
Abstract
Retinitis pigmentosa (RP) is a heterogeneous set of hereditary eye diseases, characterized by selective death of photoreceptor cells in the retina, resulting in progressive visual impairment. Approximately 20–40% of RP cases are autosomal dominant RP (ADRP). In this study, a Chinese ADRP family previously localized to the region between D1S2819 and D1S2635 was sequenced via whole-exome sequencing and a variant c.1345C > G (p.R449G) was identified in PRPF3. The Sanger sequencing was performed in probands of additional 95 Chinese ADRP families to investigate the contribution of PRPF3 to ADRP in Chinese population and another variant c.1532A > C (p.H511P) was detected in one family. These two variants, co-segregate with RP in two families respectively and both variants are predicted to be pathological. This is the first report about the spectrum of PRPF3 mutations in Chinese population, leading to the identification of two novel PRPF3 mutations. Only three clustered mutations in PRPF3 have been identified so far in several populations and all are in exon 11. Our study expands the spectrum of PRPF3 mutations in RP. We also demonstrate that PRPF3 mutations are responsible for 2.08% of ADRP families in this cohort indicating that PRPF3 mutations might be relatively rare in Chinese ADRP patients.
Similar content being viewed by others
Introduction
Retinitis pigmentosa (RP) refers to a heterogeneous set of hereditary retinal degenerative disorders which are responsible for the blindness of more than 1.5 million people worldwide and affect about 1 in 1000 people in China. RP is characterized by a selective death of photoreceptors that are light-sensing cells in the retina, resulting in progressive visual impairment1,2,3.There are three modes of Mendelian inheritance in RP—autosomal-dominant RP (ADRP), autosomal-recessive RP (ARRP), and X-linked RP (XLRP)1. Approximately 20–40% of RP cases are ADRP and mutations in over 20 genes are known to cause ADRP. Amongst ADRP causative genes are an unusual class— pre-mRNA splicing genes4— eight of which are ubiquitous core snRNP proteins (PRPF3, PRPF8, PRPF31, PRPF4, SNRNP200, and PRPF6) and splicing factors (RP9 and DHX38). Mutations in those genes ubiquitously expressed and essential for splicing, have so far been reported to cause a disease that displays retina-specific phenotype5.
PRPF3 (RP18, OMIM 601414) gene spans approximately 32 kb at chromosome 1q216, contains 16 exons and encodes a protein of 683 amino acids in length with a calculated molecular weight of 77 kDa7, which is a human homologue of the yeast U4/U6-associated splicing factor Prp3. Only three clustered PRPF3 mutations, c.1482C > T (p.T494M), c.1478C > T (p.P493S) and c.1466C > A (p.A489D), have been identified thus far in RP in several populations. The mutation T494M is the most frequently detected substitution in PRPF3 while P493S occurs rather sporadically5. All the mutations are in the exon11 (c.1427–1526) of PRPF3 and previous surveys failed to identify mutations outside of this exon. Therefore, only the region c.1427–1526 was screened for testing PRPF3 mutations in some reported studies8,9. In this work, we identified two novel PRPF3 mutations, c.1345C > G (p.R449G) and c.1532A > C (p.H511P), in two Chinese families with ADRP. Furthermore, our study demonstrates that PRPF3 mutations are responsible for 2.08% of ADRP families in our cohort indicating that PRPF3 mutation might be relatively rare in Chinese patients with ADRP.
Results
Clinical evaluations of ADRP families
The pedigree of Family 020001 indicates a dominant inheritance pattern of three generations (Fig. 1). Medical history of all the affected individuals shows that early onset of night blindness is at age 4 to 10 years old, with subsequent loss of far peripheral vision after 20 years. Clinical details of that family were previously described10. Additional 95 Chinese families had a primary diagnosis of RP based on clinical descriptions provided by the referring clinicians at the time of enrollment. The inheritance pattern of those RP families is AD (Fig. 1).

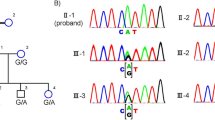
Pedigrees of Family 020001 and Family 020021.
Probands are pointed out by arrow. Circles indicate females, and squares, males. Filled symbols indicate affected patients, and empty symbols, healthy controls. Individuals who were genotyped are marked with an asterisk.
Mutation analysis
We selectively performed whole-exome sequencing (WES) on the proband (II: 4) of Family 020001 previously mapped to the region between D1S2819 and D1S263510 (95.53Mb-159.17 Mb). An average of 9.71 Gb of sequence data were obtained, and 99.94% of bases originated from the targeted exome, resulting in a mean coverage of 115-fold. More than 98.94% of the targeted exons were covered more than 10-fold. We employed the following filters to filter the exome results: total variants → heterozygous variants → variants that are absent in the 1000 Genomes Project, 1000G_ASN, esp6500si_all, and dbSNP137 → variants that are coding or splicing → variants that are predicted to be pathogenic. Following the filteration procedures described above, only one heterozygous missense variant in PRPF3 (NM_004698: exon10: c.1345C > G: p.R449G) was detected to be located within the linkage region between D1S2819 and D1S263510, which would lead to the change of amino acid from Arginine to Glycine at residue 449 (p.R449G). Sanger sequencing confirmed this variant (Fig. 2a). The variant was predicted to be probably damaging with a score of 0.980 by PolyPhen-2, a score of 0.05 by SIFT, and a score of −6.698 by PROVEAN. Intra-familial co-segregation analysis of Family 020001 further revealed that this heterozygous missense variant was present in all the affected and absent in normal family members, which further confirmed that the variant was a potential ADRP causing mutation for Family 020001.

Mutations identified in Family 020001 and Family 020021.
(a) Sequence chromatograms showing the identified mutations and their wild type form. (b) Schematic representation of the linear location of our identified PRPF3 mutations and previously reported mutations (red star) in context of genome (upper) and protein (below). (c) Orthologous protein sequence alignment of PRPF3 from human (H. sapiens), chimpanzees (P. troglodytes), cattle (B. taurus), Chicken (G. gallus), Norway rat (R. norvegicus), house mouse (M. musculus), African clawed frog (X. laevis), zebra fish (D. rerio), yeast (S. cerevisiae), and Pombe yeast (S. pombe). Completely conserved residues across all species aligned are shaded with black. Residues boxed in red show the conservation of residues identified mutations in this study.
To investigate the contribution of PRPF3 to ADRP in Chinese population, Sanger sequencing of PRPF3 was performed in the probands of additional 95 Chinese ADRP families. We identified another novel variant c.1532A > C in exon12 of PRPF3 gene in Family 020021 (Fig. 2a), which would lead to the change of amino acid from Histidine to Proline at residue 511 (p.H511P). This variant co-segregated in the family with RP and was predicted to be probably damaging with a score of 1.000 by PolyPhen-2, a score of 0.00 by SIFT, and a score of −9.785 by PROVEAN.
These two mutations, c.1345C > G (p.R449G) and c.1532A > C (p.H511P), were further confirmed to be absent in 200 unrelated ethnically matched healthy controls as well as in database of probably benign variation. Both mutations are absent either in existing database of disease-causing mutations or in the reported literatures and thus considered to be novel.
Multiple orthologous sequence alignment (MSA) revealed that p.R449G and p.H511P were highly conserved from human to yeast (Fig. 2c). The residues R449 and H511 in human PRPF3 correspond to the residues R245 and H308 in Prp3 of Saccharomyces cerevisiae S288c (NP_010761.3), respectively, by MSA of PRPF3 from Homo sapiens to yeast. Prp3 structure from PDB (Protein Data Bank): 3JCM (Cryo-em Structure of the yeast spliceosomal U4/U6.U5 Tri-snrnp) shows that amino acids R245 and H308 of Prp3 play a key role in Prp3 binding to RNA duplex11 suggesting that changes in those two residues may impair the structure and function of spliceosomal U4/U6.U5 Tri-snrnp (Fig. 3a).

Structural model of the wild type Prp3 in the spliceosomal U4/U6.U5 tri-snRNP from S. cerevisiae. The residues (words in red) shown in the figures are completely conserved with the mutated residues in human PRPF3 by MSA.
The positions of those residues in human PRPF3 indicated in the figures, which correspond to the same residues in Prp3 of Saccharomyces cerevisiae S288c (NP_010761.3) respectively by MSA of PRPF3 from Homo sapiens to yeast. (a) Both amino acid H (yellow) and R (yellow) play a key role in Prp3 (purple)-U4/U6 snRNA duplex (brown) interaction.(b) The residues (pointed out by red open arrows)of Prp3 (purple) are important in protein-protein interaction.
Discussion
PRPF3 gene is one of pre-mRNA splicing genes and located in the region 150.29–150.33 Mb on chromosome 1q216.Though PRPF3 is essential for splicing and ubiquitously expressed in almost every cell of the body, mutations in the gene have so far been reported to cause a disease that displays retina-specific phenotype RP. So far, only three clustered PRPF3 mutations, c.1482C > T (p.T494M)7,9,12,13,14,15,16, c.1478C > T (p.P493S)7,9 and c.1466C > A (p.A489D)17, have been reported in RP in several populations.
T494M is a common mutation of PRPF3 in ADRP as it has also been found in English, Danish, Spanish, American, Japanese, Korean, and Swiss ADRP families. P493S occurs in a sporadic RP case from UK or Germany and in a Caucasian ADRP family from American. A489D was reported in a Spanish ADRP family (Table 1). All these three mutations are in the exon 11 (c.1427–1526) of PRPF3, for which only the region c.1427–1526 was screened for testing PRPF3 mutations in reported studies including the original linkage paper about Family 020001 of this study, because previous surveys failed to find mutations outside of this exon8,9. The mutation c.1345C > G (p.R449G) is in the exon 10 and c.1532A > C (p.H511P) is in the exon 12, outside of the region frequently analyzed for detecting PRPF3 mutation. In our study, the identification of two novel PRPF3 mutations expands the spectrum of PRPF3 mutations in RP and suggests that more PRPF3 mutations might be found if the region outside of the exon 11 is screened. PRPF3 mutations associated with RP are reported in different populations but there is no report about PRPF3 mutation in Chinese population. This is the first report of the identification of PRPF3 mutation in the Chinese population. Total of 96 Chinese ADRP families were recruited in our study and two PRPF3 mutations were identified in two of the families. PRPF3 mutations are responsible for 2.08% of ADRP families in this cohort indicating that PRPF3 mutations might be relatively rare in Chinese patients with ADRP.
PRPF3 gene encodes a 683aa protein7, which is a human homologue of the yeast U4/U6-associated splicing factor Prp318. PRPF3 protein contains one PWI motif19, one PRP3 domain and a DUF1115 domain (Fig. 2b)20. Three previously reported mutations and two novel mutations in this study all locate in PRP3 domain (Fig. 2b). MSA revealed that those five residues mutated in RP patients were highly conserved across all the species (Fig. 2c). Structural analysis of the wild type Prp3 in the yeast spliceosomal U4/U6.U5 Tri-snrnp (PDB: 3JCM) showed that the yeast counterparts of human Prp3 residues R449 and H511 buried into two successive grooves of RNA duplex, suggesting their necessity in U4/U6 snRNA duplex interaction11 (Fig. 3a), which suggest that two novel mutations R449G and H511P probably impair binding of PRPF3- RNA duplex because both glycine and proline have propensity to break α-helical structure of PRPF3. In contrast, yeast counterparts of human PRPF3 residuesT494, P493 and A489 are clustered in the C-terminal conserved region, which play an important role in protein-protein interaction5 (Fig. 3b), which is mentioned the previous report. Our study may provide alternative insights into the relevant pathogenesis for RP.
In summary, we, for the first time, reported PRPF3 mutation spectrum in the Chinese population and demonstrated that PRPF3 mutations are responsible for 2.08% of ADRP families in this cohort indicating that PRPF3 mutation might be relatively rare in Chinese patients with ADRP. Two novel mutations, c.1345C > G (p.R449G) and c.1532A > C (p.H511P), were identified in our study, which expands the spectrum of PRPF3 mutations in RP. Structure analysis provides alternative insights into the etiology of RP. However, future research is still warranted to establish the pathogenic mechanism underlying how novel PRPF3 mutations identified in this study would cause RP.
Methods
Study Subjects and Clinical Examinations
The Institutional Review Board (IRB) of Tongji Eye Institute of Tongji University School of Medicine (Shanghai, China) and the Center for Gene Diagnosis, Zhongnan Hospital of Wuhan University (Wuhan, China) approved this study and the whole procedure of this study adhered to the tenets of the Declaration of Helsinki. All participating family members provided informed written consent that has been endorsed by the respective IRBs and is consistent with the tenets of the Declaration of Helsinki.
A three-generation Chinese Family 020001 with ADRP was recruited for this study. The routine and ophthalmologic examination were performed on all patients including visual acuity tests, the condition of the fundus, the extent of the visual field, and flash electroretinogram after informed consent form was obtained. Genotyping and linkage analysis were completed by Yuan et al.10. 7 affected and 6 unaffected individuals from this family were collected 5 ml blood samples. We also recruited another cohort of 200 unrelated and ethnically matched individuals, and an additional 95 ADRP families. Total genomic DNA was isolated with DNA extraction kits (TianGen, Beijing, China) according to the manufacturer’s protocol.
Whole-exome sequencing and data analysis
Genomic DNA (5 μg) from the proband (individual II: 4) of Family 020001 was sent to Biotechnology Corporation (Shanghai, China) for whole-exome capture followed by sequencing. The whole exome was captured using Agilent SureSelect Human All Exon Kit according to the manufacturer’s instructions. Briefly, samples were first prepared as Illumina sequencing libraries, which were then enriched for the desired target according to the Illumina Exome Enrichment protocol. Then, each captured library was loaded on a HiSeq 2500 platform, where paired-end sequencing was conducted with read lengths of 100 bp, providing an average coverage depth of at least 100× for each sample. Data were aligned to UCSC Genome Browser build hg19 by the Burroughs Wheeler Aligner. Local realignment was performed with the Genome Analysis Toolkit IndelRealigner and variants were called with SAM tools. Variants were filtered against 1000 Genomes Project, 1000G_ASN, esp6500si_all, and dbSNP137. Copy-number variations (CNVs) and structural variants from WES data were also assessed and bioinformatic prediction was based on BAM files analyzed by the Integrative Genomics Viewer (IGV) and CoNIFER.
Sanger sequencing and in silico analysis
In all members of Family 020001 with available DNA samples, the mutation was validated by Sanger sequencing as previously described15. The primers of all coding exons including flanking sequence of RPRF3 were designed by the web-based version of the Primer3 program (Table S1). Samples from another cohort of 200 unrelated ethnically matched controls and the probands of additional 95 ADRP families were also sequenced to detect variations in PRPF3.
Both variants were analyzed to determine the likelihood of pathogenicity with the following procedure. First, existing databases of disease-causing mutations (e.g. the Human Gene Mutation Database at http://www.hgmd.org) were searched for previous reports of the variant. Secondly, databases of probably benign variation such as dbSNP (http://www.ncbi.nlm.nih.gov/snp/), the 1000 Genomes database 9 (http://browser.1000genomes.org/index.html), and the NHLBI Exome Sequencing Project database (http://evs.gs.washington.edu/EVS) were also examined to determine if the variant was found in healthy controls. Third, evolutionary conservation of the amino acids region including mutation was assessed via the GeneDoc program (www.cris.com/~ketchup/genedoc.shtml) through alignment of the PRPF3 orthologous protein sequences of the following species: Homo sapiens (NP_004689.1), Pan troglodytes (NP_001233355.1), Bos taurus (NP_001039516.1), Gallus gallus (NP_001026561.1), Rattus norvegicus (NP_001102029.1), Mus musculus (NP_001303680.1), Xenopus laevis(NP_001085273.1). Danio rerio (NP_991311.2), Saccharomyces cerevisiae S288c (NP_010761.3) and Schizosaccharomyces pombe 972h- (NP_594560.1). Fourth, SIFT (Sorting Intolerant from Tolerant, http://www.sift.jcvi.org/), PolyPhen-2 (Polymorphism Phenotyping v2, http://www.genetics.bwh.harvard.edu/pph2/) and PROVEAN (Protein Variation Effect Analyzer, http://provean.jcvi.org/) online servers were used to detect the potential pathogenic impacts of the mutation. Fifth, the positions of the amino acids found mutated in RP, corresponds to their positions in yeast Prp3 (NP_010761.3) respectively by sequence alignment of PRPF3 from Homo sapiens and S. cerevisiae. The high resolution Cryo-em structure of the yeast spliceosomal U4/u6.u5 Tri-snrnp (PDB 3JCM) was used for analysis the potential effect of those mutations.
Additional Information
How to cite this article: Zhong, Z. et al. Two novel mutations in PRPF3 causing autosomal dominant retinitis pigmentosa. Sci. Rep. 6, 37840; doi: 10.1038/srep37840 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Hartong, D. T., Berson, E. L. & Dryja, T. P. Retinitis pigmentosa. The Lancet 368, 1795–1809 (2006).
Xu, L., Hu, L., Ma, K., Li, J. & Jonas, J. B. Prevalence of retinitis pigmentosa in urban and rural adult Chinese: The Beijing Eye Study. Eur J Ophthalmol 16, 865–6 (2006).
Bird, A. C. Retinal photoreceptor dystrophies LI. Edward Jackson Memorial Lecture. Am J Ophthalmol. 119, 543–62 (1995).
Liu, M. M. & Zack, D. J. Alternative splicing and retinal degeneration. Clin Genet 84, 142–9 (2013).
Ruzickova, S. & Stanek, D. Mutations in spliceosomal proteins and retina degeneration. RNA Biol 1–9 (2016).
Heng, H. H., Wang, A. & Hu, J. Mapping of the human HPRP3 and HPRP4 genes encoding U4/U6-associated splicing factors to chromosomes 1q21.1 and 9q31-q33. Genomics 48, 273–5 (1998).
Chakarova, C. F. et al. Mutations in a third member of pre-mRNA splicing factor genes, implicated in autosomal dominant retinitis pigmentosa. Hum Mol Genet 11, 87–92 (2002).
Sullivan, L. S. et al. Prevalence of Mutations in eyeGENE Probands With a Diagnosis of Autosomal Dominant Retinitis Pigmentosa. Invest Ophthalmol Vis Sci 54, 6255–61 (2013).
Sullivan, L. S. et al. Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: a screen of known genes in 200 families. Invest Ophthalmol Vis Sci 47, 3052–64 (2006).
Yuan, Y., Zhou, X., Wang, F., Yan, M. & Ding, F. Evidence for a novel autosomal dominant retinitis pigmentosa linked to chromosome 1p22.1-q12 in a Chinese family. Curr Eye Res 36, 154–67 (2011).
Wan, R. et al. The 3.8 A structure of the U4/U6.U5 tri-snRNP: Insights into spliceosome assembly and catalysis. Science 351, 466–75 (2016).
Vaclavik, V., Gaillard, M. C., Tiab, L., Schorderet, D. F. & Munier, F. L. Variable phenotypic expressivity in a Swiss family with autosomal dominant retinitis pigmentosa due to a T494M mutation in the PRPF3 gene. Molecular Vision 16, 467–475 (2010).
Marti’nez-Gimeno, M. et al. Mutations in the Pre-mRNA Splicing-Factor GenesPRPF3,PRPF8, andPRPF31in Spanish Families with Autosomal Dominant Retinitis Pigmentosa. Investigative Opthalmology & Visual Science 44, 2171 (2003).
Kim, C. et al. Microarray-based mutation detection and phenotypic characterization in Korean patients with retinitis pigmentosa. Mol Vis 18, 2398–410 (2012).
Audo, I. et al. Development and application of a next-generation-sequencing (NGS) approach to detect known and novel gene defects underlying retinal diseases. Orphanet J Rare Dis. 7, 8 (2012).
Wada, Y., Itabashi, T., Sato, H. & Tamai, M. Clinical features of a Japanese family with autosomal dominant retinitis pigmentosa associated with a Thr494Met mutation in the HPRP3 gene. Graefes Arch Clin Exp Ophthalmol 242, 956–61 (2004).
Gamundi, M. J. et al. Transcriptional expression of cis-acting and trans-acting splicing mutations cause autosomal dominant retinitis pigmentosa. Hum Mutat 29, 869–78 (2008).
Agafonov, D. E. et al. Molecular architecture of the human U4/U6.U5 tri-snRNP. Science 351, 1416–20 (2016).
Blencowe, B. J. & Ouzounis, C. A. The PWI motif: a new protein domain in splicing factors. Trends Biochem Sci 24, 179–80 (1999).
Marchler-Bauer, A. et al. The Conserved Domain Database (CDD). Nucleic Acids Res. 43 (Database issue): D222–D226 (2015).
Acknowledgements
The authors thank all patients and their family members for their participation. We also thank Dr. Cindy Yang and Dr. Jingbo Xiao for helpful comments. This work was supported by the National Key Basic Research Program of China (973 Program 2015CB964601); National Natural Science Foundation of China (81371062, 31501014, 81472024, and 81270365); and Thousand Youth Talents Program of China (to J.C.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Z.Z. (Zilin Zhong) and M.Y. contributed equally to this report. Study design: Z.Z. (Zilin Zhong), F.Z. and J.C. the samples and performed the experiments: Z.Z. (Zilin Zhong), M.Y., L.H., Z.W., W.S. and F.Z. Data interpretation and analysis: Z.Z. (Zilin Zhong), F.Z., Z.Z. and J.C. Manuscript preparation: Z.Z. (Zilin Zhong) and J.C. All authors have read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhong, Z., Yan, M., Sun, W. et al. Two novel mutations in PRPF3 causing autosomal dominant retinitis pigmentosa. Sci Rep 6, 37840 (2016). https://doi.org/10.1038/srep37840
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep37840
This article is cited by
-
TMEM43 promotes pancreatic cancer progression by stabilizing PRPF3 and regulating RAP2B/ERK axis
Cellular & Molecular Biology Letters (2022)
-
Long-term clinical course of 2 Japanese patients with PRPF31-related retinitis pigmentosa
Japanese Journal of Ophthalmology (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
