
Abstract
While cerebrospinal fluid (CSF) biomarkers for Creutzfeldt-Jakob disease (CJD) are established and partly included in the diagnostic criteria, no blood biomarkers are available. Here, we assessed the utility of serum neurofilament light chain (NF-L) and tau protein in comparison to CSF markers (NF-L and phosphorylated NF heavy chain (pNF-H), tau, S100B, 14-3-3) and prion conversion assay (real-time quaking induced conversion (RT-QuIC)) for sporadic and genetic CJD. Importantly, a Gerstmann-Sträussler-Scheinker mutation carrier in the asymptomatic phase and at disease onset was included as well. Both NF-L and tau were markedly increased in CJD serum, reaching similar or even better performance as in CSF (sensitivity and specificity for serum NF-L 100% and 85.5%, and for serum tau 84.6% and 96.2%, respectively). Serum S100B showed high sensitivity as well (84.2%), but lower specificity (63%). CSF neurofilaments were increased before symptom onset, while prion seeding assay was negative. Just before a clinical diagnosis could be made, all CSF markers and NF-L in the serum were increased and CSF prion conversion assay was positive. The data suggest that neurofilaments are sensitive and specific blood markers for the diagnosis of genetic and sporadic CJD and might represent promising tools to predict disease onset.
Similar content being viewed by others

Introduction
Prion diseases are transmissible and fatal neurodegenerative diseases neuropathologically characterized by the presence of misfolded prion proteins, which are exceptionally resistant to routine decontamination procedures. The availability of an easy, early, and reliable test in prion disorders is therefore a relevant issue both for public health reasons and for establishing diagnosis, but also for differential diagnosis with other treatable conditions.
The diagnosis of sporadic and genetic CJD can be supported by fluid biomarkers, including 14-3-3 proteins, tau proteins, neurofilaments and S100B in cerebrospinal fluid (CSF)1,2,3,4,5,6. Potential blood markers comprise acute-phase proteins7 and S100B8. Among the mentioned candidate markers, only 14-3-3 in the CSF was included into the diagnostic criteria by the WHO.
While for a long time the detection of misfolded prion protein in body fluids failed, some approaches have eventually been successful9 and high diagnostic accuracy was reported for the recently developed real-time quaking induced conversion (RT-QuIC) prion conversion assay which uses minute amounts of the β-structure-rich insoluble conformer of the prion protein (PrPSc) in CSF as seeds10,11 while other studies report lower diagnostic performance12. Further, it is unclear when the misfolded prion protein appears or reaches the detectable limit in CSF of CJD patients. However, for disease onset, especially in genetic cases, blood markers are needed with the perspective to have an objective read-out for therapeutic trials. Presently, no validated blood biomarker for the differential diagnosis and prediction of onset exists.
In the present study we therefore addressed the questions whether we can measure these biomarkers with newly established assays in blood, of which diagnostic value they are, and how early in the disease process these markers appear.
Results
A summary of the marker concentrations, cut-off levels, and diagnostic performance in terms of sensitivity and specificity as well as the positive and negative predictive values for the discrimination between CJD and controls is given in Table 1.
Surrogate marker concentrations in serum
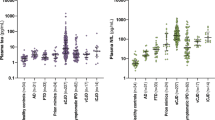
Boxplots of marker levels in serum of the diagnostic groups are shown in Fig. 1A–C. NF-L serum concentrations were higher in both sCJD and gCJD compared to DCo or Co. At 44.7 pg/ml cut-off 100% sensitivity and 85.5% specificity was reached for discrimination between CJD and controls.

Marker concentrations, ROC diagrams, and RT-QuIC results for CJD and controls.
Boxplots show the serum levels of NF-L (A), tau (B), and S100B (C), as well as the CSF levels of NF-L (D), tau (E), and pNF-H (F). Displayed are the results for CJD patients (indicated separately for sCJD and gCJD) and from demented (DCo) and non-demented controls (Co). The concentrations of analytes determined in serum and CSF from a gCJD patient at presymptomatic and early symptomatic disease stage are indicated by green and red circles, respectively. Boxes give median values with interquartile ranges, whiskers indicate the concentration range. Three and two asterisks indicate statistically significant differences of p < 0.0001 and p < 0.01, respectively. The diagnostic performance of markers is illustrated by ROC curves for serum (G) and CSF (H). Except for S100B, of which levels were determined in DCo only, ROC analyses were conducted considering all CJD measures (i.e. sCJD and gCJD) versus all controls (i.e. DCo and Co). The results of RT-QuIC are shown in (I). Displayed are the mean fluorescence signals with SD detected in the CSF of sCJD patients (n = 6), gCJD patients (n = 4), and controls (n = 6). Separately shown are the results for 3 replicate measurements of the gCJD mutation carrier in the asymptomatic disease stage (orange) and at disease onset (red).
Serum tau was significantly different in sCJD, but not gCJD, and controls. At 2.2 pg/ml cut-off, CJD was recognized with 84.6% sensitivity and 96.2% specificity.
The concentrations determined for S100B in serum samples differed significantly between all CJD and DCo (p = 0.0006), but post hoc test after Kruskal-Wallis test (p = 0.0465) revealed no significant differences between sCJD, gCJD, and DCo (Fig. 1C).
At the presymptomatic stage serum tau and S100B were similar to control levels, whereas NF-L tended to be elevated, and at symptom onset serum markers were clearly increased (green and red circles respectively in Fig. 1A–C).
ROC analysis of serum marker concentrations of all CJD versus all controls yielded comparable estimates for the AUC for NF-L and tau (Fig. 1G).
Surrogate marker concentrations in CSF
Boxplots of the marker levels in CSF samples of the diagnostic groups are shown in Fig. 1D–F.
The levels of NF-L and pNF-H in CSF samples were strongly increased in CJD compared to both control groups. CSF tau concentrations were significantly increased in CJD as well, however, due to low levels in an E200K and the insert 4 × 24 carrier, the difference between gCJD and DCo was not significant.
At the presymptomatic disease stage, CSF tau was normal and NF-L and especially pNF-H were already elevated above the 75% quartile of controls. At disease onset all CSF markers were increased. See green and red circles in Fig. 1D–F, respectively.
In the ROC analyses pNF-H, tau, and NF-L showed similar performance (Fig. 1H).
Prion seeding activity in CSF
A summary of RT-QuIC results is shown in Fig. 1I. In 10/14 CJD CSF samples the RT-QuIC assay was positive (71.4% sensitivity). False negative results were obtained for one sample from a patient with M/M codon 129 polymorphism and an NF-L serum just above the cut-off level, and for 3 gCJD samples from patients with E200 K and E196 K mutations. These 3 patients were positive for CSF 14-3-3 and above the cut-off for all other markers, with the exception that the E200 K mutation carrier had a tau CSF level clearly below the diagnostic threshold. CSF samples from 6 non demented control cases failed in the RT-QuIC assay (100% specificity). After a prolonged amplification time, in 2 out of 6 control CSF samples a signal emerged after 48 h and 96 h, respectively, resulting in a signal less than 10% of the signal obtained with CJD samples after 5 days of measurements. In the CSF from the presymptomatic gCJD patient no seeding activity could be detected. At the onset of first symptoms the signal was positive with minimally delayed starting point of about 3 h and a maximum similar to that determined in the other positive CJD samples.
Discussion
Today, improvement of CJD diagnosis is attempted in two ways: (1) There is a search for surrogate markers in the serum of CJD patients, and (2) Methods are developed to amplify the minute amounts of abnormal PrP present in body fluids for a specific diagnosis.
In the present study we show that the known biomarkers NF-L and tau measured in serum samples parallel the increase found in CSF, yielding similar statistical significance for differentiation. NF-L, which also is elevated in motor neuron disease13,14,15, is found at even higher levels in CJD patients. pNF-H which was found to be increased also in amyotrophic laterla sclerosis (ALS) plasma by in house assays16,17, yielded excellent diagnostic performance in CSF in our cohort. Additionally, the analysis of the serum from a presymptomatic gCJD patient provides first evidence for pNF-H to be elevated already before symptoms appear and clinical diagnosis can be made.
If serum NF-L qualifies as marker for broad differential diagnoses has to be examined in future studies including especially patients with rapidly progressive neurodegenerative dementia. As recent data point to a generally strong correlation between CSF and blood levels of NF-L and proteopathic lesions18, high diagnostic power for the discrimination of CJD and at least neurodegenerative diseases is likely to be expected.
Serum tau can now be detected more sensitively and is shown for the first time to be increased not only in sCJD19 but also in gCJD. Differences between gCJD and controls, however, were less marked compared to NF-L and pNF-H.
The time point at which the biomarkers increase in CSF and also in serum is a mostly open question. It can be hypothesized that the analyzed markers are increased early in CJD as they indicate neuroaxonal degeneration. We found the presymptomatic stage of one CJD mutation carrier characterized by normal serum and CSF tau and serum S100B. Neurofilaments, especially pNF-H in the CSF, already show a trend for increased levels and might therefore represent candidate markers for the onset of the disease as it was shown for asymptomatic ALS gene carrier15. At symptom onset of the gCJD patient, all markers are clearly increased in serum and CSF to the range of the CJD cohort.
Similar results were obtained with the specific approach, the RT-QuIC, which has been established for CJD diagnostics from CSF12,20,21 and urine22, however, failed to be successful in serum until now. Seeding activity could not be detected in the CSF from the presymptomatic phase, but in parallel to the appearance of first CJD symptoms.
Taken together, we show that serum NF-L and serum tau can be used in the diagnosis of both sCJD and gCJD and provide preliminary evidence that neurofilament levels tend to be increased presymptomatically.
Methods
Patients
This study was conducted according to the principles expressed in the Declaration of Helsinki. Collection and analysis of samples were approved by the Ethics Committees of the Medical Faculties of the University Göttingen (approval number 100305) and Ulm (approval number 20/10). All patients or their next relatives in case of severe dementia gave written informed consent to their participation in the study.
In total, our retrospective study included 103 patients that were seen in the general outpatient clinic and the outpatient memory clinic of the Department of Neurology in Ulm and the surveillance unit for transmissible spongiform encephalopathies in the Department of Neurology in Göttingen.
All methods were performed in accordance with the relevant guidelines and regulations.
CSF was obtained by lumbar puncture, centrifuged, aliquoted and stored within 2–48 h at −80 °C until analysis. Serum was processed likewise and stored within 2 h at −80 °C.
Serum and CSF samples of 43 CJD patients were analyzed, of which 4 had clinically diagnosed “probable CJD” (neuropathology missing) and 39 were neuropathologically verified, including 33 sporadic CJD (sCJD) and 9 genetic CJD (gCJD: E200 K (n = 3), V210I (n = 2), E196 K, insert 5 × 24 (n = 3), and insert 4 × 24). Additionally, samples from a 50 year old GSS patient (P102L) at the presymptomatic disease stage and two years later at onset of symptoms before clinical diagnosis were included. Control groups comprised 40 patients without signs of dementia (Co), and 20 demented patients (DCo) (Alzheimer’s disease n = 12, mild cognitive impairment n = 4, frontotemporal dementia n = 3, normal pressure hydrocephalus n = 1).
For demographic characteristics of the diagnostic groups see Table 1.
Laboratory markers
Serum NF-L and serum tau protein were measured with ultrasensitive Single molecule array (Simoa) assays23,24,25.
Samples were analyzed for S100B with electrochemiluminescence immunoassay (Elecsys S100B, Roche, Penzberg, Germany)8, and with ELISAs for NF-L (IBL, Hamburg, Germany), pNF-H (Biovendor, Heidelberg, Germany), and Tau (Fujirebio, Hanover, Germany) according to manufacturer’s specifications. Mean inter-assay CV for the ELISAs was <20%14.
Real-time quaking-induced conversion (RT-QuIC) was carried out as described elsewhere21.
Statistics
Statistical analyses were performed using graphpad prism 5 software. Standard measures of diagnostic test validity such as sensitivity, specificity, and predictive values accompanied by their 95% confidence intervals (CI) were calculated for varying biomarker cut-off levels. The optimal cut-off level for dichotomizing values was selected as the situation maximizing the Youden index26. The receiver operating characteristics (ROC) curve is used for a graphical visualization of the impact of the variation in the cut-off values. Two-tailed unpaired Mann-Whitney t-test and Kruskal-Wallis test at a significance level of 5% were used to determine statistical differences between two groups or more groups, respectively. Dunn’s post hoc comparison was applied following Kruskal-Wallis test in case of significant differences.
Additional Information
How to cite this article: Steinacker, P. et al. Neurofilaments in blood and CSF for diagnosis and prediction of onset in Creutzfeldt-Jakob disease. Sci. Rep. 6, 38737; doi: 10.1038/srep38737 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Chohan, G. et al. The role of cerebrospinal fluid 14-3-3 and other proteins in the diagnosis of sporadic Creutzfeldt-Jakob disease in the UK: a 10-year review. J Neurol Neurosurg Psychiatry 81, 1243–1248 (2010).
Hamlin, C. et al. A comparison of tau and 14-3-3 protein in the diagnosis of Creutzfeldt-Jakob disease. Neurology 79, 547–552 (2012).
Otto, M. et al. S-100 protein concentration in the cerebrospinal fluid of patients with Creutzfeldt-Jakob disease. J Neurol 244, 566–570 (1997).
Otto, M. et al. Tau protein and 14-3-3 protein in the differential diagnosis of Creutzfeldt-Jakob disease. Neurology 58, 192–197 (2002).
Skillback, T. et al. Diagnostic performance of cerebrospinal fluid total tau and phosphorylated tau in Creutzfeldt-Jakob disease: results from the Swedish Mortality Registry. JAMA Neurol 71, 476–483 (2014).
van Eijk, J. J., van Everbroeck, B., Abdo, W. F., Kremer, B. P. & Verbeek, M. M. CSF neurofilament proteins levels are elevated in sporadic Creutzfeldt-Jakob disease. J Alzheimers Dis 21, 569–576 (2010).
Fratini, F. et al. Increased levels of acute-phase inflammatory proteins in plasma of patients with sporadic CJD. Neurology 79, 1012–1018 (2012).
Otto, M. et al. Diagnosis of Creutzfeldt-Jakob disease by measurement of S100 protein in serum: prospective case-control study. BMJ 316, 577–582 (1998).
Sobrova, P., Ryvolova, M., Adam, V. & Kizek, R. Capillary electromigration based techniques in diagnostics of prion protein caused diseases. Electrophoresis 33, 3644–3652 (2012).
McGuire, L. I. et al. Real time quaking-induced conversion analysis of cerebrospinal fluid in sporadic Creutzfeldt-Jakob disease. Ann Neurol 72, 278–285 (2012).
Orru, C. D. et al. Rapid and sensitive RT-QuIC detection of human Creutzfeldt-Jakob disease using cerebrospinal fluid. MBio 6 (2015).
Park, J. H. et al. Real-Time Quaking-Induced Conversion Analysis for the Diagnosis of Sporadic Creutzfeldt-Jakob Disease in Korea. J Clin Neurol 12, 101–106 (2016).
Lu, C. H. et al. Neurofilament light chain: A prognostic biomarker in amyotrophic lateral sclerosis. Neurology 84, 2247–2257 (2015).
Steinacker, P. et al. Neurofilaments in the diagnosis of motoneuron diseases: a prospective study on 455 patients. J Neurol Neurosurg Psychiatry 87, 12–20 (2016).
Weydt, P. et al. Neurofilament levels as biomarkers in asymptomatic and symptomatic familial amyotrophic lateral sclerosis. Ann Neurol 79, 152–158 (2016).
Lu, C. H. et al. Plasma neurofilament heavy chain levels and disease progression in amyotrophic lateral sclerosis: insights from a longitudinal study. J Neurol Neurosurg Psychiatry 86, 565–573 (2015).
Boylan, K. et al. Immunoreactivity of the phosphorylated axonal neurofilament H subunit (pNF-H) in blood of ALS model rodents and ALS patients: evaluation of blood pNF-H as a potential ALS biomarker. J Neurochem 111, 1182–1191 (2009).
Bacioglu, M. et al. Neurofilament Light Chain in Blood and CSF as Marker of Disease Progression in Mouse Models and in Neurodegenerative Diseases. Neuron 91, 494–496 (2016).
Noguchi-Shinohara, M., Hamaguchi, T., Nozaki, I., Sakai, K. & Yamada, M. Serum tau protein as a marker for the diagnosis of Creutzfeldt-Jakob disease. J Neurol 258, 1464–1468 (2011).
Cramm, M. et al. Characteristic CSF prion seeding efficiency in humans with prion diseases. Mol Neurobiol 51, 396–405 (2015).
Shi, S., Mitteregger-Kretzschmar, G., Giese, A. & Kretzschmar, H. A. Establishing quantitative real-time quaking-induced conversion (qRT-QuIC) for highly sensitive detection and quantification of PrPSc in prion-infected tissues. Acta Neuropathol Commun 1, 44 (2013).
Shi, S. et al. Quantitative Real-Time Quaking-Induced Conversion Allows Monitoring of Disease-Modifying Therapy in the Urine of Prion-Infected Mice. J Neuropathol Exp Neurol 74, 924–933 (2015).
Kuhle, J. et al. Comparison of three analytical platforms for quantification of the neurofilament light chain in blood samples: ELISA, electrochemiluminescence immunoassay and Simoa. Clin Chem Lab Med 54, 1655–1661 (2016).
Oliver, J. M. et al. Serum Neurofilament Light in American Football Athletes over the Course of a Season. J Neurotrauma 33, 1784–1789 (2016).
Randall, J. et al. Tau proteins in serum predict neurological outcome after hypoxic brain injury from cardiac arrest: results of a pilot study. Resuscitation 84, 351–356 (2013).
Youden, W. J. Index for rating diagnostic tests. Cancer 3, 32–35 (1950).
Acknowledgements
We are indebted to the participants and their families. We thank all the physicians who referred patients to our clinic. This work was supported by the Federal Ministry of Education and Research, Germany, Competence Net Neurodegenerative Dementias (project: FTLDc 01GI1007A), EU Joint Programme - Neurodegenerative Disease (JPND) (BiomarkAPD (01ED1203F), Sophia (01ED1202A), PreFrontAls (01ED1512)) European Union (NADINE EU-FP7, 246513, Anteprion EU-FP6 ORPHA257930), VINNOVA, the Swedish Research Council and the European Research Council (grant #681712).
Author information
Authors and Affiliations
Contributions
All authors made substantial contributions to conception and design, and/or acquisition of data, and/or analysis and interpretation of data. The first draft was written by PS and MO, followed by critical revision of all authors. All authors gave final approval of the version to be submitted. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. Individual contributions: Study conception and design: P.S., K.B., H.Z., M.O. Acquisition, analysis, and interpretation of data: P.S., K.B., S.H., S.S., V.R., P.O., A.G., J.K., D.S., H.Z., M.O. Writing of the manuscript P.S., K.B., H.Z., M.O. Critical revision of the manuscript P.S., K.B., S.H., S.S., V.R., P.O., A.G., J.K., D.S., H.Z., M.O. K.B. and H.Z. are co-founders of Brain Biomarker Solutions in Gothenburg AB, a GU Venture-based platform company.
Ethics declarations
Competing interests
KB and HZ are co-founders of Brain Biomarker Solutions in Gothenburg AB, a GU Venturebased platform company.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Steinacker, P., Blennow, K., Halbgebauer, S. et al. Neurofilaments in blood and CSF for diagnosis and prediction of onset in Creutzfeldt-Jakob disease. Sci Rep 6, 38737 (2016). https://doi.org/10.1038/srep38737
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep38737
This article is cited by
-
The natural history study of preclinical genetic Creutzfeldt-Jakob Disease (CJD): a prospective longitudinal study protocol
BMC Neurology (2023)
-
Validation of Plasma and CSF Neurofilament Light Chain as an Early Marker for Sporadic Creutzfeldt-Jakob Disease
Molecular Neurobiology (2022)
-
Diagnostic and prognostic value of plasma neurofilament light and total-tau in sporadic Creutzfeldt-Jakob disease
Alzheimer's Research & Therapy (2021)
-
Evaluation of plasma tau and neurofilament light chain biomarkers in a 12-year clinical cohort of human prion diseases
Molecular Psychiatry (2021)
-
Amyloid-beta modulates the association between neurofilament light chain and brain atrophy in Alzheimer’s disease
Molecular Psychiatry (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
