N-dimethyl leucine (DiLeu) reagents via LC-ESI-MS/MS and CE-ESI-MS/MS. The Analyst 140, 467–475 (2015).
Suwanabol, P. A. et al. TGF-beta and Smad3 modulate PI3K/Akt signaling pathway in vascular smooth muscle cells. American journal of physiology. Heart and circulatory physiology 302, H2211–2219 (2012).
DiRenzo, D. M. et al. A crosstalk between TGF-beta/Smad3 and Wnt/beta-catenin pathways promotes vascular smooth muscle cell proliferation. Cellular signalling 28, 498–505 (2016).
Brito, P. M. et al. Resveratrol inhibits the mTOR mitogenic signaling evoked by oxidized LDL in smooth muscle cells. Atherosclerosis 205, 126–134 (2009).
Yoshida, T., Kaestner, K. H. & Owens, G. K. Conditional deletion of Kruppel-like factor 4 delays downregulation of smooth muscle cell differentiation markers but accelerates neointimal formation following vascular injury. Circulation research 102, 1548–1557 (2008).
Goel, S. A. et al. High-throughput screening identifies idarubicin as a preferential inhibitor of smooth muscle versus endothelial cell proliferation. PloS one 9, e89349 (2014).
Kleinedler, J. J., Foley, J. D., Orchard, E. A. & Dugas, T. R. Novel nanocomposite stent coating releasing resveratrol and quercetin reduces neointimal hyperplasia and promotes re-endothelialization. Journal of controlled release: official journal of the Controlled Release Society 159, 27–33 (2012).
Aires, V. et al. Resveratrol metabolites inhibit human metastatic colon cancer cells progression and synergize with chemotherapeutic drugs to induce cell death. Molecular nutrition & food research 57, 1170–1181 (2013).
Yurdagul, A. Jr. et al. Resveratrol promotes endothelial cell wound healing under laminar shear stress through an estrogen receptor-alpha-dependent pathway. American journal of physiology. Heart and circulatory physiology 306, H797–806 (2014).
Baur, J. A. & Sinclair, D. A. Therapeutic potential of resveratrol: the in vivo evidence. Nature reviews. Drug discovery 5, 493–506 (2006).
Park, S. J. et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 148, 421–433 (2012).
Nwachukwu, J. C. et al. Resveratrol modulates the inflammatory response via an estrogen receptor-signal integration network. eLife 3, e02057 (2014).
Sajish, M. & Schimmel, P. A human tRNA synthetase is a potent PARP1-activating effector target for resveratrol. Nature 519, 370–373 (2015).
Li, H. X. et al. Kruppel-like factor 4 promotes differentiation by transforming growth factor-beta receptor-mediated Smad and p38 MAPK signaling in vascular smooth muscle cells. The Journal of biological chemistry 285, 17846–17856 (2010).
Cheng, Y. et al. MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circulation research 105, 158–166 (2009).
Chan, A. Y. et al. Resveratrol inhibits cardiac hypertrophy via AMP-activated protein kinase and Akt. The Journal of biological chemistry 283, 24194–24201 (2008).
Liu, M. et al. Resveratrol inhibits mTOR signaling by promoting the interaction between mTOR and DEPTOR. The Journal of biological chemistry 285, 36387–36394 (2010).
Acknowledgements
NIH grants R01HL-068673, R01HL129785, and T32 training Grant HL-110853 (to K.C.K.); NIH grants R21EB019558, R01HL093282, and T32HL110853 (to WLM); NIH grant S10RR029531 and a Vilas Distinguished Achievement Professorship with funding provided by the Wisconsin Alumni Research Foundation and University of Wisconsin-Madison School of Pharmacy (to L.L.); AHA award 16PRE30160010 (to B.W.); grant from National Natural Science Foundation of China (NSFC, No. 81471858, to J.J.K.); NIH grants R01 HL133665 and R01EY022678, and a Wisconsin Alumni Research Foundation (WARF) Accelerator award (to L.-W. G.). We thank Sarah Franco, Dr. Bo Liu, and Dr. Jue Zhang for helpful comments. This work was also supported by University of Wisconsin ICTR-Clinical and Type 1 Translational Research Pilot Program (UL1TR000427 to K.C.K.). This funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Y.Z., B.W., K.C.K., W.L.M., J.K. and L.-W.G. designed experiments. Y.Z., T.T., B.W., A.K., M.Z., B.B., G.U., Y.S., D.D., S.A.G., Y.-F.Z., C.L., D.A.R., and X.S. carried out experiments and prepared figures. Y.Z. and L.-W.G. wrote the manuscript; Y.Z., X.S., L.L., W.L.M., K.C.K., J.K. and L.-W.G. critically reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhu, Y., Takayama, T., Wang, B. et al. Restenosis Inhibition and Re-differentiation of TGFβ/Smad3-activated Smooth Muscle Cells by Resveratrol. Sci Rep 7, 41916 (2017). https://doi.org/10.1038/srep41916
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep41916
This article is cited by
-
Resveratrol as an Anti-inflammatory Agent in Coronary Artery Disease: A Systematic Review, Meta-Analysis and Meta-Regression
Chinese Journal of Integrative Medicine (2024)
-
Hyperinsulinemia-induced KLF5 mediates endothelial angiogenic dysfunction in diabetic endothelial cells
Journal of Molecular Histology (2019)
-
Analysis of Combined Transcriptomes Identifies Gene Modules that Differentially Respond to Pathogenic Stimulation of Vascular Smooth Muscle and Endothelial Cells
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
