
Abstract
The liver is the central metabolic organ and exhibits fundamental functions in haematological traits. Hepatic expression, haematological, plasma biochemical, and body composition traits were assessed in a porcine model (n = 297) to establish tissue-specific genetic variations that influence the function of immune-metabolism-correlated expression networks. At FDR (false discovery rate) <1%, more than 3,600 transcripts were jointly correlated (r = |0.22–0.48|) with the traits. Functional enrichment analysis demonstrated common links of metabolic and immune traits. To understand how immune and metabolic traits are affected via genetic regulation of gene expression, eQTLs were assessed. 20517 significant (FDR < 5%) eQTLs for 1401 transcripts were identified, among which 443 transcripts were associated with at least one of the examined traits and had cis-eQTL (such as ACO1 (6.52 × 10−7) and SOD1 (6.41 × 10−30). The present study establishes a comprehensive view of hepatic gene activity which links together metabolic and immune traits in a porcine model for medical research.
Similar content being viewed by others

Introduction
The liver is of particular interest given its vital roles in maintaining homeostasis and health and regulating nutrient utilization. Accordingly, insights into the regulation of liver expression profiles potentially have implications for traits associated with physical and metabolic integrity. Traits related to fatness and body composition are important not only as economic factors in pork production but also because of their association with serious diseases in humans1,2. Pigs share many similarities with humans in terms of their physiology and genome and therefore provide a good model for medical research including studies on the transplantation of organs3.
Accordingly, we used expression analyses in the liver to identify genes associated with plasma haematological and biochemical traits that might serve as biomarkers of “liver functioning” and as surrogate traits for immune and metabolic status. However, variations in complex traits depend not only on the expression profile of single hepatic genes but also on networks of genes and transcripts. Therefore, we further performed a weighted gene co-expression network analysis (WGCNA) to systematically assess the pathways in which co-expressed genes are interconnected. Thus, we obtained a more precise set of parameters that might serve as either key biomarkers of acute metabolic and immune status mediated through hepatic functions, or as predictors of genetically determined metabolic and immune capabilities.
Expression-QTL (eQTL) analysis integrates gene expression levels and genome-wide genotyping information to identify genetic variations associated with changes in gene expression. Variations in complex traits largely reflect polymorphisms affecting regulatory sequences, rather than coding sequences. Knowledge of the position of the analyzed genes and markers enables the differentiation of cis- and trans-eQTLs4. We have previously shown that the detection of trait-dependent expressed genes in a relevant tissue facilitates the identification of genes associated with complex traits, such as muscle and meat properties and coping behaviors; these genes represent strong candidate genes5,6. Studies of eQTLs in the human liver have led to the identification of genes associated with clinical phenotypes and provide a foundation for pharmacogenomics7.
Given the central role of the liver in controlling the homeostasis of the metabolic and immune systems, body composition and all fat traits, we hypothesized that the variation of these traits might largely reflect genes and metabolic pathways active in the liver. Herein, we characterized hepatic transcription profiles and the genetic regulation of their expression in association with immune and metabolic traits in pigs. The analyses of trait-correlated hepatic expression, co-expression interactions and eQTL-detection provide a common link for haematological, biochemical, and clinical-chemical biomarkers of hepatic functions contributing to metabolic homeostasis, innate defense and resilience. This knowledge provides a rational basis not only for understanding pig physiology but also for pig models used in human medical research.
Results
Hepatic trait-correlated expression
The expression levels of 24904 liver transcripts in 297 individuals were correlated with biomarkers of immune and metabolic status, including haematological, biochemical and body composition traits. All measurements and descriptions of these traits are shown in Supplementary Table 1. Haematological traits refer to three components: leukocytes (white blood cell count: WBC, lymphocyte count: LYM), erythrocytes (red blood cell count: RBC, hemoglobin concentration: HGB, hematocrit level: HCT, mean corpuscular volume: MCV, mean corpuscular volume: MCH, mean corpuscular hemoglobin concentration: MCHC, red distribution width: RDW), and platelets (platelets: PLT, mean platelet volume: MPV, plateletcrit: PCT), and the respective cell counts are markers of immune and/or inflammatory responses8,9. At a significance threshold of an FDR < 1%, we detected 5387 transcripts showing correlations with at least one of the 12 examined haematological traits. The correlation between expression levels and haematological traits ranged from |0.22–0.48|. Most transcripts were correlated with leukocytes and erythrocytes, and a few transcripts were associated with platelets.
To further refine the functional annotation of the sets of genes showing trait-correlated expression, their assignment to canonical pathways was explored using the Ingenuity Pathway Knowledge Base. Biological functions associated with trait-correlated expression are displayed in Fig. 1A and Supplementary Table 2A. Acute phase response signaling and hepatic fibrosis/hepatic stellate cell activation were correlated with LYM and WBC. Most of the erythrocyte-related traits were correlated with oxidative phosphorylation, mitochondrial dysfunction and Ephrin A signaling. The coagulation system and complement system were correlated with platelet-related traits.

Canonical pathways of transcripts correlated with (A) haematological traits, (B) biochemical traits, and (C) body composition phenotype. Heatmap displaying the correlation of transcripts related to canonical pathways with phenotypes; intensity of color indicates significance from light to dark.
The phenotypes of 8 biochemical traits (albumin: ALB, ammonia nitrogen: NH3, blood urea nitrogen: BUN, total cholesterol: TCHO, triglyceride: TG, glucose: GLU, inorganic phosphorus: IP, creatinine: CREA) were used to examine correlations with the expression profiles of the liver. At a significance level of an FDR < 1%, 6321 transcripts were correlated with a minimum of one biochemical trait at r = |0.22–0.41|. Canonical pathways such as acute phase response signaling showed the closest association with CREA, TCHO and BUN. TCHO-, TG- and GLU-correlated genes were associated with common canonical pathways of PXR/RXR activation, FXR/RXR activation and TR/RXR activation (Fig. 1B; Supplementary Table 2B).
Body composition traits were examined to characterize metabolic end product phenotypes. In total, 11 traits, including 6 fat traits (fat depth at shoulder: FDS, fat depth at tenth rib: FDTR, loin fat depth at loin: FDL, average back fat: ABF, fat area: FA, Intramuscular fat content: MLDIMF), 3 muscle traits (loin eye area: LEA, protein content: MLDP, muscle to fat ratio: MFR), body weight (BW) and body length (BL), were used. At a significance level of an FDR < 1%, 2571 transcripts were correlated with at least one body composition trait at r = |0.22–0.44|. The list of transcripts correlated with fat traits was associated with the canonical pathways of LPS/IL-1-mediated inhibition of RXR function, TR/RXR activation, LXR/RXR activation, PXR/RXR activation and xenobiotic metabolism signaling. For the muscle traits, the list of correlated transcripts was enriched for IGF-1 signaling, alanine biosynthesis II, alanine degradation III, phenylalanine degradation I and glucocorticoid receptor signaling (Fig. 1C; Supplementary Table 2C).
Altogether, 10064 out of 24904 transcripts were correlated with at least one of the surrogate traits of immune and metabolic status, with considerable overlap among the groups of traits (Fig. 2A). In fact, 2661 transcripts were common between haematological and biochemical traits, and 1282 transcripts were common to biochemical and body composition phenotype traits, while 795 transcripts were shared between haematological and body composition phenotype traits, and 528 transcripts were common to all immune and metabolic status traits. The numbers of transcripts that were only associated with haematological, biochemical and body composition traits were 2459, 2906 and 1022, respectively.

Venn diagrams displaying numbers of trait-correlated transcripts.
(A) Number of liver transcripts correlated with haematological and biochemical traits as well as body composition traits. (B) Number of liver transcripts correlated with haematological and biochemical traits as well as body composition traits with cis eQTL effect.
Hepatic co-expression and trait-associated co-expression modules
We explored transcriptional changes not only at the level of individual genes but also in terms of gene interactions. Thus, a WGCNA was performed using the transcriptome data from 24904 liver transcripts. Five modules were highly correlated with phenotypes, as shown in Fig. 3. The co-expressed transcripts in each module were assigned to canonical pathways. The magenta module was highly correlated with leucocyte counts, whereas the green module was highly correlated with erythrocyte counts. The top highly connected hub genes in the magenta module were SOCS3, LOC100154449, JUNB, BTG2 and IL4R. The transcripts in the magenta module were associated with acute phase response signaling, whereas the transcripts in the green module were core components of the mitochondrial oxidative phosphorylation complexes encoded by the mitochondrial genome (Fig. 4). The highly connected hub genes in the green module were MT-ATP6, MT-ND4L, MT-CO3, MT-ATP8 and MT-ND5. Platelet counts were modestly correlated with the red module, which was enriched for genes belonging to HIPPO signaling. The highly connected hub genes in the red module were LOC102159151, LOC102159016, CD86, LOC100511343 and MAB21L3. Body composition traits associated with muscle mass were correlated with the purple module, whereas fat traits were more correlated with the cyan module. The 85 transcripts in the purple module were enriched in PXR/RXR activation, in contrast with the 65 transcripts in the cyan module, which encoded many genes associated with cholesterol biosynthesis (Fig. 4). The most connected hub genes in the cyan module were ACACA, ACSS2, EBP, GPAM, GPAT and THRSP. Biochemical traits such as ALB, GLU and TG were correlated with the purple module. PPP1R3C, G6PC, PPP1R3B, SLC25A25, SGK1 were the most connected genes in the purple module.

Correlation matrix of module eigengene values obtained for mRNAs and phenotypes.
Weighted gene co-expression network analysis (WGCNA) groups genes into modules based on patterns of gene co-expression. Each of the modules was labelled with a unique color as an identifier. 5 modules show highly significant correlation with body composition, biochemical and haematological traits. Within each cell, upper values are correlation coefficients between module eigengenes and the traits; lower values are the corresponding p-values.

Canonical pathways of the modules whose eigenvalues correlated with the surrogate traits.
Heatmap displaying the correlation of modules of trait-correlated and co-expressed transcripts with canonical pathways; intensity of color indicates significance from light to dark.
Hepatic eQTLs
A numerical summary of the whole-genome association study of gene expression levels in the liver (eQTL) is shown in Table 1. In total, 20517 significant eQTLs, corresponding to 1401 probe sets, reached the threshold of an FDR < 5% (p < 10−7). At this significance level, 11366 SNPs were associated with the expression of 1075 annotated transcripts, and 6865 eQTLs were identified as cis, belonging to 1028 probe sets (814 annotated transcripts) (Supplementary Table 3).
Hepatic cis-eQTL and plasma haematological traits
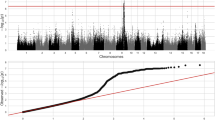
The expression levels of 341 transcripts were significantly correlated with haematological traits associated with SNPs, revealing 2,439 eQTLs. Focusing on cis regulation, 808 cis-eQTLs were identified from 219 transcripts that were significantly correlated (FDR < 1%) with one of haematological traits (Supplementary Table 4). For traits associated with red blood cells we identified 47 annotated transcripts with eQTLs showing trait-correlated expression with RBCs, including 6 for HCT, 12 for HGB, 27 for MCV, 9 for MCH, 22 for MCHC and 1 for RDW loci. Superoxide dismutase 1 (SOD1) was one of the transcripts that were highly negatively correlated with HCT and RBC and exhibited a cis-eQTL with a p-value ranging from 6.08 × 10−10 to 6.41 × 10−30 (Fig. 5A). SNP ASGA012109 located in linkage disequilibrium regions of 205–206 Mb of chromosome 13 (Fig. 5B) was highly associated with SOD1 levels (Fig. 5C). Most of the annotated transcripts (72 transcripts) were correlated with the LYM and WBC. In total, we identified 5 annotated transcripts correlated with MPV and 2 annotated transcripts correlated with PLT and PCT. All liver transcripts with cis-eQTLs whose expression levels were correlated with haematological traits are presented as a network in Fig. 6.

Genome wide association of SOD1 transcript level.
(A) Manhattans plot of cis-eQTL of SOD1 and highly associated with SNP ASGA0102109 at 205.6 Mb of pig chromosome 13 (SSC13). (B) Linkage disequilibrium regions range from 205–206 Mb of SSC13 while SOD1 star position at 205.6 Mb. (C) Genotype of ASGA0102109 loci show associated with SOD1 transcript levels.

A network of all liver hepatic transcripts with cis-eQTLs and expression levels correlated with haematological traits.
Each node represents a haematological traits (blue) connected with various transcripts (grey). Some of the transcripts are connected with more than one node.
Hepatic cis-eQTL and plasma biochemical traits
There were 447 transcripts correlated with biochemical traits at a significant level of a 1% FDR, which were also associated with 3554 eQTLs, including 1148 cis-eQTL of 289 transcripts (Supplementary Table 5). Most of the transcripts were correlated with IP (120) followed by BUN (73) and CREA (62). We identified 24 transcripts correlated with ALB, 29 transcripts correlated with GLU, 33 transcripts correlated with TG, 12 transcripts correlated with TCHO and only 4 transcripts correlated with NH3. Some transcripts were correlated with more than one biochemical trait. A corresponding network of transcripts is shown in Fig. 7.

A network of all liver hepatic transcripts with cis-eQTLs and expression levels correlated with biochemical traits.
Each node represents a biochemical traits (green) connected with various transcripts.
Hepatic cis-eQTLs and body composition
A total of 152 transcripts whose expression levels were correlated with body composition traits presented 1582 eQTLs, including 645 cis-eQTLs of 106 transcripts (Supplementary Table 6). Most of the transcripts were correlated with carcass traits (body weight (172) and length (80)) at slaughter. The number of transcripts correlated with fat/muscle traits ranged from 6–104. A network of these transcripts is shown in Fig. 8.

A network of all liver hepatic transcripts with cis-eQTLs and expression levels correlated with body composition traits.
Each node represents a body composition traits (red) connected with various transcripts (grey). Some of the transcripts are connected with more than one node.
Genetic regulation of liver transcripts contributes to common immune and metabolic complex traits
The 1028 transcripts with cis-eQTL primarily belonged to the canonical pathways of LPS/IL-1 mediated inhibition of RXR function, xenobiotic metabolism signaling and glutathione-mediated detoxification. A total of 443 of the 1028 transcripts were correlated with one of these surrogate traits of immune and metabolic status with considerable overlap among the group of traits (Fig. 2B). A total of 114 transcripts overlapped between haematological and biochemical traits, while 46 transcripts were shared between biochemical traits and the body composition phenotype, and only 24 transcripts overlapped between haematological traits and the body composition phenotype. Among the 443 transcripts, 13 were associated with the all types of traits belonging to 10 annotated genes, including TLR5, KCTD2, SLC16A1, ACTR3B, SORL1, C5orf4, ABHD14B, TMSB10, MASP2 and LIPC.
Discussion
The results of the present study provide a comprehensive dissection of the liver transcriptional landscape corresponding to clinical immune and metabolic markers. Furthermore, this is the first report of markers of immune and metabolic status and body composition correlated with hepatic mRNA expression profiles. Additionally, the co-expression of whole transcripts was analyzed using well-designed and characterized Snowball arrays10. WGCNA groups genes with similar profiles into modules and hub nodes that play important roles in many networks, and highly connected hub genes play an important role in biology and regulation as well11,12. Many hub genes identified in the present study, such as SOCS3 and JUNB in the magenta module, have been reported as a key transcriptional modulators of macrophage phagocytosis and activation13. Hub genes in the purple module such as PPP1R3B and PPP1R3C act as glycogen phosphorylases in liver glycogen metabolism14. Other hub genes in the cyan module, such as ACACA, GPAM, EPB and ACSS2, play a significant role in lipid metabolism.
We assessed whole-genome expression in the liver to establish tissue-specific genetic variations that influence the function of immune-metabolism-correlated genes. The liver is a heterogeneous tissue with different cell types; more than 80% are hepatocytes, and the minor populations are cholangiocytes, stellate cells, endothelial cells and several hematopoietic cells. Variation in transcript abundance is possibly due to either variation of cell population or the change of cell functions. However, the tissue sampling was done from an identical anatomic site of healthy mature livers with a normal cell composition. Moreover, hepatic cell composition is stable and this study addresses the link of genetic variation and transcript abundance, with the first being independent from cell type. The variations represent the causal link between SNPs, gene expression and immune-metabolic status. To determine the genetic impact on expression, we focused on cis-eQTLs by considering SNPs located in genomic windows near the transcripts. These regions include promoters, enhancers and UTRs encompassing transcription factor-binding sites and regulatory elements15,16.
Genetic regulation of liver transcripts and immune status
Erythrocytes
A potential role of erythrocytes as surrogate biomarkers of hepatic mitochondrial oxidative status in diverse oxidative conditions has been reported in an animal model17. Previous studies have reported that defects in enzymes involved in hepatic heme biosynthesis play a significant role in mitochondrial energetic metabolism, as shown in the Hmbs (−/−) mouse model, where mitochondrial respiratory chain complexes I, II and, III are significantly reduced18. Here, we demonstrated that hepatic energetic metabolism was correlated with haematological traits such as MCV, MCH and RBC.
In addition, the majority of transcripts correlated with erythrocyte traits showing significant cis-eQTL, such as Aconitase 1 (ACO1), Superoxide dismutase 1 (SOD1), and solute carrier family 19 member 2 (SLC19A2) were also related to the oxidative stress. ACO1 is a bifunctional, cytosolic protein involved in the control of iron homeostasis and the TCA cycle for energy metabolism19. In the present study, the transcript levels of ACO1 were correlated with erythrocytes traits (RBC, MCH and MCV) with highly significant cis-eQTLs. Notably, iron homeostasis plays an essential role in the oxidative status of the cell, as pathological accumulation of iron might lead to the generation of additional reactive oxygen species (ROS) and oxidative stress20,21. SOD1 is one of the most important antioxidants and has a crucial impact on the lifespan and quantity of red blood cells in peripheral blood, as shown in Sod1-deficient (knockout) mice22. Accordingly Sod1 knockout mice are unable to compensate for oxidative stress triggers, resulting in dysfunction of the proteasomal system and accelerated accumulation of damaged proteins. Mutation of the mouse Sod1 gene (knockout) decreases the number of erythrocytes and quantity of peripheral blood red blood cells22. A recent study reported that oxidative-stress triggers dysfunction of the proteasomal system and accelerates the accumulation of damaged proteins, leading to a shortened lifespan of RBCs and, hence, anemia in Sod1-deficient mice23. In the present study SOD1 was found to be highly negatively correlated with HCT and RBC and exhibited a cis-eQTL. The associated SNPs were located between 727 bp to -496 bp from the start codon of SOD1. SLC19A2 encodes a thiamine transporter protein. Thiamine also plays a role in reducing cellular oxidative stress via bridging the energy-producing glycolytic and pentose phosphate metabolic pathways, which is critical for generating ferric-reducing/antioxidant chemical-reducing power in cells24,25. Mutation of the mouse Slc19a2 gene (homozygous knockout) also decreases the number of erythrocytes in mice26. In the present study, the cis-eQTL of SLC19A2 was positively correlated with RBC and negatively correlated with LYM, consistent with a previous report. Apparently, the role of the liver in controlling the oxidative status of mitochondria (i.e. balancing metabolism induced production of ROS and the immediate detoxification of reactive intermediates) has a crucial effect on erythrocyte traits.
We identified significant associations of genetic variants with ribosomal protein gene and erythrocyte traits. RPL27A have a strong cis-eQTL (1.09 × 10−25–1.41 × 10−11) and its expression levels were found to be correlated with erythrocyte traits, such as RBC and MCV. A group of pathologies have been associated with ribosome defects, among which Diamond-Blackfan anemia (DBA) is the best studied, showing common clinical features including anemia, low reticulocyte counts, and macrocytic erythrocytes27,28,29. Hematocrit level (HCT) and RBC were shown to be highly correlated with Rho guanine nucleotide exchange factor 10 (ARHGEF10), and SNPs located within the 1 Mb windows of this gene were associated with its expression levels in the present study. The ARHGEF10 gene was significantly associated with atherothrombotic stroke, which occurs when a blood clot forms on an atherosclerotic plaque within a blood vessel and blocks blood flow30. The SNP rs4376531 affects ARHGEF10 transcriptional activity, reflecting differences in Sp1-binding affinity30. We demonstrated a relationship between erythrocytes and the genetically regulated expression levels of ARHGEF10, but the function remains elusive.
Leukocytes
A major function of the liver is to manufacture and secrete substances that are transported via the systemic circulation. Indeed, local inflammatory or injurious processes in the liver, e.g., after partial hepatectomy and during hepatic fibrosis, largely affect leukocyte-mediated activity throughout the body31. Kupffer cells, which are specialized macrophages in liver, are activated upon liver infection and injury, and leukocytes, which release pro-inflammatory cytokines such as tumor necrosis factor (TNF), and reactive oxygen32, also regenerate IL-6 and IL-10.
We identified SAA1, which encodes serum amyloid A (SAA), a major acute-phase protein primarily synthesized and secreted in the liver, as a strong cis-eQTL and demonstrated that the expression of SAA1 was correlated with LYM. SAA has been reported as a chemoattractant for phagocytes and mast cells33,34, exhibits cytokine-like properties35 and induces T lymphocyte migration and adhesion to endothelial cells36 during the acute-phase response. LBP encodes a protein involved in the acute-phase immunologic response to Gram-negative bacterial infections and is primarily induced in the liver37. A previous study indicated that mutations in the lipopolysaccharide-binding proteins impair innate immunity through reduced binding to LPS and lipopeptides and reduced cytokine responses and concentrations in pneumonia38. Here, we identified SNPs surrounding the LBP transcript that were associated with its expression levels and LBP expression levels were correlated with the LYM. Thus, not only structural variation but also the expression of LBP might play a significant role in innate immune response. SORL1, also known as LR11, encodes a mosaic protein belonging to the vacuolar protein sorting 10 (VPS10) domain-containing receptor family and the low-density lipoprotein receptor (LDLR) family. Soluble LR11 (sLR11) is released through proteolytic shedding. Circulating sLR11 is a biomarker for atherosclerosis, coronary stenosis, diabetic retinopathy and acute leukemia39,40,41,42. A role for CD9 in the shedding of LR11 in leukocytes has also been reported43. We identified a cis-eQTL for SORL1 and showed negative correlation of its expression with LYM. TNFRSF11B (tumor necrosis factor receptor superfamily member 11b) encodes a protein belonging to the TNF-receptor superfamily. Studies in a mouse counterpart also suggested that this protein and its ligand play a role in lymph node organogenesis and vascular calcification44,45. A cis-eQTL of TNFRSF11B was also identified in the present study, and its transcription levels were highly correlated with WBC (r = 0.329, p = 1.85 × 10−8).
Platelets
Platelets are small anucleate blood elements involved in hemostasis and thrombosis and immune responses46. The intravenous injection of low levels of lipopolysaccharide (LPS) into mice induces the accumulation of platelets in the liver, which eventually return to the circulation or undergo degradation47. The complement system is involved in this degradation. The interactions between platelets and the complement system are complex; platelets can activate complement and vice-versa48. In the present study, only a few liver transcripts were correlated with platelets, and most of these transcripts belonged to the coagulation system and the complement system. Mannose-binding lectin (MBL2) encodes mannose-binding protein and is capable of activating the complement pathway. The expression of complement factors during infection is tissue specific, and complement expression in the liver results from a systemic acute-phase response to infection49. Polymorphisms in MBL2 have been associated with susceptibility to infectious diseases and immune traits as well as complement activity50,51,52. In the present study, a significant positive correlation between platelets and MBL2 transcript levels (PCT and PLT, r = 0.25 p = 4.19 × 10−5) was detected. Although some reports have emphasized a role of platelet interactions with the complement system, the direct role of MBL2 remains unknown53,54. The genetic regulation of MBL2 transcripts might affect platelet traits and, ultimately, immune status. The α1,2-fucosyltransferase I (FUT1) enzyme is important for the biosynthesis of membrane glycoproteins that play a role in various inflammatory reactions55. In pigs, FUT1 is associated with resistance to E.coli F18 infection and the expression of various defense pathways56,57. In the present study, FUT1 was shown to exhibit a cis-eQTL (p = 2.2 × 10−12), and its transcripts levels were significantly positively correlated with platelets (MPV, r = 0.234, p = 9.11 × 10−5).
Genetic regulation of liver transcripts and metabolic status
Hepatic genes are involved in a variety of physiological processes. Many transcripts can be regulated through nuclear hormone receptors including retinoid X receptor α (RXRα), liver X receptor (LXR), farnesoid X receptor (FXR), retinoic acid receptor (RAR), constitutive androstane receptor (CAR), pregnane X receptor (PXR) and peroxisome proliferator-activated receptor (PPAR). One peculiar characteristic of some of these receptors is their ability to regulate not only the metabolic system but also the innate and adaptive immune systems58,59.
We observed a highly significant enrichment of transcripts associated with LXR/RXR, PXR/RXR and FXR/RXR activation among those transcripts correlated with biochemical and body composition traits, consistent with a previous study on liver expression relation to body composition in other pig breeds6. These pathways play an important regulatory role in the metabolism of carbohydrates, fatty acids and cholesterol60.
Methylmalonic aciduria cblB type (MMAB) encodes a protein that catalyzes the conversion of vitamin B12 to adenosylcobalamin. MMAB affects TG levels through adenosylcobalamin and methylmalonyl-CoA mutase61. MMAB has been associated with TG levels62. In the present study, the transcripts levels of MMAB were found to be negatively correlated with TG and MMAB exhibited a cis-eQTL.
BUN is the major nitrogenous product of protein and amino acid catabolism and reflects the balance status of amino acids. The protein is often used as an indicator of kidney and liver function. We identified transcripts with cis-eQTL, such as SORL1, LTBP1, and ABHD14B whose expression was correlated with fat traits and BUN. Low BUN indicates a good balance of amino acids and suggests relatively low urea synthesis and hydration in the liver and a relatively high dietary protein efficiency63. Moreover, genetically regulated transcripts such as SORL1, LTBP1 and ABHD14B play a significant role in both metabolic and immune processes. These transcripts might be pleiotropic genes whose genetic pathways are common to obesity, amino acid balance and immune traits.
There is increasing evidence that the complement system plays an important role in diabetes. In particular, mannose-binding lectin (MBL) levels are elevated in diabetes patients64. As previously described, MBL2 transcript levels are significantly positively correlated with platelets, and negatively correlated with creatinine levels. MASP2 encodes a member of the peptidase S1 family of serine proteases and cleaves complement components C2 and C4 to generate C3 convertase in the lectin pathway of the complement system. Polymorphisms in MASP2 genes are associated with protein serum levels and functional activity and with susceptibility to or protection against infectious diseases65. Significantly higher MASP2 levels are found in children and adults with type 1 diabetes mellitus66. In the present study, the expression levels of MASP2 were found to be positively correlated with leukocytes and negatively correlated with ALB, BUN, TCHO and MFR, further supporting the role of MASP2 in immune and metabolic processes. Thymosin beta 10 (TMSB10) was shown to be correlated with haematological, biochemical and body composition traits, and the transcript levels of TMSB10 were positively correlated with leukocytes and negatively correlated with erythrocytes, ALB, BUN and all fat traits. This finding is consistent with previous reports of the correlation of TMSB10 with insulin-stimulated sprouting and adipose tissue expansion67.
Conclusions
In summary, the analysis of transcript profiles together with information on their genetic regulation provide a new resource for understanding genotype-phenotype mapping associated with hepatic gene expression and the physiological processes of immune, metabolic and body composition traits. Analyses of genetically regulated transcripts and the correlations of liver transcript levels with immune and metabolic as well as body composition traits enable the complexity of this system to be captured and visualized. The liver transcripts whose biological and genetically pathways are common to immune and metabolic status were demonstrated. Our analyses of trait-correlated hepatic expression and eQTL detection complement genome-wide association studies for immune and metabolic traits. This detailed analysis highlighted numerous candidate genes common to both systems.
Material and Methods
Animals, sample collection and phenotype measure
Animal care and tissue collection procedures followed the guidelines of the German Law of Animal Protection, and the experimental protocol was approved by the Animal Care Committee of the Leibniz Institute for Farm Animal Biology. Performance-tested pigs of German Landrace pigs were used for GWAS of liver transcript levels (n = 297). Liver samples were collected from pigs at an average age of 170 days. Veterinary inspection of the carcasses and organs after slaughter confirmed lack of any impairments, disease symptoms and pathological signs to avoid any bias of blood phenotypes. The haematological and biochemical traits were determined using an automated analyzer device (ABX Pentra 60 HORIBA, Montpellier, France; (Fuji DriChem 4000i, FujiFilm, Minato, Japan).
SNP genotypes
Genotyping was performed using the PorcineSNP60 BeadChip (Illumina Inc., San Diego, CA, USA) per manufacturer’s SNP Infinium HD assay protocol. In brief, DNA was amplified, fragmented, and hybridized to the PorcineSNP60 BeadChip containing 62163 locus-specific 50-mers. Single-base extension of captured oligos incorporated labels that were detected by Illumina iScan, and images were subsequently converted to intensity data. Intensity data were normalized and assigned a cluster position, genotype, and quality score with GenomeStudio software (Illumina Inc.). Samples with call rates <99% were removed. Markers with low minor-allele frequency (<5%) as well as those that strongly deviated from Hardy-Weinberg equilibrium (p < 0.0001) were also excluded. The average call rate for all samples was 99.8% ± 0.2. The markers of the 60 K chip were mapped to the porcine reference genome, Sscrofa 10.2.
mRNA microarray analysis
Total RNA was isolated from the liver of 297 animals using TRI Reagent (Sigma, Taufkirchen, Germany). The RNA was amplified using Ambion WT Expression Kit (Affymetrix). Subsequently, 5.5 μg of the resulting cDNA was fragmented and labeled using the Affymetrix Terminal Labeling Kit. The fragmented cDNA was hybridized to the microarray using the Affymetrix Hybridization, Wash and Stain Kit and Affymetrix standard protocols.
Porcine Snowball Microarrays (Affymetrix) containing 47,880 probe-sets were used to determine the expression profile. Expression Console software was used for robust multichip average (RMA) normalization and the detection of present genes by applying the DABG (detection above background) algorithm. Further filtering was done by excluding transcripts with low signals and probes that were present in less than 80% of the samples. 24,909 probes passed the quality filtering and were used for further analyses. Expression data are available in the Gene Expression Omnibus public repository (GEO accession number GSE83932: GSM2221843-GSM2222139).
Data pre-processing
After quality control and filtering the expression data were further pre-processed to account for systemic effects. Mixed-model analyses of variance using JMP Genomics (SAS Institute) were used for adjustment. The genetic similarity matrix between individuals was computed as identity by descent of each pair for the k-matrix and used as a random effect. For control of population stratification, top principal components (PC) which explain variation of more than 1% were considered as covariates. In total 17 PCs were included as covariates. Gender was considered as a fixed effect, and carcass weight was used as a covariate. The residuals were retained for further analysis.
eQTL of mRNA
Analyses of eQTLs were conducted using the R-package ‘Matrix eQTL´ for testing the association between each SNP and residual of transcript abundancies by modeling the effect of genotype as least squares model (Shabalin; 2012). ‘Matrix eQTL’ performs a separate test for each gene - SNP pair and corrects for multiple comparisons by calculating FDR68. Annotation and localization of SNP sites and probe-sets (Ensembl_Sscrofa_10.2) allowed discrimination of cis- and trans-regulation. We defined an eQTL as ‘cis’ if an associated SNP was located within an area less than 1 Mb from the probe set/gene. The associations of transcript levels with haematological, biochemical and end production traits were evaluated estimating spearman coefficient of correlation (r) and corrected for multiple comparisons by calculating FDR68.
Weighted Gene Co-expression Network Analysis (WGCNA)
A weighted gene co-expression network was constructed using normalized gene expression data of 297 livers with the blockwise Modules function of the WGCNA package in R69. The blockwise Modules function allows the entire dataset of 24,909 probe-sets to be utilised in the construction of the weighted gene co-expression network12. Module–trait associations were estimated using the correlation between the module eigengene and the phenotype. Within each module, the intramodular connectivity of each gene was evaluated using two methods, defined as module membership (MM) and the soft connectivity (K). Module membership (MM) is defined as the correlation of expression profile (xi) and each module eigengene (ME),  The intramodular soft connectivity (K) is defined as,
The intramodular soft connectivity (K) is defined as,  which is the sum of all pairwise adjacencies of a gene to all other genes in the module. Genes within each module were then ranked using both the absolute value of module membership and the intramodular soft connectivity, which enables further identification of key players in the regulation network, defined as hub genes.
which is the sum of all pairwise adjacencies of a gene to all other genes in the module. Genes within each module were then ranked using both the absolute value of module membership and the intramodular soft connectivity, which enables further identification of key players in the regulation network, defined as hub genes.
Additional Information
How to cite this article: Ponsuksili, S. et al. Genetically regulated hepatic transcripts and pathways orchestrate haematological, biochemical and body composition traits. Sci. Rep. 6, 39614; doi: 10.1038/srep39614 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Reilly, J. & Kelly, J. Long-term impact of overweight and obesity in childhood and adolescence on morbidity and premature mortality in adulthood: systematic review. Int J Obes 35, 891–898 (2011).
Freedman, D., Ogden, C. & Kit, B. Interrelationships between BMI, skinfold thicknesses, percent body fat, and cardiovascular disease risk factors among U.S. children and adolescents. BMC Pediatr 15, 188 (2015).
Ekser, B., Markmann, J. & Tector, A. Current status of pig liver xenotransplantation. Int J Surg 23, 240–246 (2015).
Doss, S., Schadt, E., Drake, T. & Lusis, A. Cis-acting expression quantitative trait loci in mice. Genome Res 15, 681–691 (2005).
Ponsuksili, S., Murani, E., Trakooljul, N., Schwerin, M. & Wimmers, K. Discovery of candidate genes for muscle traits based on GWAS supported by eQTL-analysis. Int J Biol Sci 10, 327–337 (2014).
Ponsuksili, S., Murani, E., Brand, B., Schwerin, M. & Wimmers, K. Integrating expression profiling and whole-genome association for dissection of fat traits in a porcine model. J Lipid Res 52, 668–678 (2011).
Glubb, D., Dholakia, N. & Innocenti, F. Liver expression quantitative trait loci: a foundation for pharmacogenomic research. Front Genet 3, 153 (2012).
Müller, M. & Brem, G. Disease resistance in farm animals. Experientia 47, 923–934 (1991).
Okada, Y. et al. Genome-wide association study for C-reactive protein levels identified pleiotropic associations in the IL6 locus. Hum Mol Genet 20, 1224–1231 (2011).
Freeman, T. et al. A gene expression atlas of the domestic pig. BMC Biol 10, 90 (2012).
Liu, J., Jing, L. & Tu, X. Weighted gene co-expression network analysis identifies specific modules and hub genes related to coronary artery disease. BMC Cardiovasc Disord 16, 54 (2016).
Ponsuksili, S. et al. Identification of common regulators of genes in co-expression networks affecting muscle and meat properties. PLoS One 10, e0123678 (2015).
Gordon, P., Okai, B., Hoare, J., Erwig, L. & Wilson, H. SOCS3 is a modulator of human macrophage phagocytosis. J Leukoc Biol 100, 771–780 (2016).
Agius, L. Role of glycogen phosphorylase in liver glycogen metabolism. Mol Aspects Med 46, 34–45 (2015).
Lawrenson, K. et al. Cis-eQTL analysis and functional validation of candidate susceptibility genes for high-grade serous ovarian cancer. Nat Commun 22, 8234 (2015).
Smemo, S. et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 507, 371–375 (2014).
Valdecantos, M., Pérez-Matute, P., Prieto-Hontoria, P., Sánchez-Campayo, E., Moreno-Aliaga M. & Martínez J. Erythrocyte antioxidant defenses as a potential biomarker of liver mitochondrial status in different oxidative conditions. Biomarkers 16, 670–678 (2011).
Homedan, C. et al. Acute intermittent porphyria causes hepatic mitochondrial energetic failure in a mouse model. Int J Biochem Cell Biol 51, 93–101 (2014).
Tong, W. & Rouault, T. Metabolic regulation of citrate and iron by aconitases: role of iron-sulfur cluster biogenesis. Biometals 20, 549–564 (2007).
Farooq, M., Pracheil, T., Dong, Z., Xiao, F. & Liu, Z. Mitochondrial DNA instability in cells lacking aconitase correlates with iron citrate toxicity. Oxid Med Cell Longev 493536 (2013).
Galaris, D. & Pantopoulos, K. Oxidative stress and iron homeostasis: mechanistic and health aspects. Crit Rev Clin Lab Sci 45, 1–23 (2008).
Hadjur, S. et al. Defective hematopoiesis and hepatic steatosis in mice with combined deficiencies of the genes encoding Fancc and Cu/Zn superoxide dismutase. Blood 98, 1003–1011 (2001).
Homma, T., Kurahashi, T., Lee, J., Kang, E. & Fujii, J. SOD1 deficiency decreases proteasomal function, leading to the accumulation of ubiquitinated proteins in erythrocytes. Arch Biochem Biophys 583, 65–72 (2015).
Berdanier, C. Advanced Nutrition: Micronutrients. CRC Press LLC (1997).
Frederikse, P., Farnsworth, P. & Zigler, J. J. Thiamine deficiency in vivo produces fiber cell degeneration in mouse lenses. Biochem Biophys Res Commun 258, 703–707 (1999).
Fleming, J. et al. Male infertility and thiamine-dependent erythroid hypoplasia in mice lacking thiamine transporter Slc19a2. Mol Genet Metab 80, 234–241 (2003).
Armistead, J. & Triggs-Raine, B. Diverse diseases from a ubiquitous process: the ribosomopathy paradox. FEBS Lett 588, 1491–1500 (2014).
Danilova, N. & Gazda, H. Ribosomopathies: how a common root can cause a tree of pathologies. Dis Model Mech 8, 1013–1026 (2015).
James, A., Wang, Y., Raje, H., Rosby, R. & DiMario, P. Nucleolar stress with and without p53. Nucleus 5, 402–426 (2014).
Matsushita, T. et al. Functional SNP of ARHGEF10 confers risk of atherothrombotic stroke. Hum Mol Genet 19, 1137–1146 (2010).
Moshage, H. Cytokines and the hepatic acute phase response. J Pathol 181, 257–266 (1997).
Billings, R., Lauterburg, B., Thurman, R., Uetrecht, J. & Wendel, A. Liver Toxicity Mediated by Leukocytes and Kupffer Cells Chapter. Pharmacological Sciences: Perspectives for Research and Therapy in the Late 1990s. Birkhäuser Basel (1995).
Badolato, R. et al. Serum amyloid A is a chemoattractant: induction of migration, adhesion, and tissue infiltration of monocytes and polymorphonuclear leukocytes. J Exp Med 180, 203–209 (1994).
Olsson, N., Siegbahn, A. & Nilsson, G. Serum amyloid A induces chemotaxis of human mast cells by activating a pertussis toxin-sensitive signal transduction pathway. Biochem Biophys Res Commun 254, 143–146 (1999).
Patel, H., Fellowes, R., Coade, S. & Woo, P. Human serum amyloid A has cytokine-like properties. Scand J Immunol 48, 410–418 (1998).
Xu, L. et al. A novel biologic function of serum amyloid A. Induction of T lymphocyte migration and adhesion. J Immunol 155, 1184–1190 (1995).
Schumann, R. et al. The lipopolysaccharide-binding protein is a secretory class 1 acute-phase protein whose gene is transcriptionally activated by APRF/STAT/3 and other cytokine-inducible nuclear proteins. Mol Cell Biol 16, 3490–3503 (1996).
Eckert, J. et al. The crystal structure of lipopolysaccharide binding protein reveals the location of a frequent mutation that impairs innate immunity. Immunity 39, 647–660 (2013).
Jiang, M. et al. Ang II-stimulated migration of vascular smooth muscle cells is dependent on LR11 in mice. J Clin Invest 118, 2733–2746 (2008).
Sakai, S. et al. Circulating soluble LR11/SorLA levels are highly increased and ameliorated by chemotherapy in acute leukemias. Clin Chim Acta 413, 1542–1548 (2012).
Takahashi, M., Bujo, H., Shiba, T., Jiang, M., Maeno, T. & Shirai, K. Enhanced circulating soluble LR11 in patients with diabetic retinopathy. Am J Ophthalmol 154, 187–192 (2012).
Zhu, Y. et al. LR11, an LDL receptor gene family member, is a novel regulator of smooth muscle cell migration. Circ Res 94, 752–758 (2004).
Tsukamoto, S. et al. Tetraspanin CD9 modulates ADAM17-mediated shedding of LR11 in leukocytes. Exp Mol Med 46 (2014).
Kong, Y., Boyle, W. & Penninger, J. Osteoprotegerin ligand: a common link between osteoclastogenesis, lymph node formation and lymphocyte development. Immunol Cell Biol 77, 188–193 (1999).
Stolina, M. et al. Continuous RANKL inhibition in osteoprotegerin transgenic mice and rats suppresses bone resorption without impairing lymphorganogenesis or functional immune responses. J Immunol 179, 7497–7505 (2007).
Lam, F., Vijayan, K. & Rumbaut, R. Platelets and their interactions with other immune cells. Compr Physiol. Compr Physiol 5, 1265–1280 (2015).
Shibazaki, M., Kawabata, Y., Yokochi, T., Nishida, A., Takada, H. & Endo, Y. Complement-Dependent Accumulation and Degradation of Platelets in the Lung and Liver Induced by Injection of Lipopolysaccharides. Infect Immun 67, 5186–5191 (1999).
Peerschke, E., Yin, W. & Ghebrehiwet, B. Complement activation on platelets: implications for vascular inflammation and thrombosis. Mol Immunol 47, 2170–2175 (2010).
Peña, J. et al. Multiplexed digital mRNA profiling of the inflammatory response in the West Nile Swiss Webster mouse model. PLoS Negl Trop Dis 8, e3216 (2014).
Filho, R. et al. High frequency of variant alleles of the mannose-binding lectin 2 (MBL2) gene are associated with patients infected by hepatitis B virus. Viral Immunol 23, 449–453 (2010).
Xu, H., Zhao, M., Wan, T., Song, G., He, J. & Chen, Z. Association between Mannose-binding lectin gene polymorphisms and hepatitis B virus infection: a meta-analysis. PLoS One 8, e75371 (2013).
Phatsara, C. et al. Molecular genetic analysis of porcine mannose-binding lectin genes, MBL1 and MBL2, and their association with complement activity. Int J Immunogenet 34, 55–63 (2007).
Chatterjee, M. & Geisler, T. Inflammatory Contribution of Platelets Revisited: New Players in the Arena of Inflammation. Semin Thromb Hemost 42, 205–214 (2016).
Saggu, G., Cortes, C., Emch, H., Ramirez, G., Worth, R. & Ferreira, V. Identification of a novel mode of complement activation on stimulated platelets mediated by properdin and C3(H2O). J Immunol 190, 6457–6467 (2013).
Amin, M. et al. A key role for Fut1-regulated angiogenesis and ICAM-1 expression in K/BxN arthritis. Ann Rheum Dis 74, 1459–1466 (2015).
Bao, W. et al. Microarray analysis of differential gene expression in sensitive and resistant pig to Escherichia coli F18. Anim Genet 43, 525–534 (2012).
Meijerink, E. et al. A DNA polymorphism influencing alpha(1,2)fucosyltransferase activity of the pig FUT1 enzyme determines susceptibility of small intestinal epithelium to Escherichia coli F18 adhesion. Immunogenetics 52, 129–136 (2000).
Huang, W. & Glass, C. Nuclear receptors and inflammation control: molecular mechanisms and pathophysiological relevance. Arterioscler Thromb Vasc Biol 30, 1542–1549 (2010).
Francis, G., Fayard, E., Picard, F. & Auwerx, J. Nuclear receptors and the control of metabolism. Annu Rev Physiol 65, 261–311 (2003).
Ahuja, H., Szanto, A., Nagy, L. & Davies, P. The retinoid X receptor and its ligands: versatile regulators of metabolic function, cell differentiation and cell death. J Biol Regul Homeost Agents 17, 29–45 (2003).
Maiti, N., Widjaja, L. & Banerjee, R. Proton transfer from histidine 244 may facilitate the 1,2 rearrangement reaction in coenzyme B(12)-dependent methylmalonyl-CoA mutase. The Journal of biological chemistry 274, 32733–32737 (1999).
Jiao, H. et al. Pathway-Based Genome-Wide Association Studies for Plasma Triglycerides in Obese Females and Normal-Weight Controls. PLoS One 10, e0134923 (2015).
Hahn, J., Biehl, R. & Baker, D. Ideal digestible lysine level for early-finishing and late-finishing swine. J Anim Sci 73, 773–784 (1995).
Zhang, N. et al. Association of levels of mannose-binding lectin and the MBL2 gene with type 2 diabetes and diabetic nephropathy. PLoS One 8, e83059 (2013).
Beltrame M. et al. MBL-associated serine proteases (MASPs) and infectious diseases. Mol Immunol 67, 85–100 (2015).
Jenny, L., Ajjan, R., King, R., Thiel, S. & Schroeder, V. Plasma levels of mannan-binding lectin-associated serine proteases MASP-1 and MASP-2 are elevated in type 1 diabetes and correlate with glycaemic control. Clin Exp Immunol 180, 227–232 (2015).
Gealekman, O. et al. Control of adipose tissue expandability in response to high fat diet by the insulin-like growth factor-binding protein-4. J Biol Chem 289, 18327–18338 (2014).
Benjamini, Y. & Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J R Stat Soc Series B Stat Methodol 57, 289–300 (1995).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008).
Acknowledgements
The authors thank Annette Jugert, Hannelore Tychsen, Joana Bittner and Nicole Gentz for excellent technical help.
Author information
Authors and Affiliations
Contributions
S.P. drafted the manuscript and analyzed the data. S.P. and K.W. conceived the study. F.H. helped analyzed the data. F.H., E.M. and N.T. helped in drafting the manuscript. K.W. contributed to data interpretation and helped in drafting the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Ponsuksili, S., Trakooljul, N., Hadlich, F. et al. Genetically regulated hepatic transcripts and pathways orchestrate haematological, biochemical and body composition traits. Sci Rep 6, 39614 (2016). https://doi.org/10.1038/srep39614
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep39614
This article is cited by
-
Genetic regulation and variation of expression of miRNA and mRNA transcripts in fetal muscle tissue in the context of sex, dam and variable fetal weight
Biology of Sex Differences (2022)
-
Epigenome-wide skeletal muscle DNA methylation profiles at the background of distinct metabolic types and ryanodine receptor variation in pigs
BMC Genomics (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
