
Abstract
Majority of novel X-ray crystal structures of proteins are currently solved using the anomalous diffraction signal provided by selenium after incorporation of selenomethionine instead of natural methionine by genetic engineering methods. However, selenium can be inserted into protein crystals in the form of selenourea (SeC(NH2)2), by adding the crystalline powder of selenourea into mother liquor or cryo-solution with native crystals, in analogy to the classic procedure of heavy-atom derivatization. Selenourea is able to bind to reactive groups at the surface of macromolecules primarily through hydrogen bonds, where the selenium atom may serve as acceptor and amide groups as donors. Selenourea has different chemical properties than heavy-atom reagents and halide ions and provides a convenient way of phasing crystal structures of macromolecules.
Similar content being viewed by others

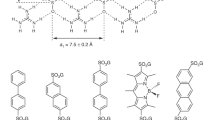

Introduction
Because of the availability of large number of structures of macromolecules stored in the Protein Data Bank1 (PDB), the majority of X-ray crystal structures of proteins and nucleic acids are nowadays solved by the Molecular Replacement technique. However, the crystal structures containing molecules for which there is no sufficiently similar atomic model available, have to be solved by the “special atom” method. The initial phasing of diffraction data is then based on the isomorphous signal of heavy atoms or the anomalous signal of certain atoms present in crystals of the native molecules or introduced into investigated crystals2.
The “classic” approach, used since the early days of protein crystallography, involves derivatization of native crystals by prolonged soaking in solutions or co-crystallization with various reagents containing heavy metals3, such as Hg, Pt, Au etc. Variations of this approach involve, for example, the use of the heavy-metal clusters4, especially suitable for structures of very large macromolecular complexes, the gaseous xenon or krypton pressurized into native crystals5, or the short soaking in salts of halides6 (Br or I). It is also possible to obtain useful anomalous phasing signal from sulfur of Cys and Met naturally occurring in proteins7,8,9 or from phosphorus in nucleic acids10.
The currently most widely used approach is based on the introduction of selenomethionine into proteins by genetic engineering methods11. Selenium has the X-ray K absorption edge at a wavelength of 0.979 Å and exhibits a significant anomalous signal, which can be very conveniently used for phasing by the Multi- or Single-wavelength Anomalous Diffraction (MAD12 or SAD13) approaches at any of the available synchrotron beam lines. Selenium can be also chemically introduced into nucleic acids14,15.
However, the tests have shown that it is possible to effectively introduce selenium into native crystals by soaking them in the presence of selenourea (SeU, SeC(NH2)2), Fig. 1. This simple compound, similar to well known urea (Supplementary Fig. 1), penetrates through the crystal solvent channels and binds to certain functions at the surface of biomolecules, in analogy to the heavy-atom or halide derivatization. The interactions of SeU at the macromolecular surface are different than of the hitherto utilized compounds. The two amide groups of SeU may serve as donors in hydrogen bonds formed with various acceptors, such as carbonyl or carboxyl functions of various amino acids, hydroxyl groups of Thr and Tyr residues or suitable atoms of nucleic acids. On the other hand, the Se atom in the central selenocarbonyl moiety of SeU may accept hydrogen bonds from various donors, provided by amides, hydroxyl groups and protonated amines. SeU can interact with solvent water molecules through both of these ways. Figure 2 illustrates some examples how SeU binds to proteins and a DNA oligomer.

Selenourea binding sites illustrated by anomalous difference maps.
(a) Lysozyme, 9 binding sites; (b) thaumatin, 13 binding sites; (c) trypsin, 10 binding sites; (d) CFP, 12 binding sites; (e) HPP, 1 binding site; (f) DDD, 3 binding sites. Macromolecules are shown as cartoon. SeU are shown as ball-and-stick with selenium colored pink, carbon colored green, and nitrogen colored blue. Anomalous difference maps are shown as pink mesh at 4 σ level.

Selenourea-macromolecules interactions.
(a) Lysozyme, sites 1 and 3; (b) lysozyme, site 2; (c) thaumatin, sites 6, 7, and 8; (d) trypsin, sites 4 and 5; (e) CFP, sites 1 and 3; (f) CFP, site 10; (g) HPP, site 1; (h) DDD, sites 1 and 2. Atoms and bonds are shown as ball-and-stick and valences of bonds are also illustrated. Water molecules involved in interactions are shown as red balls. Color scheme of macromolecules: C, gray or yellow (if symmetry equivalent); N, blue; O, red. 2Fo-Fc density maps of SeU are shown as blue mesh at 1σ level. Hydrogen bonds are shown as black dashed lines.
The use of SeU as a provider of anomalous signal for the SAD phasing has been examined on crystals of several proteins and a nucleic acid. These crystals may either be soaked for a few minutes in the appropriate cryoprotecting solution complemented with SeU powder or a pinch of the powderized SeU may be added directly to the crystallization drops containing native crystals. The latter approach has the advantage of not diluting or significantly modifying the content and concentration of the original crystallization medium. The amount of SeU powder added into mother liquor or cryo-solution is about 5% in volume.
The SeU molecule is small, of the size smaller than most of the heavy-metal complexes used for classic derivatization of proteins, and in analogy to small halide ions, rapidly diffuse through the solvent channels of macromolecular crystals. It can be used in a wide pH range, at least from 4 to 9. To prevent SeU from the potential oxidation of the Se atom in solution, it may advisable to add a reducing agent, such as sodium sulfite (Na2SO3) or tris(2-carboxyethyl)phosphine (TCEP). The high concentrations of urea are routinely used for denaturation of proteins, but no adverse effects were observed after relatively short exposure of protein crystals to SeU.
Similar to halide ions, but opposite to heavy-atom complexes, SeU does not require any chemical modification or hydrolysis before binding to hydrophilic groups at the surface of macromolecules. In contrast to the Cl−, Br− and I− ions that can only serve as acceptors of hydrogen bonds, SeU is a potent donor of hydrogen bonds through its two amide groups. In addition, the lone electron pairs of the selenium atom may act as acceptors.
The examples shown in the Methods section ascertain that SeU can be successfully and conveniently used as a practical and easily applicable vehicle for phasing novel crystal structures of macromolecules by the SAD (or MAD) approach through the anomalous signal of selenium.
Methods
The proteins selected for testing the SeU as a phasing vehicle were: HEW lysozyme16, thaumatin17, bovine trypsin18, cyan fluorescent protein (CFP)19 and histidinol phosphate phosphatase (HPP)20. In addition, a B-DNA Dickerson-Drew dodecamer (DDD)21 was used.
The information about proteins and their crystals selected for testing the SeU phasing is presented in Supplementary Table 1.
SeU sub-packaging
SeU was purchased from Sigma (98%, Product Number: 230499) packed in brown glass bottle under argon due to air and moisture sensitivity. The shell around SeU was already oxidized to dark -selenium22. SeU was subpackaged into 1.5 mL Eppendorf tubes, where each tube only contained a small amount of SeU crystalline powder in order to reduce the frequency of exposing to the environment.
Protein expression, purification, and crystallization
Lysozyme
Lyophilized powder of lysozyme from chicken egg white was obtained from Sigma (Product Number: L4919) and used without further purification. Lysozyme was dissolved in 10 mM pH 4.6 citrate buffer at a concentration of 40 mg/mL and mixed 1:1 with the well solution consisting of 25% (w/v) PEG3350 and 50 mM citrate buffer pH 4.0. Crystals appeared after two days at room temperature in sitting-drops. The powder of SeU was picked up by 2 μL pipette tip and directly transferred into the mother liquor. After 10 min, the color of mother liquor turned to slightly brown due to the oxidation of SeU (Supplementary Fig. 2). A crystal was fished out and washed with the well solution containing additional 20% (v/v) 2-methyl-2,4-pentanediol (MPD), then flash frozen in liquid nitrogen.
Thaumatin
Thaumatin was purchased from Sigma (Product Number: T7638) and used without further purification. Thaumatin was dissolved in 50 mM HEPES buffer pH 7.0 at a concentration of 35 mg/mL. Crystals appeared after two days after mixing 1 μL protein solution and 1 μL well solution containing 750 mM sodium/potassium tartrate, 100 mM citrate buffer pH 6.5 using hanging-drop vapor diffusion method at room temperature. The SeU powder was added into the cryo-solution consisting of well solution supplemented with 20% (v/v) glycerol and 50 mM Na2SO3. Native thaumatin crystals were transferred from mother liquor into cryo-solution containing SeU and Na2SO3 and soaked for 5 min, then vitrified in liquid nitrogen.
Trypsin
Bovine trypsin was purchased from Sigma (Product Number T9935) and used without further purification. The complex of trypsin and benzamidine contained 30 mg/mL trypsin and 5 mg/mL benzamidine in 50 mM Tris-HCl buffer pH 7.0. Crystals appeared after three days using hanging-drop vapor diffusion method at room temperature after mixing the trypsin-benzamidine complex with reservoir consisting of 20% (w/v) PEG8000, 200 mM ammonium sulfate, 100 mM citrate buffer pH 6.5 at 1:1 ratio. The SeU powder was added into cryo-solution consisting of 30% (w/v) PEG3350, 20% (v/v) MPD, 50 mM Tris-HCl pH 7.0, and 50 mM Na2SO3. Crystals were fished out from the drop and transferred into cryo-solution, soaked for 5 min, then flash frozen in liquid nitrogen.
CFP
The clone for CFP overexpression was acquired from the Midwest Center for Structural Genomics, with the CFP open reading frame ligated into pMCSG68 vector. The pMCSG68 vector introduces the N-terminal His6-tag, followed by the Tobacco Etch Virus (TEV) protease cleavage site preceding the genuine sequence of the expressed protein. Overexpression was carried out in BL21 Gold E. coli cells (Agilent Technologies). The bacteria were cultured with shaking at 210 rpm in LB media supplemented with 150 μg/mL ampicillin at 37 °C until the A600 reached 1.0. The cultures were cooled down to 18 °C and the CFP production was induced by addition of isopropyl-D-thiogalactopyranoside to the final concentration of 0.5 mM. The protein expression was carried out for 18 h and then the cultures were centrifuged at 3,500 g for 20 min at 4 °C. Cell pellet from 1 L culture was resuspended in 35 mL of binding buffer (50 mM Tris-HCl pH 8.0; 500 mM NaCl; 20 mM imidazole; 1 mM TCEP) and stored at −80 °C. The samples were thawed and the cells were disrupted by sonication using bursts of total duration of 4 min, with appropriate intervals for cooling. Cell debris was pelleted by centrifugation at 25,000 g for 30 min at 4 °C. The supernatant was applied to a column packed with 10 mL of HisTrap HP resin (GE Healthcare), plugged into the Vacuum Manifold (Promega) connected to a vacuum pump. After binding, the column was washed five times with 50 mL of the binding buffer and His6-tagged CFP was eluted with 20 mL of elution buffer (50 mM Tris-HCl pH 8.0; 500 mM NaCl; 300 mM imidazole; 1 mM TCEP). The His6-tag was cleaved with TEV protease (final concentration 0.2 mg/mL) and the excess of imidazole was removed by dialysis (overnight at 4 °C) at the same time. The solution was mixed with HisTrap HP resin to remove the cleaved His6-tag and the remaining His6-tagged TEV protease. The flow-through was collected, concentrated to 3.5 mL and applied on a HiLoad Superdex 200 16/60 column (GE Healthcare) equilibrated with a buffer composed of 25 mM Tris-HCl pH 8.0, 200 mM NaCl and 1 mM TCEP. Homogenous fractions of CFP monomer was collected and concentrated to 15.6 mg/mL. Crystallization screening was carried out by robot (Mosquito) using sitting-drop vapor diffusion method. Crystallization was manually optimized by sitting-drop method at room temperature. The best crystals were obtained after four days at room temperature by mixing 1uL CFP solution with 1 μL well solution consisting 16% (w/v) PEG 3350, 50 mM citric acid, and 50 mM bis-tris propane buffer pH 5.0. The SeU crystalline powder was added into cryo-solution containing 30% (w/v) PEG 3500, 20% (v/v) MPD, and 50 mM potassium phosphate buffer pH 7.5, then CFP crystals were transferred from mother liquor to cryo-solution. After a 5 min soak with SeU, crystals were flash frozen in liquid nitrogen.
HPP
The HPP protein expression, purification, and crystallization were described elsewhere20. In short, the crystals of HPP were grown in 15% (w/v) PEG 3350, 0.2 M diammonium hydrogen phosphate buffer pH 8.0 at room temperature. Crystals appeared in one week and were kept in the hanging drop for about half year. SeU powder was directly dropped into the mother liquor with HPP crystals for 10 min, then soaked crystals were transferred to paratone-N to remove the surface water and vitrified in liquid nitrogen.
DDD
The B-DNA Dickerson-Drew dodecamer d(CGCGAATTCGCG)2 was purchased from Eurofins MWG Operon (Huntsville, USA) and used without further purification. The DDD solution at 2 mM concentration was incubated at 60 °C for 10 min, then slowly cooled down to room temperature. Crystals appeared after two days at room temperature using sitting-drop vapor diffusion method by mixing DDD solution with precipitant consisting of 40 mM sodium cacodylate pH 7.0, 12 mM spermine tetrachloride, 80 mM NaCl, 10%(v/v) MPD at ratio 1:1. The well solution contained 35% (v/v) MPD. The SeU powder was added into cryo-solution consisting 40% (v/v) MPD and 50 mM Tris-HCl buffer pH 7.0 to generate SeU saturated solution. The DDD crystals were transferred from mother liquor to the SeU saturated cryo-solution and soaked for 1 min, then rapidly transferred to a goniostat under a stream of gaseous nitrogen at 100 K delivered by an Oxford Cryosystems cryocooler at the beamline.
X-ray diffraction data collection and processing
All diffraction data were collected at a wavelength corresponding to slightly higher energy than the as absorption edge of Se, at the SER-CAT 22-ID/BM beamlines of the Advanced Photon Source (Argonne National Laboratory, USA). The diffraction data from lysozyme, thaumatin, trypsin, and CFP crystals were collected with the 180° total rotation range, 1° per image. The diffraction data of HPP were collected with 100° range with half degree per image and DDD diffraction data were acquired with 360° range and 2° per image. All data sets were processed by HKL-2000 and with the “auto-correction”23 option in scaling. The “no merge original index” option was used to generate alternative, unmerged set of data, intended only to calculate correlation coefficient of anomalous difference for two random half set (CCano) by phenix.anomalous_signal24. For lysozyme and thaumatin, the data were scaled separately within 45°, 90°, 180° rotation range to analyse the strength of anomalous signal at different multiplicity. Similarly, the data sets of trypsin and CFP were scaled with 90° and 180° rotation range. The data of HPP were only scaled with 100° rotation range. Regarding DDD, the data were scaled within 90°, 180°, and 360° rotation range. The plots of CCano versus resolution showed strong anomalous signals for data sets of lysozyme, thaumatin, trypsin, CFP, and DDD, even at low multiplicity (Supplementary Fig. 3). The anomalous signal of HPP was relatively weak. The statistics of diffraction data at maximal redundancy are listed in Supplementary Table 1.
Substructure determination, density modification, model building and structure refinement
SHELXD25 was used for anomalous substructure determination for all data with 1,000 phase trials except for HPP with the number of trials increased to 10,000. The correlation coefficients between observed and calculated normalized anomalous differences within all data (CCall) and 30% of reflections which were not used during the dual-space refinement (CCweak) with increasing multiplicity are illustrated in Supplementary Fig. 4 and Supplementary Table 2. The substructure refinement, density modification and the initial chain tracing were carried out by SHELXE25, which provided high quality density maps clearly showing the side chain groups of the autotraced poly-Ala backbones for lysozyme, thaumatin, trypsin, and CFP. Model building was carried out by ARP/wARP26, which built more than 90% of total amino acids for lysozyme, thaumatin, trypsin, and CFP (Supplementary Table 2). Chain tracing and model building of HPP did not yield interpretable density map. After phenix.autosol and phenix.autobuild27, 233 residues were built out of total 277 residues of HPP. Nine base-pairs were successfully built for DDD by phenix.autobuild with the calculated phasing and density map from SHELXE. The final models were refined by REFMAC528 with occupancies refined for each SeU molecule. Water molecules were added by COOT29. The statistics of structure refinement are also listed in Supplementary Table 1. The overall structures were illustrated by PyMoL30.
Additional Information
Accession codes: Protein Data Bank (PDB) codes: 5T3F (SeU-lysozyme), 5T3G (SerU-thaumatin), 5T3H (SeU-trypsin), 5T3I (SeU-CFP), 5T3J (SeU-HPP), 5T3L (SeU-DDD).
How to cite this article: Luo, Z. Selenourea: a convenient phasing vehicle for macromolecular X-ray crystal structures. Sci. Rep. 6, 37123; doi: 10.1038/srep37123 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Berman, H. M. et al. Nucleic Acids Res. 28, 235–242 (2000).
Blundell, T. & Johnson, L. N. Protein crystallography, pp. 183–239. London: Academic Press (1976).
Blake, C. C. F. et al. A historical perspective: how the structure of lysozyme was actually determined, In International Tables for Crystallography, vol. F, pp. 745–772. Kluwer Academic Publishers, Dordrecht (2001).
Dauter, Z. Use of polynuclear metal clusters in protein crystallography. Compt. Rend. Chimie 8, 1808–181 (2005).
Prangé, T. et al. Exploring hydrophobic sites in proteins with xenon or krypton. Proteins 30, 61–73 (1998).
Dauter, Z., Dauter, M. & Rajashankar, K. R. Novel approach to phasing proteins: derivatization by short cryo-soaking with halides. Acta Crystallogr. D56, 232–237 (2000).
Hendrickson, W. A. & Teeter, M. M. Structure of the hydrophobic protein crambin determined directly from the anomalous scattering of sulfur. Nature 290, 107–113 (1981).
Dauter, Z. et al. Can anomalous signal of sulfur become a tool for solving protein crystal structures? J. Mol. Biol. 289, 83–92 (1999).
Liu, Q. & Hendrickson, W. A. Robust structural analysis of native biological macromolecules from multi-crystal anomalous diffraction data. Acta Crystallogr. D69, 1314–1332 (2013).
Dauter, Z. & Adamiak, D. A. Anomalous signal of phosphorus used for phasing DNA oligomer: importance of data redundancy. Acta Crystallogr. D57, 990–995 (2001).
Hendrickson, W. A., Horton, J. R. & LeMaster, D. M. Selenomethionyl proteins produced for analysis by multiwavelength anomalous diffraction (MAD): a vehicle for direct determination of three dimensional structure. EMBO J. 9, 1665–1672 (1990).
Hendrickson, W. A. Determination of macromolecular structures from anomalous diffraction of synchrotron radiation. Science 254, 51–58 (1991).
Wang, B. C. Resolution of phase ambiguity in macromolecular crystallography. Methods Enzymol. 115, 90–112 (1985).
Wilds, C. J. et al. Selenium-assisted nucleic acid crystallography: use of phosphoroselenates for MAD phasing of a DNA structure. J. Am. Chem. Soc. 124, 14910–14916 (2002).
Lin, L., Sheng, J. & Huang, Z. Nucleic acid X-ray crystallography via direct selenium derivatization. Chem. Soc. Rev. 40, 4591–4602 (2011).
Blake, C. C. F. et al. How the structure of lysozyme was actually determined. International Tables for Crystallography Vol. F, pp. 745–772 (2001).
Ko, T. P., Day, J., Greenwood, A. & McPherson, A. Structures of three crystal forms of the sweet protein thaumatin. Acta Crystallogr. D50, 813–825 (1994).
Renatus, M., Bode, W., Huber, R., Stürzebecher, J. & Stubbs, M. T. Structural and functional analyses of benzamidine-based inhibitors in complex with trypsin: implications for the inhibition of factor Xa, tPA, and urokinase. J. Med. Chem. 41, 5445–5456 (1998).
Lelimousin, M. et al. Intrinsic dynamics in ECFP and cerulean control fluorescence quantum yield. Biochemistry 48, 10038–10046 (2009).
Ruszkowski, M. & Dauter, Z. Structural studies of Medicago truncatula histidinol phosphate phosphatase from inositol monophosphatase superfamily reveal details of penultimate step of histidine biosynthesis in plants. J. Biol. Chem. 291, 9960–9973 (2016).
Wing, R. et al. Crystal structure analysis of a complete turn of B-DNA. Nature 287, 755–758 (1980).
Mishra, B., Hassan, P. A., Priyadarsini, K. I. & Mohan, H. Reactions of biological oxidants with selenourea: formation of redox active nanoselenium. J. Phys. Chem. B 109, 2718–12723 (2005).
Otwinowski, Z. & Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997).
Terwilliger, T. C. et al. Can I solve my structure by SAD phasing? Anomalous signal in SAD phasing. Acta Crystallogr. D72, 346–358 (2016).
Sheldrick, G. M. A short history of SHELX. Acta Crystallogr. A64, 112–122 (2008).
Langer, G., Cohen, S. X., Lamzin, V. S. & Perrakis, A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nature Protocols 3, 1171–1179 (2008).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D66, 213–221 (2010).
Murshudov, G. N., Vagin, A. A. & Dodson, E. J. Refinement of Macromolecular Structures by the Maximum-Likelihood method Acta Crystallogr. D53, 240–255 (1997).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D66, 486–501 (2010).
The PyMOL Molecular Graphics System, Version 1.6 Schrödinger, LLC. http://www.pymol.org.
Acknowledgements
This work was supported by the Intramural Research Program of the National Cancer Institute. The help of Zbigniew Dauter in preparation of this manuscript is gratefully appreciated. The instruction for CFP expression and purification and growth of HPP crystals by Milosz Ruszkowski are appreciated. Provision of thaumatin and trypsin crystals by Miroslawa Dauter is gratefully acknowledged.
Author information
Authors and Affiliations
Ethics declarations
Competing interests
The author declares no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Luo, Z. Selenourea: a convenient phasing vehicle for macromolecular X-ray crystal structures. Sci Rep 6, 37123 (2016). https://doi.org/10.1038/srep37123
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep37123
This article is cited by
-
Structural and mechanistic insights into the bifunctional HISN2 enzyme catalyzing the second and third steps of histidine biosynthesis in plants
Scientific Reports (2021)
-
Hydrogen Bonds with Chalcogens: Looking Beyond the Second Row of the Periodic Table
Journal of the Indian Institute of Science (2020)
-
Crystal Structure of MpPR-1i, a SCP/TAPS protein from Moniliophthora perniciosa, the fungus that causes Witches’ Broom Disease of Cacao
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
