
Abstract
Endozoicomonas bacteria are globally distributed and often abundantly associated with diverse marine hosts including reef-building corals, yet their function remains unknown. In this study we generated novel Endozoicomonas genomes from single cells and metagenomes obtained directly from the corals Stylophora pistillata, Pocillopora verrucosa, and Acropora humilis. We then compared these culture-independent genomes to existing genomes of bacterial isolates acquired from a sponge, sea slug, and coral to examine the functional landscape of this enigmatic genus. Sequencing and analysis of single cells and metagenomes resulted in four novel genomes with 60–76% and 81–90% genome completeness, respectively. These data also confirmed that Endozoicomonas genomes are large and are not streamlined for an obligate endosymbiotic lifestyle, implying that they have free-living stages. All genomes show an enrichment of genes associated with carbon sugar transport and utilization and protein secretion, potentially indicating that Endozoicomonas contribute to the cycling of carbohydrates and the provision of proteins to their respective hosts. Importantly, besides these commonalities, the genomes showed evidence for differential functional specificity and diversification, including genes for the production of amino acids. Given this metabolic diversity of Endozoicomonas we propose that different genotypes play disparate roles and have diversified in concert with their hosts.
Similar content being viewed by others


Introduction
Many multi-cellular organisms rely on a diverse microbiome to provide important nutritional, protective and developmental functions. These include the transformation of proteins into forms digestible by the host1,2, synthesis of essential vitamins, minerals or amino acids3,4, priming of the host immune system5,6, xenobiotic degradation7,8, and protection against pathogens9,10. In higher order vertebrates, such as humans, the microbiome fulfilling these niches is extremely complex and consists of thousands of species and functions, forming an intricate web of interactions11. Invertebrates can also form complex symbioses with many microbial partners that provide critical functions for the host. For example, the Hawaiian bobtail squid, Euprymna scolopes, uses bioluminescence as a predator-avoidance mechanism through colonization of its light organ by the bacterium, Vibrio fischeri12. In several beetle species, gut microbes are used to detoxify harmful substances, such as caffeine, or to aid in the digestion of nutrient-poor tissue, thereby contributing to adaptive divergence and niche expansion1,8. In an example of the protective ability of the microbiome, symbionts of the nematode worm, Caenorhabditis elegans, have rapidly evolved mechanisms to protect the host against attacks from invading pathogenic bacteria10. The overarching picture that emerges from these and other studies is that animals (and plants) are considered holobionts or metaorganisms that live in close association with a species-specific and diverse microbiome13.
Despite these advances in our understanding of the importance of bacterial symbionts to hosts, the function of the overwhelming majority of identified bacteria remains to be determined. One globally distributed group of symbiotic bacteria without a known function are from the genus Endozoicomonas (Gammaproteobacteria; Oceanospirillales; see Neave et al. for review14). These bacteria associate with a wide variety of marine hosts, including corals15,16,17,18,19, and other cnidarians20,21, sponges22,23, gorgonians24,25,26, molluscs27,28, worms29, fish30,31, and tunicates32,33. Despite these associations with numerous hosts in oceans worldwide, the functional role of Endozoicomonas remains unclear. Dimethylsulfoniopropionate (DMSP) breakdown has been suggested as a potential role26,34, however, sequenced Endozoicomonas genomes lack DSMP metabolic pathways35. Endozoicomonas may also participate in a nutritional symbiosis, where the bacteria produce extracellular enzymes to degrade complex organic carbon sources that can then be used by the host25, as occurs with Oceanospirillales bacteria and deep-sea Osedax worms2. Another possibility is that Endozoicomonas interact with the algal symbiont Symbiodinium, either in a mutualistic or antagonistic relationship36,37, although Endozoicomonas are also commonly found in organisms without photosymbionts38. Endozoicomonas may also produce antimicrobial compounds to deter invading pathogenic microbes39, which has been seen for other coral-associated bacteria40. In contrast to these beneficial scenarios, the only observations of Endozoicomonas with marine vertebrates have been with diseased fish in aquaculture facilities. For example, E. elysicola formed cysts on the gills of cobia, Rachycentrum canadum, causing epitheliocystis and mass mortalities31. Moreover, a novel species of Endozoicomonas was responsible for epitheliocystis in the sharpsnout bream, Diplodus puntazzo30. These opposing functions suggest that Endozoicomonas have multiple roles in their many hosts, and members from this genus may opportunistically transition through different symbiotic relationships, i.e., mutualistic, commensalistic, and parasitic.
Despite the abundance of Endozoicomonas symbionts, only three complete Endozoicomonas genomes are publically available, including E. elysicola, E. montiporae, and E. numazuensis, isolated from a sea slug, coral, and a sponge, respectively35,41, therefore providing a limited understanding of their functional gene repertoire. The relatively slow pace of Endozoicomonas genome sequencing may be attributed to the difficulty in obtaining cultured isolates from host tissue. Here we used culture-independent methods of genome sequencing, including metagenomic binning and single cell genomics, to obtain a further four Endozoicomonas genomes from the reef-building corals Stylophora pistillata, Pocillopora verrucosa, and Acropora humilis. Comparative genomics was subsequently used to collectively interrogate the seven available genomes in order to better understand their shared and distinct functional characteristics. We found that the Endozoicomonas genomes were enriched for genes associated with transporter activity, particularly carbon sugar transport, as well as cell secretion and transposase activity, suggesting that Endozoicomonas have a potential role in the upcycling of carbohydrates or the supply of proteins to the host. The enrichment in transposase activity may help Endozoicomonas to quickly adapt to a new host or take advantage of a new niche. Apart from these commonalities, we also determined the set of taxon-specific genes. Functional enrichment of these species-specific gene sets indicates niche specialization of different Endozoicomonas genotypes. This is the first study to comparatively analyse Endozoicomonas genomes and provides important functional insight into this enigmatic genus.
Results
Genome sequencing and assembly
Metagenomic binning was used to obtain 81.0% of the Endozoicomonas genome from Acropora humilis and 89.7% of the Endozoicomonas genome from Pocillopora verrucosa, with low contamination levels for both genomes (Supp. Fig. 1; Supp. Table 1). The genome from P. verrucosa in a number of cases contained two copies of expected single copy genes (Supp. Fig. 1; heterogeneity = 2), which was caused by the presence of two Endozoicomonas strains that were unable to be separated during the binning process. Difficulties in separating closely related strains is often encountered using metagenomic binning42, and for this reason, we restricted our analyses to functional gene content rather than genome size or synteny comparisons to avoid confounding the results.
Using single cell genomics, two distinct strains of Endozoicomonas cells were also recovered from the coral Stylophora pistillata, designated here as “Type A” and “Type B”. In this case, however, the extraction of single bacterial cells allowed for the two Endozoicomonas strains to be sequenced independently. By sequencing and co-assembling 10 identical cells of Type A, 60.2% of the genome was recovered with very little contamination. For Type B, three identical cells were co-assembled, recovering 75.9% of the genome with low contamination (Supp. Fig. 1).
Several limitations to the techniques employed here were experienced, as is commonly encountered, including incomplete genome recoveries, difficulties in separating closely related strains and relatively fragmented genome bins (Supp. Fig. 1; Table 1). For these reasons, our analysis focused on core gene sets or techniques using relative measures rather than absolute (e.g., percent of genes coding for functions, rather than number of genes), thereby minimising the influence of these inherent issues.
Endozoicomonas core genome phylogeny
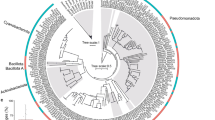
A “core” and “accessory” Endozoicomonas pan-genome was calculated using all seven genomes (i.e. three that were previously available and four generated in this study) to show regions of genomic similarity and dissimilarity (Fig. 1A). The core set (n = 301) was then used to construct a super-alignment and phylogenetic tree (Fig. 1B). In some cases host phylogeny reflected symbiont phylogeny. For example, the corals P. verrucosa and S. pistillata belong to the same coral family (Pocilloporidae), and their symbionts were closely related (Fig. 1B). Moreover, the Endozoicomonas genomes obtained from the same coral species (S. pistillata Type A and Type B) were very closely related; in fact, their core amino acid sequences had an average similarity of 97.4%. Interestingly however, the Endozoicomonas genomes did not always cluster according to host phylogeny. For example, the Endozoicomonas symbiont of the coral A. humilis shared a branch with E. numazuensis, a sponge symbiont, and was not closely related to the other coral symbionts (Fig. 1B). The remaining Endozoicomonas genomes, E. elysicola, a sea slug symbiont, and E. montiporae, a coral symbiont, did not align closely with any of the other genomes (Fig. 1B).

Endozoicomonas pan-genome showing (A) core and accessory genes, and (B) phylogenetic relationship of the Endozoicomonas genomes based on core protein sequences. In (A), genes shared between genomes are indicated by overlapping segments and the outermost track indicates genome size (million base pairs). In (B), the scale bar indicates the mean number of substitutions per site and confidence from 1000 bootstrap replicates are shown on the branches.
Molecule transport and genetic recombination are enriched in Endozoicomonas genomes
To determine the functional signatures that characterise the genus Endozoicomonas, Gene Ontology (GO) terms were compared between Endozoicomonas and other related members of the Oceanospirillales, plus more distantly related Vibrio, Wolbachia and Shewanella bacteria (Tables 2 and 3). We chose these bacterial groups because they contain relatively well-studied symbiotic bacteria and a large number of sequenced genomes. The following comparisons, however, may only be relevant for these particular bacterial groups. Many of the most enriched GO terms were associated with the generic transport of molecules, such as organic substance transport, carbohydrate transport, and single-organism transport. In addition, more than twice the number of genes involved in phosphoenolpyruvate-dependent sugar phosphotransferase (PTS; used for the uptake and phosphorylation of specific extracellular carbohydrates), were detected in Endozoicomonas compared to other Oceanospirillales bacteria (Table 3). When the genes that comprise the PTS system were examined, 62% of the specific binding components targeted lactose and cellobiose. Another enriched process in Endozoicomonas bacteria compared to other Oceanospirillales bacteria was dicarboxylic acid transport, which allows for the movement of these molecules within cells and across membranes. Possibly related to this, secretion processes, in particular protein secretion, were significantly enriched in the Endozoicomonas genomes compared to other bacteria (Table 3). Another enriched process that may be related to genome adaptability, was transposition (including DNA-mediated) and DNA recombination (Table 3).
Endozoicomonas strains show signs of functional specificity
The Endozoicomonas genomes were compared to each other using high-level functions from the RAST subsystem classification, and this corroborated that the Endozoicomonas genomes coded for similar high-level functions, although several potential strain-specific functions were detected (Fig. 2; Supp. Table 2). For example, the Endozoicomonas from the coral P. verrucosa contained more genes for cofactors, vitamins, prosthetic groups, pigments and RNA metabolism, compared to the others. Interestingly, Endozoicomonas Type B from the coral S. pistillata coded for ~50% more cofactors, vitamins, prosthetic groups, pigments than the very closely related Type A from the same coral (Fig. 2). Within this functional group, 64% of the genes were for riboflavin and folate biosynthesis. In addition, Type A had more genes for DNA metabolism, while on the other hand, the Type B strain had more genes for protein metabolism (Fig. 2; Supp. Table 2). All of the Endozoicomonas genomes devoted much of their functional repertoire to carbohydrate metabolism (~10%), however, E. elysicola, a sea slug symbiont, had a particularly high percentage (~15%; Fig. 2).

Percentage of Endozoicomonas genes annotated into high level functions within the RAST (Rapid Annotation using Subsystem Technology) subsystem classifications.
Another category containing a large number of genes was amino acids and derivatives (Figs 2 and 3). This category was examined in more detail due to the interesting possibility that the symbionts produce essential amino acids that cannot be synthesized by the host4. Strain variability was seen in the genes encoding arginine, the urea cycle, and polyamines (Fig. 3; Supp. Table 3). In particular, E. numazuensis and Endozoicomonas from A. humilis had very few genes in this category, however, all other genomes were well represented. Moreover, there were further functional divisions within this group. A number of the genomes distributed functions between arginine biosynthesis (E. elysicola (33%), E. montiporae (44%), Endozoicomonas from P. verrucosa (44%)) and degradation (E. elysicola (46%), E. montiporae (48%), Endozoicomonas from P. verrucosa (45%)). In contrast, the two genomes from S. pistillata, Types A and B, did not code any genes for arginine biosynthesis, instead encoding more than 80% of the genes for arginine degradation. Similarly, Types A and B from S. pistillata did not encode any genes for branched chain amino acids (Fig. 3; Supp. Table 3), while the other genomes in this category coded for isoleucine, leucine, and valine biosynthesis and degradation. Another interesting amino acid category was alanine, serine, and glycine. In this case, Types A and B from S. pistillata coded almost 50% more alanine and serine biosynthesis genes than the other genomes (Fig. 3).

Percentage of Endozoicomonas genes in the RAST (Rapid Annotation using Subsystem Technology) amino acids and derivatives classification.
Discussion
This study compared the genomes of Endozoicomonas associated with corals, a sponge and a sea slug obtained from isolates and cultivation-independent metagenomics and single cell sorting. The sequencing and availability of these Endozoicomonas genomes from a diverse range of hosts, environments, and ecologies provides a solid foundation for understanding the functional diversity of Endozoicomonas, and our analysis provides new insight about their genomic similarities and functional characteristics.
By comparing the phylogenetic relationships of the genomes, patterns of co-diversification between host and symbiont emerged, which has been found for other Endozoicomonas symbionts. For example, La Rivière et al. found that Endozoicomonas-like symbionts in gorgonians had similar phylogenetic relationships to their hosts43, suggesting the co-divergence of host and symbiont. Here, the related corals Stylophora pistillata and Pocillopora verrucosa had symbionts that were also related, potentially indicating co-diversification between host and symbionts. However, symbionts from the other two coral species, Acropora humilis and Montipora aequituberculata, were not closely related, suggesting that co-diversification if occurring is more complicated and may depend on other factors. For example, Neave et al. found that the brooding coral S. pistillata contained Endozoicomonas genotypes specific to well-defined geographic areas, while the spawning coral P. verrucosa shared Endozoicomonas genotypes across large geographic scales44. Accordingly, differences in the mode of symbiont transmission (i.e. horizontal or vertical) may determine if the symbiont will co-evolve with the host, and account for some of the differences observed here.
The Endozoicomonas genomes were enriched for genes involved in the transport of molecules, and genes for the secretion of proteins, when compared to other Oceanospirillales bacteria and more distantly related bacterial groups including some symbionts. This enrichment in transport and secretion may relate to the transfer of organic molecules between the symbiont and host, or alternatively, between individuals of Endozoicomonas within the cyst-like structures that they typically form30,44. Of particular interest, dicarboxylic acid transporters were enriched in the Endozoicomonas genomes, which has been seen in other symbioses, such as the well-known legume-Rhizobium symbiosis45. In this case, the plant exchanges carbon photosynthates in the form of dicarboxylic acid for fixed nitrogen in the form of ammonia, which is produced by the symbiotic bacteria46. In fact, dicarboxylic acid is the primary carbon source for these symbionts46. A similar symbiosis may be at work here between Endozoicomonas bacteria and the photosynthate-producing Symbiodinium algae. Although none of the Endozoicomonas genomes have the genes for fixing nitrogen directly, E. elysicola, E. numazuensis, and E. montiporae, all have several forms of nitrate reductases, allowing the conversion of nitrate to nitrite and the conversion of nitrite to ammonia, which could then be secreted. Indeed, nitrogen cycling is discussed as one of the key regulatory processes in coral holobiont functioning47. Alternatively, the ammonia may be further transformed by the bacteria into useful amino acids. In fact, all of the Endozoicomonas genomes contained pathways for the assimilation of ammonia through the synthesis of glutamine and glutamate. Interestingly, in symbioses between pea aphids and Buchnera bacteria, glutamine and glutamate are the only precursors required for the synthesis of all other essential amino acids by the Buchnera symbionts4,48. The Endozoicomonas genomes contained complete pathways for the synthesis of a variety of amino acids, including alanine, aspartate, cysteine, glycine, homocysteine, homoserine, leucine, lysine, methionine, serine, and threonine. The genomes differed, however, in their capacity to produce these amino acids, which may indicate strain-specific functions. Although the production of essential amino acids may be a role for Endozoicomonas symbionts, more research into each specific symbiotic system is required. First steps may include the sequencing of the host genome to determine if essential amino acid biosynthesis pathways are absent.
The Endozoicomonas genomes were also enriched for genes involved in the phosphoenolpyruvate-dependent sugar phosphotransferase (PTS) system. This system detects the nutritional requirements of the cell and regulates the phosphorylation and uptake of sugars accordingly49,50. Interestingly, the PTS system in Endozoicomonas mostly encoded for lactose and cellobiose specific subunits. Cellobiose is a basic sugar component of cellulose, which is an important constituent of plant cells, including algal cells51. This raises the interesting possibility that Endozoicomonas, which may live in symbiotic partnerships with Symbiodinium algae, consume degrading algal cells. This process may be beneficial to the host by removing unwanted algal components after cell death. Alternatively, Endozoicomonas may live parasitically on algal cells. Indeed, a previous microscopy study detected some Endozoicomonas cells in close proximity to Symbiodinium cells within a coral host44. The PTS system may also be involved in chemotaxis52 or the detection of quorum-sensing molecules53. As previously discussed, Endozoicomonas frequently form cyst-like clusters in their host30,44 and quorum sensing could provide an important communication channel between individuals. Chemotaxis for the mobile Endozoicomonas cells is also likely to be an important process, particularly for finding optimal niche microhabitats within their many hosts.
Another enriched process in the Endozoicomonas genomes was transposition (mostly DNA-mediated) and DNA recombination, which may help the species to rapidly adapt to a new host or to opportunistically transition between symbiotic lifestyles (mutualistic, commensalistic, or parasitic). A recently conducted analysis of an Endozoicomonas genome that is parasitic on the sharpsnout bream, Diplodus puntazzo, also found a high proportion of transposases, which was suggested as a mechanism for adapting to a new niche or host30. Importantly, expansion of transposases in the genome, particularly insertion sequences, is thought to be an early step in the transition of a free-living bacterium to a host-adapted lifestyle54. For example, the arthropod and nematode endosymbiont, Wolbachia, has a significantly reduced genome size with a high proportion of non-functional insertion elements55. Almost a quarter (23%) of genes in the obligate intracellular symbiont, Amoebophilus asiaticus, code for transposase genes, indicating genome degradation and adaption to its new host56. Transposases may also help symbionts by allowing the rapid evolution of mechanisms to avoid host immune responses57. Although the Endozoicomonas genomes are enriched for transposase elements, the genomes are also relatively large (about 2.8 Mbs and up to 6.3 Mbs; Table 1), suggesting that they are not undergoing streamlining. It’s possible that Endozoicomonas strains have a free-living stage, perhaps when moving between hosts, which requires the maintenance of a complete gene repertoire. Different Endozoicomonas strains are also likely to have different lifestyles, which could also influence genome structure and restructuring.
In several instances the Endozoicomonas species showed signs of functional specificity. For example, the species often differed in their ability to produce certain amino acids, which may relate to what can be consumed from the host, or which amino acids are required by the host. A particularly interesting example of functional specificity was seen in the two Endozoicomonas genotypes isolated from the same coral (Stylophora pistillata, Types A and B). These two genotypes were very closely related based on their core genome similarity (Fig. 1B), suggesting a recent speciation event. In fact, studies using traditional 3% OTU clustering of the SSU rRNA gene would be unlikely to differentiate these two strains. Nevertheless, the Type A genotype had more genes for DNA metabolism, while Type B had more genes for protein metabolism, possibly indicating niche partitioning within the coral holobiont. Moreover, Type B was enriched for the production of riboflavin and folic acid, two important B vitamins. This production of B vitamins has been seen in other relationships between corals and bacteria and may be an important process for healthy coral functioning58,59. These functional variations could indicate that the genotypes occupy two different niches within the coral, or alternatively, one genotype may be replacing the other due to the natural selection of beneficial functions. Multiple genotypes of Endozoicomonas are often detected within individual hosts, particularly in corals44. This seemingly frequent divergence of Endozoicomonas genotypes may be facilitated by the high proportion of transposases in the genomes, as discussed above.
The Endozoicomonas genomes were obtained using metagenomic binning and single cell genomics techniques due to difficulties in obtaining cultured isolates, and several advantages and shortcomings associated with the techniques were experienced. Metagenomic binning is cost effective as there are few laboratory-processing steps, which may allow more genomes to be obtained. On the other hand, the in silico binning process is only becoming established, and still requires time investment and bioinformatics training. Moreover, the binning process is complicated by the presence of closely related genotypes or abundant DNA from other organisms, such as the coral and Symbiodinium here, although this may be overcome with the development of new bioinformatics pipelines60,61,62. In this regard, a major advantage of single cell genomics is the ability to confidently isolate and sequence the genome of interest, including genomes from closely related strains. Conversely, single cell genomics can be expensive due to the specialized procedures, and isolated single cells require amplification of their DNA before sequencing (typically using multiple displacement amplification (MDA)), which can lead to amplification bias and problems with genome assembly. We experienced several of these issues, including genome incompleteness, heterogeneity, and uneven genome amplification (due to MDA) that may have non-randomly biased our genome comparison results. Thus, important genes or functions may have been missed in the incomplete Endozoicomonas genomes. Nevertheless, we believe that many of these issues were mitigated by the analysis of relative gene set abundances and by comparisons between all seven Endozoicomonas genomes with other bacterial genome sequences. Although the techniques used here are valuable for obtaining genomic information, they do not explore the complex dynamics of Endozoicomonas bacteria in situ. Future studies may use techniques such as single cell RNA-Seq63 or secondary ion mass spectrometry (SIMS)64 to refine our understanding of Endozoicomonas symbiotic relationships and their functional role within the microbiome (see Neave et al. for further discussion14).
Conclusions
Endozoicomonas bacteria frequently associate with a diverse variety of marine hosts in oceans worldwide. Despite this ubiquity, the specific functional role of Endozoicomonas symbionts is unknown. Here we used metagenomic binning and single cell genomics to increase the number of available Endozoicomonas genomes. Comparative analysis revealed that Endozoicomonas genomes are enriched for transport and secretion processes, which may be related to the transfer of carbohydrates, amino acids, and proteins between the symbiont and host. In addition, many of the enriched processes imply the transfer of molecules between other members of the holobiont. Moreover, the Endozoicomonas genomes encoded a large number of transposase genes that may be used to rapidly adapt to a new host or niche. Importantly, Endozoicomonas species show signs of functional specificity, in particular with regard to the production of amino acids which may provide insight into specific host requirements. The large functional diversity and plasticity of Endozoicomonas genomes suggests diverse functional roles.
Methods
Culture isolate sequencing
The genomes of Endozoicomonas elysicola from the sea slug Elysia ornata65, Endozoicomonas montiporae from the coral Montipora aequituberculata66, and Endozoicomonas numazuensis from the sponge cf. Haliclona spp.23 were obtained from a previous publication35.
Coral sampling
Due to unsuccessful attempts to culture Endozoicomonas from corals, we used metagenomic binning and single cell genomics to obtain Endozoicomonas genomes in a culture-independent manner. These techniques are facilitated by high abundance of the target bacterium; therefore, we used the corals Stylophora pistillata, Pocillopora verrucosa, and Acropora humilis, which harbor high concentrations of Endozoicomonas symbionts in the Red Sea19. Samples of each coral were collected in triplicate from Al Fahal Reef, which is located on the Saudi Arabian coast (22°15.100 N, 38°57.386 E). The corals were sampled using SCUBA at depths between 2 and 10 m by removing ~5 cm2 fragments with a hammer and chisel. Fragments were placed into Whirl-Pak bags (Nasco, Salida, CA, USA) underwater, brought to the surface, placed on ice and taken to the laboratory, where they were divided into samples for metagenomics (frozen to −80 °C) and single-cell sorting (processed immediately).
Metagenomic sequencing and binning
The differential coverage binning procedure outlined by Albertsen et al. was used with minor modifications to isolate Endozoicomonas genomes from other organisms in silico42. This procedure requires a minimum of 2 metagenomes, in which the target species has different abundances to generate differential coverage profiles. This differential was achieved by sequencing an unmodified metagenome and a size-fractionated metagenome each from S. pistillata, P. verrucosa, and A. humilis. Tissue was first removed from the coral skeletons by airbrushing with cold 1× PBSE (1× phosphate buffered saline, 10 mM tri-sodium EDTA). A portion of these cells were used directly for DNA extraction to obtain the unmodified metagenome. The fractionated metagenome samples were created by vortexing the airbrushed cells for 1 min, then passing the homogenate through a 5 μm filter, and centrifuging for 15 min at 500 g67. The supernatant was collected and centrifuged for a further 20 min at 8,800 g to pellet the remaining cells, which were then resuspended in 200 μl of PBSE. The resuspension was divided into 100 μl aliquots and layered separately over 300 μl of a 26%, 22% and 15% discontinuous Nicodenz gradient (Sigma-Aldrich, St. Louis, MO, USA), before centrifugation at 21,000 g for 60 min. The top 300 μl of the suspension was expected to contain a high percentage of bacterial cells and was used for DNA extraction. Several gradients from the same colonies were required to generate sufficient DNA for sequencing. DNA was extracted from both the fractionated and unmodified samples using the DNeasy Mini Plant Kit (Qiagen Inc., Valencia, CA, USA) according to the manufacturer’s instructions. The proportion of DNA belonging to coral, Symbiodinium, and bacteria was tested using a multiplex PCR to ensure adequate recovery of bacterial DNA. The PCR was compiled using the Qiagen Mulitplex PCR kit (Valencia, CA, USA) as per the manufacturer’s instructions, with primers targeting bacterial small subunit (SSU) ribosomal RNA (rRNA) genes (27F/1492R)68, the SSU rRNA of Symbiodinium, algae (ss3Z/ss5)69, and coral mitochondria (LP16S F/R)70. Products were screened for size on a 1% agarose gel with a 1 kb ladder (Sigma-Aldrich, St. Louis, MO, USA), and samples with minimal Symbiodinium and coral contamination were used for sequencing.
Unmodified and fractionated metagenomes from the corals were sequenced using 1 lane of a 2 * 100 bp, paired-end, Illumina HiSeq run (Illumina, San Diego, CA, USA) (Supp. Table 1). Raw reads were trimmed when the quality per base dropped below 20, and Illumina adapters and reads less than 75 bps were removed using Trimmomatic v.0.3371. As per the Albertsen et al. binning procedure42, the unmodified and fractionated metagenomes were combined and assembled together using IDBA-UD v.1.1.172 with read correction enabled. To generate coverage profiles, reads from the unmodified and fractionated metagenomes were mapped separately to the combined assembly using Bowtie v.2.2.473. Tetranucleotide frequency and GC content of the assembled contigs were calculated with scripts provided by Albertsen and colleagues42. Essential single copy genes were detected with Prodigal v.2.6.274, HMMER v.3.075, and MEGAN476. Using these statistics, contigs originating from Endozoicomonas genomes were separated from other organisms in R (see Supp. Fig. 2 for example of the binning procedure and Supp. Table 1 for assembled read numbers). Often these metrics were not enough to separate the numerous coral contigs from the bacteria bins, and to increase the discriminatory power we calculated the coding region frequency per contig (expected to be high for prokaryotes, low for eukaryotes) using the earlier results from Prodigal v.2.6.274. Putative Endozoicomonas contigs were re-assembled by mapping raw reads to the contigs in Bowtie v.2.2.473, extracting any missing read pairs from the matches and assembling again with IDBA-UD v.1.1.172. A final contamination check was conducted using BLAST against NCBI’s GenBank, and contigs with identities to eukaryotes were removed. Genome completeness and contamination was determined using checkM77 and the genome assemblies were annotated using the RAST pipeline78. While this procedure yielded adequate Endozoicomonas genomes from A. humilis and P. verrucosa, it was unsuccessful in retrieving Endozoicomonas genomes with sufficient completeness from S. pistillata. For this reason, we decided to pursue single cell genomics for obtaining Endozoicomonas genomes from S. pistillata (see below).
Single cell genomics
Samples from the coral Stylophora pistillata were used for a single cell genomics procedure. Immediately after collection, tissue was airbrushed from the coral skeleton using cold PBSE. The coral slurry was divided into 1 mL aliquots, combined with 100 μl of glyTE (10 × Tris EDTA, 50% glycerol), mixed gently for 5 min at ambient temperature, and frozen in liquid nitrogen to −80 °C. Samples were then shipped on dry-ice to the Bigelow Single Cell Genomics Center (SCGC) in Boothbay, ME, USA, and sequenced as described by Stepanauskas and Sieracki79. Briefly, the homogenate was sorted using fluorescence-activated cell sorting (FACS) with the sort gate based on side scatter and SYTO-9 fluorescence, and a region was selected based on bacteria-sized particles that formed a relatively homogenous cluster (Supp. Fig. 3). It should be noted that the homogenate was relatively challenging to sort due to the high abundance of other fluorescent particles, which presumably included mitochondria, host cell debris, and other attached bacteria. The selected bacterial cells were then lysed, subjected to multiple displacement amplification (MDA), and screened using amplification of nearly full length bacterial and archaeal SSU rRNA genes followed by direct Sanger sequencing79. Of the 384 cells screened, 66 were identified as Endozoicomonas, 1 belonged to the Rhodobacteraceae, and the remaining cells did not produce high-quality sequences and therefore could not be identified. Interestingly, 2 distinct strains of Endozoicomonas were detected by SSU rRNA sequence similarity (Type A and Type B), and both were selected for whole genome sequencing. For Type A, 10 cells with identical SSU rRNA gene sequences were selected, and for Type B, 3 identical cells were selected. DNA from these cells was sequenced using 1 line of a 2 * 100 bp, paired-end, Illumina HiSeq run and raw reads were trimmed as above using Trimmomatic v.0.33 (Supp. Table 1)71. Cleaned reads from each cell type were combined and assembled using SPAdes v.3.5.080 with the single cell flag. Genome assemblies were checked for contamination using the IMG single cell pipeline81, which included BLAST similarity checks and identification of outlying contigs based on tetranucleotide frequencies. As previously, genome completeness and contamination was determined using checkM77 and the assemblies were annotated using RAST78.
Core genome analysis
The “core” Endozoicomonas genome (i.e., genes present in all genomes) was determined by clustering high quality proteins (greater than 10 amino acids in length and less than 20% stop codons) using orthoMCL82. The core gene set was extracted from the orthoMCL results using custom scripts in Python v.2.7.5. Detected core protein sequences (n = 301) were then aligned using MUSCLE v.3.8.3183 and well-aligned regions were extracted and concatenated into a super alignment with Gblocks v.0.9184. An unrooted phylogenetic tree was drawn from the super alignment using RAxML v.8.2.485 with the automatically detected best GAMMA model of rate heterogeneity. An Endozoicomonas pan-genome, showing both core and accessory genes (only present in some genomes), was drawn using Circos v.0.6986.
Endozoicomonas enrichment analysis
A gene ontology enrichment analysis87 was conducted to investigate high-level functions that characterise the genus Endozoicomonas. Functional enrichment in the Endozoicomonas genomes was tested by comparison to 19 fully sequenced genomes available in GenBank88, some of which are close relatives to the Endozoicomonas, e.g. Hahella chejuensis, and some which are more distantly related, e.g. Vibrio species (see Table 2 and Results). All genomes were downloaded and annotated with gene ontology (GO) information using InterProScan v.5.689 and enrichment analysis of the GO terms was conducted using Fisher’s exact tests in the R package topGO v.2.22.090.
Declarations
Ethics approval and consent to participate
Experimental research detailed in this study complies with institutional guidelines following KAUST Institutional Biosafety and BioEthics Committee (IBEC).
Additional Information
Data availability: Raw sequence data from this study have been deposited in the NCBI Sequence Read Archive under BioProject accession PRJNA323666. Assembled Endozoicomonas genomes are publically available in RAST (Overbeek et al. 2014) using the RAST ID#’s given in Table 1.
How to cite this article: Neave, M. J. et al. Endozoicomonas genomes reveal functional adaptation and plasticity in bacterial strains symbiotically associated with diverse marine hosts. Sci. Rep. 7, 40579; doi: 10.1038/srep40579 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
06 March 2017
The HTML version of this Article was updated shortly after publication, following a technical error that resulted in the omission of the Data Availability section. This has now been corrected in the HTML version of the paper; the PDF version was correct from the time of publication.
References
Scully, E. D. et al. Functional genomics and microbiome profiling of the Asian longhorned beetle (Anoplophora glabripennis) reveal insights into the digestive physiology and nutritional ecology of wood feeding beetles. BMC Genomics 15, 1–21 (2014).
Goffredi, S. K. et al. Genomic versatility and functional variation between two dominant heterotrophic symbionts of deep-sea Osedax worms. ISME J. 8, 908–924 (2014).
Ghanbari, M., Kneifel, W. & Domig, K. J. A new view of the fish gut microbiome: Advances from next-generation sequencing. Aquaculture 448, 464–475 (2015).
Price, D. R. G. et al. Aphid amino acid transporter regulates glutamine supply to intracellular bacterial symbionts. Proc. Natl. Acad. Sci. USA 111, 320–5 (2014).
Futo, M., Armitage, S. A. O. & Kurtz, J. Microbiota plays a role in oral immune priming in Tribolium castaneum . Front. Microbiol. 6, 1383 (2015).
Nellore, A. & Fishman, J. A. The microbiome, systemic immune function, and allotransplantation. Clin. Microbiol. Rev. 29, 191–199 (2016).
Cheng, X.-Y. et al. Metagenomic analysis of the pinewood nematode microbiome reveals a symbiotic relationship critical for xenobiotics degradation. Sci. Rep. 3, 1869 (2013).
Ceja-Navarro, J. A. et al. Gut microbiota mediate caffeine detoxification in the primary insect pest of coffee. Nat. Commun. 6, 7618 (2015).
Miljkovic, M. et al. AggLb Is the largest cell-aggregation factor from Lactobacillus paracasei subsp. paracasei BGNJ1-64, functions in collagen adhesion, and pathogen exclusion in vitro . PLoS One 10, e0126387 (2015).
King, K. C. et al. Rapid evolution of microbe-mediated protection against pathogens in a worm host. ISME J doi: 10.1038/ismej.2015.259 (2016).
Lozupone, C. A., Stombaugh, J. I., Gordon, J. I., Jansson, J. K. & Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 489, 220–230 (2012).
Brooks, J. F. et al. Global discovery of colonization determinants in the squid symbiont Vibrio fischeri . Proc. Natl. Acad. Sci. 111, 17284–17289 (2014).
McFall-Ngai, M. et al. Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. 110, 3229–3236 (2013).
Neave, M. J., Apprill, A., Ferrier-Pagès, C. & Voolstra, C. R. Diversity and function of prevalent symbiotic marine bacteria in the genus Endozoicomonas . Appl. Microbiol. Biotechnol. 100, 8315–8324 (2016).
Morrow, K. M. et al. Natural volcanic CO2 seeps reveal future trajectories for host-microbial associations in corals and sponges. ISME J. 9, 894–908 (2015).
Apprill, A., Hughen, K. & Mincer, T. Major similarities in the bacterial communities associated with lesioned and healthy Fungiidae corals. Environ. Microbiol. 15, 2063–72 (2013).
Ziegler, M. et al. Coral microbial community dynamics in response to anthropogenic impacts near a major city in the central Red Sea. Mar. Pollut. Bull. 105, 629–640 (2016).
Jessen, C. et al. In-situ Effects of eutrophication and overfishing on physiology and bacterial diversity of the Red Sea coral Acropora hemprichii . PLoS One 8, e62091 (2013).
Bayer, T. et al. The microbiome of the Red Sea coral Stylophora pistillata is dominated by tissue-associated Endozoicomonas bacteria. Appl. Environ. Microbiol. 79, 4759–4762 (2013).
Doepke, H., Herrmann, K. & Schuett, C. Endobacteria in the tentacles of selected cnidarian species and in the cerata of their nudibranch predators. Helgol. Mar. Res. 66, 43–50 (2012).
Cleary, D. F. R., Becking, L. E., Polónia, A. R. M., Freitas, R. M. & Gomes, N. C. M. Jellyfish-associated bacterial communities and bacterioplankton in Indonesian Marine lakes. FEMS Microbiol. Ecol. doi: 10.1093/femsec/fiw064 (2016).
Fiore, C. L., Labrie, M., Jarett, J. K. & Lesser, M. P. Transcriptional activity of the giant barrel sponge, Xestospongia muta Holobiont: molecular evidence for metabolic interchange. Front. Microbiol. 6, 364 (2015).
Nishijima, M., Adachi, K., Katsuta, A., Shizuri, Y. & Yamasato, K. Endozoicomonas numazuensis sp. nov., a gammaproteobacterium isolated from marine sponges, and emended description of the genus Endozoicomonas Kurahashi and Yokota 2007. Int. J. Syst. Evol. Microbiol. 63, 709–14 (2013).
Bayer, T. et al. Bacteria of the genus Endozoicomonas dominate the microbiome of the Mediterranean gorgonian coral Eunicella cavolini . Mar. Ecol. Prog. Ser. 479, 75–84 (2013).
La Rivière, M., Roumagnac, M., Garrabou, J. & Bally, M. Transient shifts in bacterial communities associated with the temperate gorgonian Paramuricea clavata in the Northwestern Mediterranean Sea. PLoS One 8, e57385 (2013).
Ransome, E., Rowley, S. J., Thomas, S., Tait, K. & Munn, C. B. Disturbance to conserved bacterial communities in the cold water gorgonian coral Eunicella verrucosa . FEMS Microbiol. Ecol. 90, 404–416 (2014).
Beinart, R., Nyholm, S., Dubilier, N. & Girguis, P. Intracellular Oceanospirillales inhabit the gills of the hydrothermal vent snail Alviniconcha with chemosynthetic, γ-proteobacterial symbionts. Environ. Microbiol. Rep. 6, 656–664 (2014).
Roterman, Y. R., Benayahu, Y., Reshef, L. & Gophna, U. The gill microbiota of invasive and indigenous Spondylus oysters from the Mediterranean Sea and northern Red Sea. Environ. Microbiol. Rep. 7, 860–867 (2015).
Forget, N. L. & Juniper, K. S. Free-living bacterial communities associated with tubeworm (Ridgeia piscesae) aggregations in contrasting diffuse flow hydrothermal vent habitats at the Main Endeavour Field, Juan de Fuca Ridge. Microbiologyopen 2, 259–75 (2013).
Katharios, P. et al. Environmental marine pathogen isolation using mesocosm culture of sharpsnout seabream: striking genomic and morphological features of novel Endozoicomonas sp. Sci. Rep. 5, 17609 (2015).
Mendoza, M. et al. A novel agent (Endozoicomonas elysicola) responsible for epitheliocystis in cobia Rachycentrum canadum larvae. Dis. Aquat. Organ. 106, 31–7 (2013).
Dishaw, L. J. et al. The gut of geographically disparate Ciona intestinalis harbors a core microbiota. PLoS One 9, e93386 (2014).
Cahill, P. L., Fidler, A. E., Hopkins, G. A. & Wood, S. A. Geographically conserved microbiomes of four temperate water tunicates. Environ. Microbiol. Rep. doi: 10.1111/1758-2229.12391 (2016).
Pike, R. E., Haltli, B. & Kerr, R. G. Endozoicomonas euniceicola sp. nov. and Endozoicomonas gorgoniicola sp. nov., bacteria isolated from the octocorals, Eunicea fusca and Plexaura sp. Int. J. Syst. Evol. Microbiol. 63, 4294–4302 (2013).
Neave, M. J., Michell, C. T., Apprill, A. & Voolstra, R. Whole-genome sequences of three symbiotic Endozoicomonas strains. Genome Announc. 2, e00802–14 (2014).
Morrow, K. M., Moss, A. G., Chadwick, N. E. & Liles, M. R. Bacterial associates of two Caribbean coral species reveal species-specific distribution and geographic variability. Appl. Environ. Microbiol. 78, 6438–49 (2012).
Pantos, O., Bongaerts, P., Dennis, P. G., Tyson, G. W. & Hoegh-Guldberg, O. Habitat-specific environmental conditions primarily control the microbiomes of the coral Seriatopora hystrix . ISME J. 9, 1916–1927 (2015).
Bourne, D. G. et al. Coral reef invertebrate microbiomes correlate with the presence of photosymbionts. ISME J. 7, 1452–1458 (2013).
Bourne, D., Iida, Y., Uthicke, S. & Smith-Keune, C. Changes in coral-associated microbial communities during a bleaching event. ISME J. 2, 350–63 (2008).
Ritchie, K. B. Regulation of microbial populations by coral surface mucus and mucus-associated bacteria. Mar. Ecol. Prog. Ser. 322, 1–14 (2006).
Ding, J.-Y., Shiu, J.-H., Chen, W.-M., Chiang, Y.-R. & Tang, S.-L. Genomic insight into the host–endosymbiont relationship of Endozoicomonas montiporae CL-33(T) with its coral host. Front. Microbiol. 7, 251 (2016).
Albertsen, M. et al. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat. Biotechnol. 31, 533–8 (2013).
La Rivière, M., Garrabou, J. & Bally, M. Evidence for host specificity among dominant bacterial symbionts in temperate gorgonian corals. Coral Reefs 34, 1087–1098 (2015).
Neave, M. J. et al. Differential specificity between closely related corals and abundant Endozoicomonas endosymbionts across global scales. ISME J 11, 186–200 (2017).
Remigi, P., Zhu, J., Young, J. P. W. & Masson-Boivin, C. Symbiosis within symbiosis: evolving nitrogen-fixing legume symbionts. Trends Microbiol. 24, 63–75 (2016).
Poole, P. & Allaway, D. In Advances in Microbial Physiology 43, 117–163 (Academic Press, 2000).
Rädecker, N., Pogoreutz, C., Voolstra, C. R., Wiedenmann, J. & Wild, C. Nitrogen cycling in corals: the key to understanding holobiont functioning? Trends Microbiol. 23, 490–497 (2015).
Hansen, A. K. & Moran, N. A. Aphid genome expression reveals host-symbiont cooperation in the production of amino acids. Proc. Natl. Acad. Sci. 108, 2849–54 (2011).
Deutscher, J., Francke, C. & Postma, P. W. How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol. Mol. Biol. Rev. 70, 939–1031 (2006).
Ymele-Leki, P., Houot, L. & Watnick, P. I. Mannitol and the mannitol-specific enzyme IIB subunit activate vibrio cholerae biofilm formation. Appl. Environ. Microbiol. 79, 4675–4683 (2013).
Brady, S. K., Sreelatha, S., Feng, Y., Chundawat, S. P. S. & Lang, M. J. Cellobiohydrolase 1 from Trichoderma reesei degrades cellulose in single cellobiose steps. Nat Commun 6, 10149 (2015).
Lux, R., Jahreis, K., Bettenbrock, K., Parkinson, J. S. & Lengeler, J. W. Coupling the phosphotransferase system and the methyl-accepting chemotaxis protein-dependent chemotaxis signaling pathways of Escherichia coli . Proc. Natl. Acad. Sci. 92, 11583–11587 (1995).
Pereira, C. S. et al. Phosphoenolpyruvate phosphotransferase system regulates detection and processing of the quorum sensing signal autoinducer-2. Mol. Microbiol. 84, 93–104 (2012).
Siguier, P., Gourbeyre, E. & Chandler, M. Bacterial insertion sequences: Their genomic impact and diversity. FEMS Microbiol. Rev. 38, 865–891 (2014).
Cerveau, N., Leclercq, S., Leroy, E., Bouchon, D. & Cordaux, R. Short- and long-term evolutionary dynamics of bacterial insertion sequences: Insights from Wolbachia endosymbionts. Genome Biol. Evol. 3, 1175–1186 (2011).
Schmitz-Esser, S. et al. The genome of the amoeba symbiont ‘Candidatus Amoebophilus asiaticus’ reveals common mechanisms for host cell interaction among amoeba-associated bacteria. J. Bacteriol. 192, 1045–57 (2010).
Parkhill, J. et al. Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica . Nat Genet 35, 32–40 (2003).
Agostini, S., Suzuki, Y. & Casareto, B. Coral symbiotic complex: Hypothesis through vitamin B12 for a new evaluation. Coral Reef 11, 1–11 (2009).
Agostini, S. et al. Biological and chemical characteristics of the coral gastric cavity. Coral Reefs 31, 147–156 (2012).
Eren, A. M. et al. Anvi’o: An advanced analysis and visualization platform for ‘omics data. PeerJ 3, e1319 (2015).
Imelfort, M. et al. GroopM: an automated tool for the recovery of population genomes from related metagenomes. PeerJ 2, e603 (2014).
Alneberg, J. et al. Binning metagenomic contigs by coverage and composition. Nat Meth 11, 1144–1146 (2014).
Wang, Y. & Navin, N. E. Advances and applications of single-cell sequencing technologies. Mol. Cell 58, 598–609 (2015).
Pernice, M. & Levy, O. Novel tools integrating metabolic and gene function to study the impact of the environment on coral symbiosis. Front. Microbiol. 5, 448 (2014).
Kurahashi, M. & Yokota, A. Endozoicomonas elysicola gen. nov., sp. nov., a gamma-proteobacterium isolated from the sea slug Elysia ornata . Syst. Appl. Microbiol. 30, 202–6 (2007).
Yang, C.-S. et al. Endozoicomonas montiporae sp. nov., isolated from the encrusting pore coral Montipora aequituberculata . Int. J. Syst. Evol. Microbiol. 60, 1158–62 (2010).
Wegley, L., Edwards, R., Rodriguez-Brito, B., Liu, H. & Rohwer, F. Metagenomic analysis of the microbial community associated with the coral Porites astreoides . Environ. Microbiol. 9, 2707–19 (2007).
Sunagawa, S. et al. Bacterial diversity and White Plague Disease-associated community changes in the Caribbean coral Montastraea faveolata . ISME J. 3, 512–21 (2009).
Rowan, R. & Powers, D. Molecular genetic identification of symbiotic dinoflagellates (zooxanthellae). Mar. Ecol. Prog. Ser. 71, 65–73 (1991).
Le Goff-Vitry, M. C., Rogers, A. D. & Baglow, D. A deep-sea slant on the molecular phylogeny of the Scleractinia. Mol. Phylogenet. Evol. 30, 167–177 (2004).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Peng, Y., Leung, H. C. M., Yiu, S. M. & Chin, F. Y. L. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428 (2012).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Meth 9, 357–359 (2012).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 1–11 (2010).
Johnson, L. S., Eddy, S. R. & Portugaly, E. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics 11, 431 (2010).
Huson, D. H., Mitra, S., Ruscheweyh, H.-J., Weber, N. & Schuster, S. C. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 21, 1552–1560 (2011).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Overbeek, R. et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 42, D206–D214 (2014).
Stepanauskas, R. & Sieracki, M. E. Matching phylogeny and metabolism in the uncultured marine bacteria, one cell at a time. Proc. Natl. Acad. Sci. 104, 9052–9057 (2007).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–77 (2012).
Markowitz, V. M. et al. IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 40, D115–D122 (2012).
Li, L., Stoeckert, C. J. & Roos, D. S. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 13, 2178–2189 (2003).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 17, 540–552 (2000).
Stamatakis, A. RAxML Version 8: A tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 30, 1312–1313 (2014).
Krzywinski, M. I. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–45 (2009).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nat Genet 25, 25–29 (2000).
Benson, D. A. et al. GenBank. Nucleic Acids Res. 41, D36–D42 (2013).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Alexa, A. & Rahnenfuhrer, J. topGO: Enrichment analysis for Gene Ontology. Bioconductor (2010).
Acknowledgements
We thank the Bigelow Single Cell Genomics Center, Maine, USA, for assistance with the single cell genomics procedure. This research was supported by a KAUST-WHOI Post-doctoral Partnership Award to M.J.N. and a KAUST-WHOI Special Academic Partnership Funding Reserve Award to C.R.V. and A.A. Additional research was supported from baseline funds to C.R.V. by the King Abdullah University of Science and Technology (KAUST).
Author information
Authors and Affiliations
Contributions
M.J.N., A.A., and C.R.V. designed and conceived the study; M.J.N. and C.T.M. generated data; M.J.N., A.A., and C.R.V. analysed and interpreted data; M.J.N., A.A., and C.R.V. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Neave, M., Michell, C., Apprill, A. et al. Endozoicomonas genomes reveal functional adaptation and plasticity in bacterial strains symbiotically associated with diverse marine hosts. Sci Rep 7, 40579 (2017). https://doi.org/10.1038/srep40579
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep40579
This article is cited by
-
Coral microbiomes are structured by environmental gradients in deep waters
Environmental Microbiome (2024)
-
The coral microbiome in sickness, in health and in a changing world
Nature Reviews Microbiology (2024)
-
Probiotics reshape the coral microbiome in situ without detectable off-target effects in the surrounding environment
Communications Biology (2024)
-
A coral-associated actinobacterium mitigates coral bleaching under heat stress
Environmental Microbiome (2023)
-
Widespread occurrence of chitinase-encoding genes suggests the Endozoicomonadaceae family as a key player in chitin processing in the marine benthos
ISME Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
