
Abstract
The CRISPR-associated endonuclease Cas9 from Streptococcus pyogenes (spCas9) along with a single guide RNA (sgRNA) has emerged as a versatile toolbox for genome editing. Despite recent advances in the mechanism studies on spCas9-sgRNA-mediated double-stranded DNA (dsDNA) recognition and cleavage, it is still unclear how the catalytic Mg2+ ions induce the conformation changes toward the catalytic active state. It also remains controversial whether Cas9 generates blunt-ended or staggered-ended breaks with overhangs in the DNA. To investigate these issues, here we performed the first all-atom molecular dynamics simulations of the spCas9-sgRNA-dsDNA system with and without Mg2+ bound. The simulation results showed that binding of two Mg2+ ions at the RuvC domain active site could lead to structurally and energetically favorable coordination ready for the non-target DNA strand cleavage. Importantly, we demonstrated with our simulations that Cas9-catalyzed DNA cleavage produces 1-bp staggered ends rather than generally assumed blunt ends.
Similar content being viewed by others

Introduction
The clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) systems provide bacteria and archaea with adaptive immunity against invasive viruses and plasmids1,2,3,4. The Cas9 protein from Streptococcus pyogenes (spCas9), combined with a single guide RNA (sgRNA), a synthetic fusion of CRISPR RNA (crRNA) and trans-activating crRNA (tracrRNA), has been adapted to a most widely used toolbox for gene editing in various living cells and organisms, demonstrating great potential toward further therapeutics applications5,6,7,8.
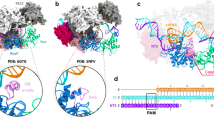
The biochemical experiments have established that double-stranded DNA (dsDNA) recognition and cleavage by Cas9 strictly require the presence of a short protospacer-adjacent-motif (PAM) in the non-target, or non-complementary, DNA strand (ntDNA) and the complementarity of the target DNA strand (tDNA) to the 10–12 nucleotide (nt) PAM-proximal “seed” region in the guide RNA6,9. Recently solved crystal structures of spCas9 reveal a bilobed architecture comprising of an α-helical recognition (REC) lobe and a nuclease (NUC) lobe (Fig. 1A)10,11,12,13,14,15,16. The NUC lobe incorporates two Mg2+-dependent nuclease domains, dubbed as RuvC and HNH, which are responsible for cutting the ntDNA and tDNA, respectively (Fig. 1B,C)6,9. The RuvC domain features a typical ribonuclease H (RNase H) fold with four functionally essential residues, D10, E762, H983 and D986 (Fig. 1B)6,12, in line with a two-metal-ion catalysis mechanism17,18,19. In contrast, the HNH domain is characterized by a ββα-metal motif with three catalytic residues, D839, H840 and N863 (Fig. 1C)6,10,12, consistent with a one-meta-ion mechanism17,18,19.

spCas9-sgRNA complexed with double strand DNA (dsDNA) substrate (A) and spCas9-catalyzed DNA hydrolysis (B–D). (A) Overall architecture of the ternary complex of spCas9, sgRNA and dsDNA. The domain organization of spCas9 is indicated in bottom-left corner. The sgRNA is colored orange, target DNA strand (tDNA) in Kelly green and non-target DNA strand (ntDNA) in cyan. (B and C) Close-up view of the catalytic center for RuvC (upper) and HNH (lower) nuclease domain. The catalytically essential residues are depicted in a stick model and colored by atom type (C, yellow; N, violet, O, red). (D) Schematic diagram showing the placement of Mg2+ pair at the interface of ntDNA and RuvC domain. S1 and S2 denote the replicated simulations in which the pair of Mg2+ were initially positioned around -3 phosphate (−3P), S5 and S6 around -4 phosphate (−4P) and S3 and S4 in between −3P and −4P (see Materials and Methods for more details). When labeling, the pro-Sp and pro-Rp oxygens of the phosphate group are abbreviated as Sp and Rp, respectively. In all panels, the 5′-NGG-3′ PAM trinucleotide in the ntDNA is highlighted in red. The putative cleavage site is denoted by a scissor shape.
A wealth of biochemical and structural information has made remarkable contributions to the mechanistic insights into RNA-guided DNA recognition and cleavage by Cas9. However, many details of the cleavage process, such as how the catalytic Mg2+ ions mediate the connections between Cas9 and DNA, remain elusive in this system11,12,16. The recent crystal structures show a closest distance of ~5.5 Å between the superimposed Mn2+ ions and the non-bridging oxygen atoms of the putative scissile phosphate on the ntDNA11,16, which is considerably larger than the typical Mg-O coordination distance of 2.1 Å for catalysis, indicating the crystalized conformation is inactive for catalysis. Intriguingly, the HNH domain assumes an inactive conformation in all the complex structures10,11,12,13,14,15,16, in which the shortest distance of the catalytic residue H840 to the opposite cleavable site on the DNA is around 14 Å, which is far beyond the range required for catalysis. To date, no active state conformation together with Mg2+ ion has been reported. We hypothesize that the catalytic Mg2+ ions induce the conformational change to an active state by facilitating the proximity of Cas9 active sites and DNA. Here, we used molecular dynamics simulations to search for the active state and to test our hypothesis.
Unlike HNH domain-mediated tDNA cleavage, determining the exact cleavage site on the ntDNA is complicated due to the additional 3′->5′ exonuclease activity of the RuvC domain6,16, especially when the protospacer 5′-end is used for direct sequencing analysis or radiolabeled for gel electrophoresis analysis2,6,9. Moreover, the target site detection methods, including direct sequencing and gel electrophoresis analysis, appear to exhibit limited resolution in terms of discrimination of 1-nt length6,9. Therefore, it is still controversial as to whether the spCas9 cuts the ntDNA at the phosphate 3 base pair (bp) upstream of the canonical 5′-NGG-3′ PAM motif (denoted as −3P) or 4 bp apart (−4P) (see Fig. 1D)6,9,10,11,20. This naturally elicits another interesting and critical question: Does Cas9 produce blunt-ended breaks or staggered-ended breaks with overhangs in the target dsDNA? Some studies reported that Cas9 cuts the ntDNA at −3P as the tDNA at +3 P, thereby leaving blunt ends9,20, whereas others argued that −4P is the ntDNA cleavage site, implying staggered ends generated by Cas910,11. Meanwhile, there exists another view that Cas9 produces either blunt or 1-bp staggered ends5,6. For genome-editing, staggered-ended DNA is more controllable than the blunt-ended DNA because the DNA repair would only occur at the staggered end rather than at either end. To determine which type of ends generated by Cas9 is critical to understand the mechanism of non-homologous end joining (NHEJ)-based gene repair following the DNA cleavage21,22,23, hence shedding lights on strategy development to enhance precise genome engineering.
Molecular dynamics (MD) simulation has emerged as a powerful tool to explore bio-macromolecule dynamics in complementary with experimental data. To investigate the mechanisms underlying Cas9-catalyzed cleavage process, here, we present the first computational simulation study on the Cas9-sgRNA-dsDNA system at the atomic level to address the following two questions: 1) Could the Mg2+ ions bridge the distance gap between the active sites of Cas9 and ntDNA? 2) Where is the cleavage site on ntDNA? Our simulation results demonstrate that introduction of two Mg2+ ions at −4P remarkably induces conformation changes to bring the active site of the RuvC domain and ntDNA into proximity, forming structurally and energetically favorable coordination in between the catalytic residues and the scissile phosphate for cleavage reaction to occur. Thus, we evidence that Cas9 generates 1-bp 5′-staggered breaks in the dsDNA.
Results
Mg2+ Ions Induce Conformational Changes to Bridge the Distance Gap between the Active Site of the RuvC Domain and the ntDNA
To probe the effect of Mg2+ on conformational change, one Mg2+ ion was placed in the HNH domain on the basis of the one-metal ion catalysis mechanism (Fig. 1C), whereas two Mg2+ ions were positioned at the interface of the RuvC domain and the ntDNA at various positions according to the two-metal-ion mechanism (Fig. 1D)17,18,19. For simplicity, S0 here represents the simulation in the absence of Mg2+; S1/S2 and S5/S6 denote the repeated simulations in which the Mg2+ pair were positioned around −3P and around −4P, respectively, whereas S3/S4 are the two simulations with the Mg2+ ions placed in between −3P and −4P (see Materials and Methods for details). Analysis of the root-mean-square deviations (RMSD) of the simulated systems revealed that the overall stability of the entire complex is largely determined by the sgRNA (Supplementary Fig. S1), as its long middle segment protrudes into the solvent, exhibiting high flexibility (Fig. 1 A). The Cas9 protein and the duplex DNA tended to reach equilibrium after dozens of nanoseconds, with RMSD fluctuating around 3 Å. The above observations are in line with a most recent study by Palermo et al.24. If not otherwise specified, all the following results were derived since 40 ns of the simulation trajectories.
We first monitored the proximity of the ntDNA and RuvC active center by measuring the distances between −3P and His983 (d-3P/His983) and between −4P and Asp10 (d-4P/Asp10) (Fig. 2A,B) for each trajectory. Without Mg2+ ions (S0), the distances between the ntDNA and the RuvC domain active residues peak at ~10 Å for both measurements (Fig. 2A,B). Compared to the control simulation (S0), addition of the Mg2+ ions obviously shifted the distance distributions of ntDNA and RuvC active center to smaller values in varying degrees regardless of the Mg2+ ions positions (S1 to S6), indicating the presence of metal ions could indeed induce the conformational change toward the active state and close the distance gap between the ntDNA and the RuvC domain as previously postulated11,12,16 and as we hypothesized. Among all simulations, the double Mg2+ binding at the −4P in S5 and S6 leads to the largest reduction in d-3P/His983 (by ~4 Å) as well as in d-4P/Asp10 (by ~6 Å) when comparing the highest peaks with that of S0 (Fig. 2A,B).

Distance distributions between the selected catalytic residues and the opposite phosphates on the target DNA (A–C) and between the two Mg2+ ions introduced to the RuvC domain (D) calculated from individual simulations. (A) Probability density for the distance between the Cγ atom of His983 on the RuvC domain and the P atom of −3 phosphate on the ntDNA. (B) Probability density for the distance between the Cγ atom of Asp10 on the RuvC domain and the P atom of -4 phosphate on the ntDNA. (C) Probability density for the distance between the Cγ atom of His840 on the HNH domain and the P atom of +3 phosphate on the tDNA. (D) Probability density for the distance between the Mg2+ pair positioned at the RuvC domain. At the upper right of each panel, a schematic diagram is given to indicate the calculated distance.
However, the HNH domain still adopted an inactive conformational state exhibiting no trend of moving close to the tDNA in all the simulations, as characterized by the distribution profiles of the distance of +3 P and His840 (d+3P/His840) that are largely overlaid with each other (Fig. 2C). Though the high intrinsic flexibility in the HNH domain11,13,16,25, our observation might result from the simulation time scale (hundreds of ns) far from sampling an activated state. More likely, in the context of dsDNA, the conformational switch of HNH domain is dependent on the completion of RuvC domain-catalyzed ntDNA cleavage accompanying by its product release13,25, which is beyond the scope of conventional MD simulation. In the following section, we focus on Mg2+-involved interactions with the RuvC domain and ntDNA.
Mg2+ Ions Binding at −4P Lead to Active State Formation for ntDNA Cleavage
Further inspection of the simulation trajectories revealed distinct binding poses of the Mg2+ pair at the interface of RuvC domain and ntDNA. In S1 and S2, the Mg2+ ion A (the one on the 3′ side of ntDNA) moves toward the −2P from its initial location at −3P (Fig. 1D, Fig. 3A and Supplementary Fig. S3A). In S3 and S4, the Mg2+ B (the one on the 5′ side of ntDNA) departs from the middle position in between −3P and −4P, entering the negatively charged center enclosed by Asp10, E762 and D986 or −4P (Fig. 1D, Fig. 3B and Supplementary Fig. S3B). Positioning Mg2+ at either −3P (S1/S2) or in between −3P and −4P (S3/S4) all led to the Mg-Mg distance larger than 6 Å (Fig. 2D). In S5 and S6, the Mg2+ pair stays around −4P (Fig. 1D, Fig. 3C and Supplementary Fig. S3C), resulting in the lowest Mg-Mg separation distance (4.3~4.7 Å) among all simulations, which is comparable to the inter-Mg2+ distance (4.1 Å) from the experimental and theoretical studies on another two-metal-ion catalytic system with two Mg2+ trapped in between the Bacillus halodurans ribonuclease H (RNase H) and an RNA/DNA hybrid (Fig. 3D and Supplementary Fig. S3D)17,26,27,28.

The representative coordination configurations involving the Mg2+ pair obtained from each set of simulations (A–C) and comparisons with the two-metal-ion catalysis by the RNase H (D). (A) The most-populated snapshot from S1. Note that the second largest cluster has a comparable population to the first one and the corresponding snapshot is present in Supplementary Fig. S2. (B) The most-populated snapshot from S3. (C) The most-populated snapshot from S5. (D) The schematic diagram of canonical two-metal-ion coordination by the RNase H. The RuvC and ntDNA residues are represented in stick model and colored by atom type and the ligand water are shown as stick and ball style. The Mg2+ pair is illustrated as cyan spheres, with the one Mg2+ on the 3′ side of the ntDNA labeled “A” and the other one on the 5′ side labeled “B”. When labeling, the pro-Sp and pro-Rp oxygens of the phosphate group are abbreviated as Sp and Rp, respectively. The green dashed line indicates the coordination bond involving Mg2+, which is defined as atom-atom distance smaller than 2.2 Å. The black dashed line denotes the hydrogen bond. The potential nucleophilic water is attached by an arrow. The average coordination distances between Mg2+ and phosphate non-bridging oxygen, protein residue oxygen, water oxygen and protein residue nitrogen are 1.9 (0.1), 2.0 (0.1), 2.1 (0.1) and 2.2 (0.0) Å, respectively, from the simulations. See also the other snapshots for S2, S4 and S6 in Supplementary Fig. S3.
In the framework of two-metal-ion mechanism, the two metal ions are coordinated by an invariable Asp (that may be a phosphate in ribozymes) and the scissile phosphate17,18,19. Among all the simulations, only the coordination composition and geometry captured in S5 and S6 (Fig. 3C; Supplementary Fig. S3C), in which the conserved Asp10 and the −4P contribute two coordination ligands to the double Mg2+, respectively, closely match those observed in the prototype RNase H system (Fig. 3D and Supplementary Fig. S3D). Unlike the other simulations (S1/S3 in Fig. 3A,B and S2/S4 in Supplementary Fig. S3A,B), in which Mg2+ ions only coordinated with one or two catalytic residues, all three acid catalytic residues (Asp10, E762 and Asp986) are involved in coordination with the two metal ions in S5 and S6. Specifically, it is interesting to note that despite the similarity in the coordination configuration, the general base His983 in S6 is directly engaged to the Mg2+ A (Supplementary Fig. 3C), whereas in S5 it is hydrogen-bonded to a water molecule that provides a ligand to the Mg2+ A coordination (Fig. S3C).
Taken together, the observations uncover formation of an active state for ntDNA cleavage stemming from the binding of two Mg2+ at –4 P. Yet it should be mentioned that compared with the canonical two-mental-ion coordination in the RNase H system, there exists a noticeable difference in terms of the scissile phosphate-mediated coordination with the two Mg2+. Both in S5 and S6, the two non-bridging oxygens (pro-Sp and pro-Rp) of −4P together form a bidentate coordination with the double Mg2+ ions (Fig. 3C and Supplementary Fig. S3C), whereas in the RNase H-substrate complex, only the pro-Sp oxygen is implicated (Fig. 3D and Supplementary Fig. S3D). This may account for why the separation of the two Mg2+ (Fig. 2D) and that of the Mg2+ B and the leaving group 3′-O in S5 and S6 (Fig. 3C and Supplementary Fig. S3C) are slightly larger than those observed in the crystal structure of RNase H complex (Fig. 3D and Supplementary Fig. S3D).
The Double Mg2+ Binding at −4P is Energetically More Favorable than Binding at Other Positions
To provide a quantitative view of the preference of the two Mg2+ ions for the scissile phosphate, we estimated the binding free energies of the Mg2+ pair at various positions via the end-point MM-GBSA approach29,30,31. Compared to the alternative MM-PBSA, MM-GBSA is computationally more efficient and has demonstrated the advantage in ranking the affinities of a series of ligands32,33,34. Given that water molecules play an important role in coordinating the catalytic ions (Fig. 3 and Supplementary Fig. S3), we consistently retained four water molecules that are closest to the two Mg2+ as part of receptor in the trajectories for MM-GBSA calculations (see Materials and Methods for details). The results showed that the binding of the Mg2+ pair at −4P in S5 and S6 is more stable than those at −3P or in between −3P and −4P in other simulations (S1–S4) (Table 1). Moreover, the binding free energy values of the set of parallel simulations are comparable in magnitude, indicating similar stability for the two coordination geometries captured in S5 and S6 (Fig. 3C and Supplementary Fig. S3C). We also calculated the non-bonded interaction energy of the Mg2+ pair with all surrounding residues and water and found a similar trend to that of MM-GBSA (Supplementary Table S1).
The biochemical experiments have evidenced the functional importance of four residues (viz. Asp10, Glu762, His983 and Asp986) at the RuvC active center for Cas9 catalytic activity6,12. We further performed per-residue free energy decomposition on these four residues to determine the contributions of individual catalytic residues to the binding of the metal ions. In S2, all of the four active residues hardly contribute to the binding of the two Mg2+, while Asp10 is the sole major contributor in S1 (Fig. 4). Compared to S3, Asp986 in S4 shows much stronger binding affinity (Fig. 4), accounting for the marked difference in binding free energy for the two simulations (Table 1). Notably, the two acid residues, Asp10 and Glu762 in S5/S6 make much larger contributions to the binding than in other simulations (Fig. 4), again suggesting most favorable binding of the Mg2+ at –4 P (Table 1). In contrast, His983 contributes marginally to stabilizing Mg2+, which is consistent with its major role as a general base activating the nucleophile17,18,19. The per-residue contribution analyses here are in line with the above structural observations (Fig. 3 and Supplementary Fig. S3).

Relative binding strength for the RuvC catalytic residues calculated from respective sets of simulations via the MM-GBSA approach.
Note that among all the simulations, the Asp10 in S5 gives the largest energetic contribution and hence is indicated 100% binding strength in the bar plot to which other residues are compared.
Discussion and Conclusions
Several crystal structures of spCas9 have been solved, however, none of these structures assume the catalytic active state. One of the reasons is that to crystalize the ternary complex of spCas9-sgRNA-dsDNA, cleavage has to be prevented by substitution of catalytic residues and/or chelation of metal ions so that the systems do not support catalysis12,16. The use of partial ntDNA (cleaved product) for complex crystallization may also impair two metal ion binding10,14,15. In the complex structure with PAM-containing partial DNA duplex (PDB code: 4UN3), for example, only a single Mg2+ is non-specially bound at the RuvC active site, forming an incomplete coordination due to lack of the involvement of phosphate group (Supplementary Fig. S4A)10. It is interesting to note that the ion A of the Mg2+ pair in our simulations spatially overlaps with that one in 4UN3 (Supplementary Fig. S4A). In the apo-Cas9 without sgRNA and dsDNA (PDB code: 4CMQ), two Mn2+ ions instead of Mg2+ were found at the RuvC active site but only at a concentration much higher than physiologically relevant11 (Supplementary Fig. S4B). Here, for the first time, we determined the catalytic active state for the RuvC domain and the ntDNA by scanning the possible scissile phosphates on the ntDNA with a pair of Mg2+. The presence of Mg2+ ions at −4P remarkably drive the RuvC domain active center to the opposite phosphate backbone (Fig. 2), leading to a reactant-like coordination primed for cleavage (Fig. 3 and Supplementary Fig. S3). We demonstrated that beyond its catalytic role, the second role of Mg2+ ions is to lead the inactive conformation of the RuvC domain and the ntDNA toward the active state for catalysis. Our results add a piece of evidence corroborating the argument that acquisition of metal ions could strength specific substrate recognition and association, as observed in many polymerases and nucleases17,18,19.
From the twin Mg2+ coordination fashions and the calculated binding free energies in different sets of simulations, we concluded that it is very unlikely for the RuvC domain to cut the ntDNA at −3P due to obvious disobedience with the two-metal-ion mechanism that entails joint coordination of the Mg2+ pair by the scissile phosphate and a conserved Asp (Fig. 3 and Supplementary Fig. S3), as well as the unfavorable binding free energy comparing to other binding conformations (Table 1). Rather, we argue that −4P is the ntDNA cleavage site based on the formation of structurally and energetically favorable coordination, which is supported by the enzymatic footprinting experiments11 and also consistent with the structural observation showing the acidic residue cluster in the RuvC domain active center is spatially closer to −4P than −3P16. To further enhance the reliability of the conclusion, we additionally performed four shorter simulations (4 × 60 ns) starting with randomly assigned velocities for each of the three binding configurations (i.e. Mg2+ pair placed at −3P, −3P/−4P and −4P, respectively) and found very similar results with respect to the two longer simulations here (2 × 200 or 2 × 300 ns) (see full details in the Supplementary Information). Since the HNH domain-mediated tDNA cleavage has been more unambiguously identified as occurring at +3P9,11, we rationalize 1-bp 5′-staggered ends initially generated by Cas9 other than generally assumed blunt ends9,20. This staggered end may be further extended to a length of interest by rationally exploiting the additional 3′->5′ exonuclease-trimming activity in the RuvC domain6,16, which could be particularly advantageous for facilitating non-homologous end joining (NHEJ)-mediated gene insertion into the mammalian genome21,22,23. Considering that the small molecules that enhance NHEJ activities have been demonstrated to enhance the CRISPR/Cas genome editing35,36, we speculate that mutations or small molecules that could modulate the cleavage site to make the 1-bp staggered end more “stagger” may also help to enhance the CRISPR/Cas9 genome editing.
In this report, we did not address the questions on the HNH domain. For example, what triggers the HNH domain to convert into an active conformation? How does the Mg2+ ion bridge the distance gap between the HNH domain and the tDNA? These intriguing questions will be investigated in our future studies. We also noticed the derived two-metal-ion coordination geometries are somewhat deviated from that by the RNase H (Fig. 3C,D and Supplementary Fig. S3C,D), though this does not qualitatively influence the conclusions drawn here. We suspect that the discrepancy is related to the additive force field, but we cannot rule out the possibility that two-metal-ion catalysis mechanism of Cas9 is slightly different from that of RNase H. These issues will be addressed in our future studies by using the polarizable force fields in the MD simulations and using the quantum mechanical/molecular mechanical (QM/MM) approaches to explore the free energy surface for catalysis reactions28.
In summary, our study indicates that binding of two catalytic Mg2+ ions at the RuvC domain catalytic center facilitates the formation of an active state for ntDNA cleavage, and, importantly, that Cas9-catalyzed target DNA cleavage produces 1-bp staggered ends rather than the blunt ends. The staggered ends have more advantages than the blunt ends in genome editing because the staggered ends help the DNA insertion in the right direction, making genome editing more efficient and more specific. The short staggered ends revealed in this study shed lights on further improvement of CRISPR/Cas9 technology to increase its efficiency and specificity.
Materials and Methods
System Setup
The initial coordinates of Cas9-sgRNA-dsDNA were taken from the Protein Data Bank (PDB code: 5F9R16). Compared to other DNA-bound crystal structures10,12,14,15, 5F9R has no missing residues and contains no mutations. Of particular note is that the dsDNA in 5F9R is trapped in a pre-cleavage state with an intact non-target strand, which is especially suitable for this study. However, catalytic metal ions are excluded from all the available DNA-bound crystal structures. Based on the two-metal-ion catalysis mechanism proposed for the RuvC domain11,12,16, two Mg2+ ions were positioned at 5′- and 3′-sides of the pre-Sp oxygen of the -3 phosphate (−3P) or −4 phosphate (−4P) (Fig. 1D and Table 2), respectively, by reference to the crystal structure of Mg2+-bound RNase H in complex with an RNA/DNA hybrid (PDB code: 1ZBI)17,26,27. Additionally, the intermediate Mg2+ locations between −3P and −4P were also taken into account, in which one Mg2+ ion was placed close to 5′-side of the pro-Sp oxygen of −3P and the other to 3′-side of the pro-Sp oxygen of −4P (Fig. 1D and Table 2). Given that other sites except −3P and −4P are evidently too far from RuvC catalytic center, we did not introduce Mg2+ ions therein (Fig. 1B,D). Meanwhile, we added one Mg2+ ion to the HNH domain following the one-metal-ion mechanism17,18,19. Note that in this crystal structure, the HNH domain active site is still considerably apart from the cleavable site on the tDNA as reflected by a distance of 17.25 Å between the His840 Cγ atom and the P atom of +3P, while the corresponding value is far higher in other target-bound structures (32~35 Å)10,12,14,15. Each Mg2+-bound complex above was solvated with TIP3P water molecules with a thickness of 13.5 Å, leading to a periodic boundary box of 139 × 124 × 187 Å3. To mimic the reaction buffer6,9, extra 16 Mg2+ were added into the water box to generate a concentration of 10 mM and the ionic strength of KCl was set to 100 mM. As a control, the original system without Mg2+ was also simulated (Table 2). The total atoms of each system add up to ~282,000.
Molecular Dynamics Simulations
All the simulations were performed by the AMBER15 pmemd engine with GPU acceleration37. The amber ff14SB force fields38 were used to describe the atomic interactions involving the protein and nucleic acids and the recently developed ion parameter sets optimized in TIP3P water39,40 were selected for the mono- and divalent ions. It is noted that none of the available non-bonded models for Mg2+ ion is able to reproduce various experimental properties simultaneously; the set of Mg2+ parameters here represent the best possible compromise targeting the experimental Mg-O distance, hydration free energy as well as coordination number40. The non-bonded interactions were truncated at 10 Å and the long-range electrostatics were treated via the particle mesh Eward summation (PME) method41 with a grid spacing of 1 Å. The covalent bonds involving hydrogen atoms were constrained employing the SHAKE algorithm42. Each simulation system was first subjected to a thorough energy minimization with the solute backbone atoms restrained, followed by slow heating from 0 K to 310.15 K and 10-ns equilibration in the isothermal-isochoric (NVT) ensemble with a time step of 1 fs. The production simulations were conducted under the isothermal-isobaric (NpT) conditions using a time step of 2 fs and were extended to at least 200 ns (Table 2), in which the temperature was maintained at 310.15 K through the Langevin thermostat and the pressure was controlled at 1.013 bar by the Monte Carlo barostat. The trajectory snapshots were saved every 10 ps. Except for the control system, two long parallel simulations (2 × 200 or 2 × 300 ns) and four additional short simulations (4 × 60 ns) with different random seeds were performed for the remaining ones (Table 2; see also Supplementary Information).
Cluster Analysis
The representative (i.e. most-populated) configurations involving the double-Mg2+ coordination were determined through the cluster analysis with the package VMD43. For each simulation trajectory, the structure ensemble of snapshots since 40 ns was grouped into four clusters by trying varying root-mean-square deviation (RMSD) cutoffs (0.6–1.4 Å), after structural alignment on the reaction interface comprising the four catalytic residues (D10, E762, H983 and D986) on the RuvC domain, the backbone of nucleotides -3 and -4 on the ntDNA and the Mg2+ pair lying between them. In the final trials, the first two clusters were found to account for >90% of total population and the snapshot closest to the centroid of largest structural ensemble was extracted for comparative analysis.
Binding Free Energy Calculations and Per-residue Energy Decomposition
The binding stability of the Mg2+ pair at respective positions was evaluated via the end-point Molecular Mechanics-Generalized Born Surface Area (MM-GBSA) approach29,30,31. Compared to the alternative Molecular Mechanics-Poisson Boltzmann Surface Area (MM-PBSA), MM-GBSA is computationally more efficient and has proved to give comparable or even better accuracy in ranking ligand affinities32,33,34. The energy terms were calculated via the program MMPBSA.py in AmberTools1444. The entropy contribution was not included here, as omission of this term does not qualitatively affect the accurate ranking of a series of ligands against the same receptor32,33,34. The snapshots used for MM-GBSA calculations were extracted since 40 ns of the production simulation at 50-ps intervals, generating 3,600 structures for S1 to S4 and 5,600 structures for S5 and S6, respectively (Table 2). In each trajectory snapshot, four water molecules closest to the Mg2+ pair were consistently kept and considered as part of the Cas9-sgRNA-dsDNA “receptor”, while the double ions were regarded as a whole “ligand”. Meanwhile, per-residue free energy decomposition was performed to estimate the binding strengths of the RuvC active residues (viz. D10, E762, H983 and D986) to the Mg2+ pair. In the final results, we reported the block averaging over even non-overlapping trajectory sections (each containing 400 snapshots) together with the standard error of the mean.
Additional Information
How to cite this article: Zuo, Z. and Liu, J. Cas9-catalyzed DNA Cleavage Generates Staggered Ends: Evidence from Molecular Dynamics Simulations. Sci. Rep. 6, 37584; doi: 10.1038/srep37584 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Barrangou, R. et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712 (2007).
Garneau, J. E. et al. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468, 67–71 (2010).
Marraffini, L. A. & Sontheimer, E. J. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat. Rev. Genet. 11, 181–190 (2010).
Wiedenheft, B., Sternberg, S. H. & Doudna, J. A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 482, 331–338 (2012).
Tsai, S. Q. & Joung, J. K. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nat. Rev. Genet. 17, 300–312 (2016).
Jinek, M. et al. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 337, 816–821 (2012).
Cong, L. et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 339, 819–823 (2013).
Mali, P. et al. RNA-Guided Human Genome Engineering via Cas9. Science 339, 823–826 (2013).
Gasiunas, G., Barrangou, R., Horvath, P. & Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 109, E2579–E2586 (2012).
Anders, C., Niewoehner, O., Duerst, A. & Jinek, M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 513, 569–573 (2014).
Jinek, M. et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 343, 1247997 (2014).
Nishimasu, H. et al. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156, 935–949 (2014).
Jiang, F., Zhou, K., Ma, L., Gressel, S. & Doudna, J. A. A Cas9-guide RNA complex preorganized for target DNA recognition. Science 348, 1477–1481 (2015).
Anders, C., Bargsten, K. & Jinek, M. Structural Plasticity of PAM Recognition by Engineered Variants of the RNA-Guided Endonuclease Cas9. Mol. Cell 61, 895–902 (2016).
Hirano, S., Nishimasu, H., Ishitani, R. & Nureki, O. Structural Basis for the Altered PAM Specificities of Engineered CRISPR-Cas9. Mol. Cell 61, 886–894 (2016).
Jiang, F. G. et al. Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science 351, 867–871 (2016).
Yang, W., Lee, J. Y. & Nowotny, M. Making and breaking nucleic acids: two-Mg2+-ion catalysis and substrate specificity. Mol. Cell 22, 5–13 (2006).
Yang, W. An equivalent metal ion in one- and two-metal-ion catalysis. Nat. Struct. Mol. Biol. 15, 1228–1231 (2008).
Yang, W. Nucleases: diversity of structure, function and mechanism. Q. Rev. Biophys. 44, 1–93 (2011).
Zetsche, B. et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 163, 759–771 (2015).
Maresca, M., Lin, V. G., Guo, N. & Yang, Y. Obligate Ligation-Gated Recombination (ObLiGaRe): Custom-designed nuclease-mediated targeted integration through nonhomologous end joining. Genome Res. 23, 539–546 (2013).
Budman, J. & Chu, G. Processing of DNA for nonhomologous end-joining by cell-free extract. EMBO J. 24, 849–860 (2005).
Guirouilh-Barbat, J. et al. Impact of the KU80 pathway on NHEJ-induced genome rearrangements in mammalian cells. Mol. Cell 14, 611–623 (2004).
Palermo, G., Miao, Y., Walker, R. C., Jinek, M. & McCammon, J. A. Striking Plasticity of CRISPR-Cas9 and Key Role of Non-target DNA, as Revealed by Molecular Simulations. ACS Cent. Sci, doi: 10.1021/acscentsci.6b00218 (2016).
Sternberg, S. H., LaFrance, B., Kaplan, M. & Doudna, J. A. Conformational control of DNA target cleavage by CRISPR-Cas9. Nature 527, 110–113 (2015).
Nowotny, M. & Yang, W. Stepwise analyses of metal ions in RNase H catalysis from substrate destabilization to product release. EMBO J. 25, 1924–1933 (2006).
Nowotny, M., Gaidamakov, S. A., Crouch, R. J. & Yang, W. Crystal structures of RNase H bound to an RNA/DNA hybrid: Substrate specificity and metal-dependent catalysis. Cell 121, 1005–1016 (2005).
Rosta, E., Nowotny, M., Yang, W. & Hummer, G. Catalytic mechanism of RNA backbone cleavage by ribonuclease H from quantum mechanics/molecular mechanics simulations. J. Am. Chem. Soc. 133, 8934–8941 (2011).
Kollman, P. A. et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 33, 889–897 (2000).
Chong, L. T., Duan, Y., Wang, L., Massova, I. & Kollman, P. A. Molecular dynamics and free-energy calculations applied to affinity maturation in antibody 48G7. Proc. Natl. Acad. Sci. USA 96, 14330–14335 (1999).
Cheatham, T. E. 3rd, Srinivasan, J., Case, D. A. & Kollman, P. A. Molecular dynamics and continuum solvent studies of the stability of polyG-polyC and polyA-polyT DNA duplexes in solution. J. Biomol. Struct. Dyn. 16, 265–280 (1998).
Sun, H. Y., Li, Y. Y., Tian, S., Xu, L. & Hou, T. J. Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys. Chem. Chem. Phys. 16, 16719–16729 (2014).
Hou, T. J., Wang, J. M., Li, Y. Y. & Wang, W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 51, 69–82 (2011).
Zuo, Z. C., Weng, J. W. & Wang, W. N. Insights into the Inhibitory Mechanism of D13-9001 to the Multidrug Transporter AcrB through Molecular Dynamics Simulations. J. Phys. Chem. B 120, 2145–2154 (2016).
Yu, C. et al. Small molecules enhance CRISPR genome editing in pluripotent stem cells. Cell Stem Cell 16, 142–147 (2015).
Davis, K. M., Pattanayak, V., Thompson, D. B., Zuris, J. A. & Liu, D. R. Small molecule-triggered Cas9 protein with improved genome-editing specificity. Nat. Chem. Biol. 11, 316–318 (2015).
Salomon-Ferrer, R., Case, D. A. & Walker, R. C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 3, 198–210 (2013).
Maier, J. A. et al. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 (2015).
Li, P., Song, L. F. & Merz, K. M. Jr. Systematic Parameterization of Monovalent Ions Employing the Nonbonded Model. J. Chem. Theory Comput. 11, 1645–1657 (2015).
Li, P., Roberts, B. P., Chakravorty, D. K. & Merz, K. M. Jr. Rational Design of Particle Mesh Ewald Compatible Lennard-Jones Parameters for +2 Metal Cations in Explicit Solvent. J. Chem. Theory Comput. 9, 2733–2748 (2013).
Darden, T., York, D. & Pedersen, L. Particle mesh Ewald: An N⋅ log (N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993).
Miyamoto, S. & Kollman, P. A. SETTLE: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 13, 952–962 (1992).
Humphrey, W., Dalke, A. & Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Model. 14, 33–38 (1996).
Miller, B. R. et al. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 8, 3314–3321 (2012).
Acknowledgements
The authors acknowledge the Texas Advanced Computing Center (TACC) at The University of Texas at Austin for providing HPC resources that have contributed to the research results reported within this paper.
Author information
Authors and Affiliations
Contributions
All the simulation work was performed by Z.C.Z. The study was conceived and supervised by J.L. Both authors contributed to analyzing the simulation results and writing the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zuo, Z., Liu, J. Cas9-catalyzed DNA Cleavage Generates Staggered Ends: Evidence from Molecular Dynamics Simulations. Sci Rep 6, 37584 (2016). https://doi.org/10.1038/srep37584
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep37584
This article is cited by
-
An Update on the Application of CRISPR Technology in Clinical Practice
Molecular Biotechnology (2024)
-
Double-strand breaks induce inverted duplication chromosome rearrangements by a DNA polymerase δ-dependent mechanism
Nature Communications (2023)
-
CRISPR-Cas Systems: Programmable Nuclease Revolutionizing the Molecular Diagnosis
Molecular Biotechnology (2023)
-
Lineage barcoding in mice with homing CRISPR
Nature Protocols (2021)
-
Lineage tracing and analog recording in mammalian cells by single-site DNA writing
Nature Chemical Biology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
