
Abstract
Leprosy is a chronic infectious and neurological disease caused by Mycobacterium leprae, an unculturable pathogen with massive genomic decay and dependence on host metabolism. We hypothesized that mitochondrial genes PARL and PINK1 would confer risk to leprosy. Thirteen tag SNPs of PARL and PINK1 were analyzed in 3620 individuals with or without leprosy from China. We also sequenced the entire exons of PARL, PINK1 and PARK2 in 80 patients with a family history of leprosy by using the next generation sequencing technology (NGS). We found that PARL SNP rs12631031 conferred a risk to leprosy (Padjusted = 0.019) and multibacillary leprosy (MB, Padjusted = 0.020) at the allelic level. rs12631031 and rs7653061 in PARL were associated with leprosy and MB (dominant model, Padjusted < 0.05) at the genotypic level. PINK1 SNP rs4704 was associated with leprosy at the genotypic level (Padjusted = 0.004). We confirmed that common variants in PARL and PINK1 were associated with leprosy in patients underwent NGS. Furthermore, PARL and PINK1 could physically interact with each other and were involved in the highly connected network formed by reported leprosy susceptibility genes. Together, our results showed that PARL and PINK1 genetic variants are associated with leprosy.
Similar content being viewed by others

Introduction
Leprosy is a chronic infectious disease which has affected mankind for more than 4,000 years1. Although the number of new cases of leprosy globally decreased to 213,899 patients in 20142, the disease is still a significant threat to public health in many parts of the world. The pathogen, Mycobacterium leprae (M. leprae), is an obligate intracellular parasite and primarily affects the skin and peripheral nerves3. When compared against its close relative, M. tuberculosis, the genome of M. leprae shows an extremely eroded evolution, which has led to nearly half of the functional genes (especially in the metabolic pathways) undergoing inactivation or pseudogenation4,5,6. This marked reduction in the number of working genes might be the primary reason why M. leprae has a long half-life in vivo and cannot be cultured in vitro. As a result, the provision of energy metabolites and nutritional products by the host has become essential to the survival of M. leprae.
Mitochondria are crucial organelles involved in the cellular energy supply, regulation of apoptotic signals and autophagy, and defenses against pathogenic microbe invasion7,8,9. Recent studies showed that the host mitochondria might play important roles in M. leprae infection. A lower expression of several mitochondrial genes was observed in nerve biopsies from leprosy patients compared to non-leprous individuals using a microarray assay10. We also found a significantly increased mtDNA copy number in lepromatous leprosy patients11 compared with controls. The mitochondrial outer membrane protein, LRRK2, has been identified by a genome-wide association study (GWAS) as one of the leprosy susceptibility genes in Han Chinese population12, and this was confirmed by our recent case-control study13 and other studies14,15, although the associated LRRK2 SNPs or their effects were different in these studies. Most recently, we provided solid evidence to show that the OPA1 gene, encoding an mitochondrial inner membrane protein, was associated with leprosy susceptibility possibly by affecting mitochondrial function and antimicrobial pathways16. All these lines of evidence support our previous hypothesis that mitochondrial function may affect host susceptibility to M. leprae and the onset of clinical leprosy11.
The presenilins-associated rhomboid-like (PARL) gene is located on chromosome 3q27 and consists of ten exons. PARL is a mitochondrial membrane protein and is a key regulator of mitochondrial integrity and function, such as mitochondrial morphology, apoptosis and glucose metabolism17,18,19. PARL can interact with OPA1 during apoptosis by regulating apoptotic cristae remodeling and cytochrome c release20. Moreover, PARL together with OPA1 can control mitochondrial morphology19 and participate in mitochondrial adaptation to heat shock21. Genetic variants in the PARL gene can influence mitochondrial content22 and susceptibility to Parkinson’s disease23, type 2 diabetes24 and LHON25, although there were some negative reports26,27.
The PTEN induced putative kinase 1 (PINK1) is a serine/threonine kinase protein that is localized in mitochondria28. PINK1 knockout mice had mitochondrial dysfunction and increased sensitivity to oxidative stress29. Moreover, PINK1 could phosphorylate Parkin, leading to the activation of E3 ligase and the NF-κB signaling pathway30. The cleavage of PINK1 was mediated by PARL and this was affected by mitochondrial membrane potential31. This scenario negatively regulated the PINK1- and PARK2/Parkin-dependent mitophagy32. Mutations in PINK1 have been reported to be associated with Parkinson’s disease28,33 and schizophrenia34, but there was a controversy27.
In this study, we aimed to investigate the possible association of genetic variants in the PARL and PINK1 genes with leprosy in Han Chinese. Our results provided several lines of evidence showing that PARL and PINK1 confer genetic susceptibility to leprosy.
Results
Association of PARL and PINK1 SNPs with leprosy per se and multibacillary patients
The minor allele frequencies (MAF) of the SNPs analyzed in this study ranged from 5.8% to 48.4% (Table 1). The power to detect an odds ratio (OR) value as 1.6 for risk allele was expected to be above 77.0% (Fig. S1). SNPs rs10937153, rs1573132 and rs607254 were not in Hardy-Weinberg equilibrium in controls (Table S1, P < 0.05) and were excluded in the following analyses. The allele and genotype frequencies of the 10 SNPs in 527 leprosy patients, 583 healthy subjects from the Yuxi Prefecture, Yunnan Province, and pooled 3093 leprosy-unaffected controls were listed in Tables 1 and 2. We constructed the linkage disequilibrium (LD) map of all the tested SNPs in the Yuxi leprosy cases, Yuxi controls and pooled leprosy-unaffected controls (Fig. 1), and observed similar LD structures for these populations. We further performed the principal component (PC) analysis for the studied populations based on the observed genotype frequencies of the 10 SNPs, together with data of the CHB, CHD, JPT, CEU populations from the HapMap data set35. The Yuxi leprosy patient, Yuxi controls and the reported controls from Hunan Province and Shanghai were clustered together, suggesting no substantial population substructure between the cases and controls (Fig. S2).

The linkage disequilibrium (LD) structures of PARL (a) and PINK1 (b) in leprosy patients and healthy controls from the Yuxi Prefecture and pooled control samples. Black squares represented high LD as measured by r2, gradually coloring down to white squares of low LD. The individual square showed the r2 value for each SNP pair (r2 value is multiplied by 100). The pooled control samples contained the reported Han Chinese without leprosy from Hunan Province (N = 984), Shanghai (N = 1526)27, and the Yuxi control samples in this study (Yuxi).
The RegulomeDB database was used to annotate the analyzed SNPs36. Except for rs2305666 and rs1043424, the other SNPs showed a signal as DNase I hypersensitivity site. SNPs rs10916840 and rs4704 were located in transcription factor binding sites, and rs1061593 showed an eQTL effect (Table 1). Two PARL SNPs showed an association with leprosy per se (rs12631031-A allele, OR = 1.189, 95% CI [1.029–1.381], P = 0.019; rs12631031, P dominant = 0.033; rs7653061, P dominant = 0.027) and MB (rs12631031-A allele, OR = 1.251, 95% CI [1.036–1.510], P = 0.020; rs12631031, P dominant = 0.019; rs7653061, P dominant = 0.023) when compared with healthy control population from the same region or with the pooled control populations (Tables 1 and 2). One PINK1 SNP (rs4704) showed an association with leprosy per se when compared with healthy control population from the Yuxi area (Pdominant = 0.033), and the association survived (TT vs. TC vs. CC, Pgenotypic = 0.004; Pdominant = 0.027) when we compared the patients with the pooled controls (Table 2).
Haplotypes were reconstructed for four PARL SNPs (rs1061593-rs2305666-rs12631031-rs7653061) and four PINK1 SNPs (rs10916832-rs10916840-rs1043424-rs4704; SNPs rs650616 and rs3738140 were excluded because these two SNPs were not genotyped in the pooled populations). There were no associations of PARL haplotypes or PINK1 haplotypes with leprosy (cases versus pooled control samples, global P-value > 0.05). We observed no significant difference of haplotype distribution frequencies between the cases and controls (Table S2).
Deep sequencing of PARL and PINK1 exons identified an association of coding variants with leprosy
To identify whether there are any other rare (allele frequency < 1%) or common variants that would confer risk to leprosy, we performed targeted gene sequencing (including the flanking region of the gene) for PARL, PINK1 and PARK2 in 80 leprosy patients from the Wenshan Prefecture, Yunnan Province, and compared to the CHB data in 1000 Genomes dataset37. Although we did not find any rare variants of PARL and PINK1 to be associated with leprosy (P > 0.05; partially due to the small sample size), one missense variant (rs3732581 [p.V212L], P = 6.434 × 10−5) and one synonymous variant (rs13091 [p.H216], P = 1.058 × 10−4) in PARL and one synonymous variant (rs45530340, [p.L63], P = 2.668 × 10−4) in PINK1 were significantly associated with leprosy per se (Table S3). It should be mentioned that the comparison might be biased as we compared the Wenshan sample to the CHB sample (103 Han Chinese from Beijing) from the 1000 Genomes dataset37 and the samples were not geographically matched. Further in silico program affiliated predication analysis showed that no missense variants in PARL, PINK1 and PARK2 were predicted to be pathogenic (Table S3).
The risk SNPs affected leprosy-related gene expression in human tissues
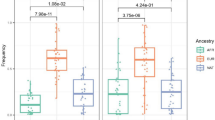
We tested the expression quantitative trait loci (eQTLs) of 34 SNPs (including 5 index tag SNPs and 21 captured PARL SNPs from the HapMap database35, and 8 tag SNPs in PINK1) in leprosy-related human blood, skin and nerve tissues from the Genotype-Tissue Expression project (GTEx, http:// www.gtexportal.org/ home/38). We found that 18 of 26 PARL SNPs were significant cis eQTLs in whole blood (P < 1.0 × 10−4). Among them, 10 of 26 SNPs were remarkably significant (P < 1.0 × 10−8). Notably, SNP rs7644746 that was tagged by the risk SNP rs7653061 reached a P value of 5.6 × 10−24 in blood (Fig. 2a). PINK1 SNP rs10916840 was a cis eQTL in skin tissue (1.5 × 10−9) based on the GTEx dataset38, whereas rs4704 was a significant trans eQTL in whole blood (3.1 × 10−30; Fig. 2b). Both SNPs affected the PINK1 mRNA expression level.

eQTL analysis of the PARL and PINK1 genes.
cis and trans eQTL of the PARL and PINK1 tag SNPs in human blood, skin and nerve tissues were identified by using the GTEx (http://www.gtexportal.org/home)38 and HaploReg dataset (http://www.broadinstitute.org/mammals/haploreg/haploreg.php)54.
The specific expression pattern of PARL and PINK1 were checked in a variety of human tissues from the BioGPS39 (http://biogps.org/#goto=welcome; Figures S3 and S4). We noticed that PARL mRNA expression level was extremely high in immune cells, but PINK1 had an extremely high mRNA expression in central nervous system. We observed a significantly differential mRNA expression of PINK1, but not PARL, in leprosy skin lesions of 66 patients from the Gene Expression Omnibus dataset (GEO; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE74481)40 (Table S4).
Protein interaction network analysis showed an active interaction of PARL with leprosy risk genes
To evaluate the protein interaction with PARL and PINK1, we used the GeneMANIA prediction server41 and identified that PARL could physically interacted and co-expressed with PINK1. PARL and PINK1 could directly or indirectly interact with many proteins, such as FXR1, NDUFB5, TRAP1 and PARK2 (Fig. 3). Note that PINK1 directly interacted with PARK2, which was identified as a leprosy risk gene in several populations42,43,44. However, our NGS analysis for the PARK2 gene revealed no association of this gene with leprosy though we observed positive associations between PARL and leprosy or between PINK1 and leprosy in this relatively small sample. This observation was consistent with a previous report for no association of PARK2 SNPs with leprosy in Han Chinese population45. However, it should be noted that our exon sequencing of the PARK2 gene did not cover its promoter region, and we could not exclude a possibility that there existed leprosy-associated SNP(s).

Protein interaction network of the PARL and PINK1 genes.
PARL can directly interact with PINK1 according to the GeneMANIA database (http://genemania.org/)41. The minimum required interaction score is >0.7 and the line thickness indicates the strength of data support.
To discern whether PARL and PINK1 participated in molecular networks that contain proteins encoded by leprosy susceptibility genes, we constructed the protein interaction network of PARL, PINK1 and the reported 228 leprosy-associated genes (Table S5; ref. 46 and references therein) by the GeneMANIA41. We found that PARL and PINK1 could physically interacted, co-expressed and genetically interacted with those proteins of the reported leprosy susceptibility genes, such as OPA1, PARK2, HLA-A, HLA-DRA, HLA-DQB, and IL10RA (ref. 46 and references therein) (Figure S5).
Discussion
Leprosy is a complex infectious and neurological disease, with impairment of both the immune and peripheral nerve systems during the infection3. Host genetic background would affect the susceptibility to leprosy, because of the genomic decay of M. leprae, and its completely parasitic mode. Genetic studies, especially recent GWAS in Han Chinese12, have suggested an important role for host genetic effect on leprosy susceptibility, although the exact mechanism was still unknown47,48. Mitochondria play important roles in cellular energy supply, cell signaling, mitophagy and anti-microbe immune responses7,8,9. It is therefore reasonable to believe that genes involved in mitochondrial function would affect the host response to microbe infection. Indeed, we recently provided evidence that genetic variants of the mitochondrial genes, like LRRK2 and OPA1 were associated with leprosy13,16, although three mitochondrial-related antimicrobial/antiviral immune genes (MAVS, MITA and MFN2) showed no evidence to be associated with leprosy49. In this study, we found that two mitochondrial genes, PARL and PINK1, conferred genetic susceptibility to leprosy per se and/or multibacillary leprosy.
Among the analyzed tag SNPs in the PARL and PINK1 genes, three SNPs (rs12631031 and rs7653061 of PARL; rs4704 of PINK1) were associated with leprosy per se and/or MB. Furthermore, deep sequencing of the PARL, PINK1 and PARK2 genes in a relatively small sample identified two PARL variants (rs3732581 [p.V212L], rs13091 [p.H216]) and one PINK1 variant (rs45530340, [p.L63]) that were associated with leprosy. We found no variant in the coding region of the PARK2 gene to be linked with leprosy. There is a possibility that promoter variant in this gene might confer risk to leprosy and this awaits future study, as the promoter region was not covered by the current exon sequencing. It was well known that genetic variation could influence gene expression50,51, therefore we performed eQTL analysis to elucidate whether these leprosy risk variants altered the PARL and PINK1 mRNA expression. We observed that two risk SNPs of PARL and all their captured SNPs were cis eQTLs for PARL mRNA expression in human blood (P value from 5.6 × 10−5 to 5.6 × 10−24), and two risk SNPs of PINK1 were cis and trans eQTLs for PINK1 mRNA expression in skin (P = 1.5 × 10−9) and blood (P = 3.1 × 10−30), respectively. Nevertheless, we only found a significantly different expression of PINK1 mRNA, but not PARL mRNA, between leprotic lesions (leprosy per se or its subtypes) and control tissues based on the re-analysis of reported datasets10,40. The exact reason for the discrepancy of PARL mRNA expression remains unknown and awaits future study.
In our recent studies, we identified an association of two mitochondrial genes (LRRK2 and OPA1) with leprosy13,16. Although there was no positive interaction among PARL, PINK1, OPA1 and LRRK2 SNPs (Table S6) based on our recently reported data13,16 and current data, the positive associations of these four genes with leprosy suggested that mitochondrial related genes should play active roles in leprosy. The protein interaction network analysis supported this speculation, as we found that the other mitochondrial genes (MCCD1, SDHD, SNCA, and VARS2) could interact with the reported leprosy susceptibility genes (Table S5). Whether these genes play their roles by directly affecting mitochondrial function, or by participating in other signaling pathways, and then affect leprosy susceptibility, is still an open question.
This study had two limitations. First, the Wenshan population analyzed by the NGS was relatively small, and we compared this population to the CHB data in 1000 Genomes dataset37, which might lead to a biased result as the samples were not well matched. For the Yuxi sample, the coverage of common PARL and PINK1 SNPs might not be sufficient. Second, we did not perform functional assays to characterize the role of PARL, PINK1 and their interactions with previously reported mitochondrial risk genes such as OPA116 and LRRK213 during M. leprae infection.
In summary, we found that common variants of the mitochondrial genes PARL and PINK1 would confer risk to leprosy per se and/or MB. Combining the reported results13,16,46 and the protein interaction network analysis, we found that PARL and PINK1 were participated in a highly connected network formed by the reported leprosy risk genes (ref. 46 and references therein). Future studies are needed to validate the association in independent populations and to explore the underlying mechanism during leprosy onset and progression.
Materials and Methods
Study subjects
This study was carried out in 1,110 individuals from the Yuxi Prefecture, Yunnan Province: 527 individuals were leprosy patients (onset age from 2 to 67 years, mean age: 24.7 ± 12.3 years; male/female ratio = 387/140; multibacillary/paucibacillary = 279/ 248); 583 individuals were healthy control subjects from the same geographic area (age from 4 to 88 years, mean age: 36.0 ± 15.5 years; male/female ratio = 365/ 218). These samples had been analyzed for potential associations of other genes with leprosy in our previous studies13,52. A total of 80 unrelated leprosy patients (38 lepromatous leprosy [LL] patients and 42 tuberculoid leprosy [TT] patients) with a family history of disease (each family has at least two leprosy patients) were collected from the Wenshan Prefecture, Yunnan Province. In brief, the diagnosis of leprosy patients was based on clinical and histopathological features, as well as the bacteriological index if available. A total of 2,510 unaffected Han Chinese from South China (including 504 schizophrenia cases and 480 healthy controls from Hunan Province and 624 schizophrenia cases and 902 healthy controls from Shanghai) that were analyzed for 5 PARL SNPs and 4 PINK1 SNPs in our recent study27 were included in this study for comparison, as we found no association between PARL and PINK1 variants and schizophrenia in these sample groups27. All healthy individuals and the reported schizophrenia patients had no history of leprosy, HIV infection, and tuberculosis. Written informed consents conforming to the tenets of the Declaration of Helsinki were obtained from each participant or the appointed guardians of the patients (for those who were unable to provide informed consent at the time of blood collection) prior to the study. The experimental methods were carried out in accordance with the approved guidelines. The institutional review board of the Kunming Institute of Zoology (KIZ) approved all experimental protocols of this study.
SNP selection, genotyping and NGS
Genomic DNA was extracted from whole blood by using the AxyPrep™ Blood Genomic DNA Miniprep Kit (Axygen, USA). We selected five PARL tag SNPs (rs1061593, rs2305666, rs10937153, rs12631031, rs7653061; Figure S6) and eight PINK1 tag SNPs (rs10916832, rs10916840, rs1043424, rs1573132, rs650616, rs607254, rs3738140, rs4704; Figure S7) that were located in a region spanning PINK1 to DDOST based on the linkage disequilibrium (LD) pattern of the analyzed genes using the international HapMap project data set (www.hapmap.ncbi.nlm.nih.gov/, Phase 3, CHB35). The potential roles of SNPs, e.g. affecting transcription factor binding sites or enacting other regulatory factor / mechanism, were estimated by referring to the RegulomeDB dataset (http://www.regulomedb.org/)36. All SNPs were genotyped in the cases and controls from the Yuxi Prefecture by using the SNaPshot assay (Table S7) as described in our previous studies27,52 at the Kunming Biological Diversity Regional Center of Instruments, KIZ. For the NGS of the PARL, PINK1 and PARK2 genes in 80 leprosy patients from the Wenshan Prefecture, we used the same approach as described in our recent study53.
PC analysis, expression and expression quantitative trait loci (eQTL) analysis
PC analysis was performed using the genotype frequencies of 10 tag SNPs (three SNPs were excluded due to the deviation of the Hardy-Weinberg equilibrium [HWE]) to show the overall clustering pattern of the 8 populations (leprosy and control populations from the Yuxi Prefecture of Yunnan Province, unaffected Han Chinese populations from Hunan Province and Shanghai27, CHB [136 Han Chinese in Beijing], CHD [109 Chinese in Metropolitan Denver, Colorado], JPT [113 Japanese in Tokyo, Japan], and CEU [113 Utah residents with Northern and Western European ancestry] populations from the HapMap database35) by using the POPSTR software (http://harpending.humanevo.utah.edu/popstr/).
We performed eQTL analysis in different human tissues by using two publically available expression data sets10,40. We first investigated whether the PARL and PINK1 variants affect gene expression in human whole blood, brain and skin tissues using the Genotype-Tissue Expression project (GTEx, http://www.gtexportal.org/home/38) data and the HaploReg dataset (http://www.broadinstitute.org/mammals/haploreg/haploreg.php)54. We also considered the overall expression profiling of these two genes in the BioGPS database (http://biogps.org/#goto=welcome)39.
We reanalyzed the largest microarray data regarding leprosy skin lesions, including 24 MB (10 mid-borderline leprosy [BB] + 10 borderline lepromatous [BL] + 4 lepromatous [LL]), 20 PB (10 tuberculoid [TT] + 10 borderline-tuberculoid [BT]), 14 type I reaction (R1), and 10 type II reaction (R2) patients, and normal skin biopsies from 9 healthy individuals. The data was retrieved from GEO under accession series GSE74481 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE74481)40.
Protein interaction network analysis
To construct the potential protein interaction network of PARL and PINK1, and to test whether the PARL and PINK1 genes interact with the other leprosy risk genes as compiled in our recent study (ref. 46 and references therein), we performed interaction network analysis by referring to a high-confidence protein interaction database GeneMANIA (http://www.genemania.org/)41.
Statistical analysis
The genotyping call rate of each tag SNP was above 98.7% in our subjects. Cases and controls were compared on the basis of the frequencies of genotypes and alleles. We randomly selected about 2% of samples for direct sequencing and confirmed 100% of consistence with the SNaPshot genotyping results. Power calculations were performed using Quanto software55. Linkage disequilibrium (LD) structure was determined using Haploview 4.2 56. Deviation from the HWE, haplotype comparison and SNP-SNP interaction were performed by using PLINK v1.07 57. The significant SNPs were further calculated by using the logistic regression, with an adjustment for sex. We predicted the potential pathogenicity of variants in the PARL, PINK1 and PARK2 genes identified by the NGS by using an in silico program affiliated prediction, following the procedure described in our recent study (ref. 53 and references therein). A P value < 0.05 was considered to be statistically significant.
Additional Information
How to cite this article: Wang, D. et al. Common variants in the PARL and PINK1 genes increase the risk to leprosy in Han Chinese from South China. Sci. Rep. 6, 37086; doi: 10.1038/srep37086 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Robbins, G. et al. Ancient skeletal evidence for leprosy in India (2000 B.C.). PloS one 4, e5669, doi: 10.1371/journal.pone.0005669 (2009).
WHO. Global leprosy update, 2014: need for early case detection. Weekly Epidemiological Record 90, 461–476 (2015).
Britton, W. J. & Lockwood, D. N. Leprosy. Lancet 363, 1209–1219 (2004).
Cole, S. T. et al. Massive gene decay in the leprosy bacillus. Nature 409, 1007–1011, doi: 10.1038/35059006 (2001).
Gómez-Valero, L., Rocha, E. P., Latorre, A. & Silva, F. J. Reconstructing the ancestor of Mycobacterium leprae: the dynamics of gene loss and genome reduction. Genome research 17, 1178–1185, doi: 10.1101/gr.6360207 (2007).
Monot, M. et al. Comparative genomic and phylogeographic analysis of Mycobacterium leprae. Nature genetics 41, 1282–1289, doi: 10.1038/ng.477 (2009).
Mayer, B. & Oberbauer, R. Mitochondrial regulation of apoptosis. News in physiological sciences 18, 89–94, doi: 10.1152/nips.01433.2002 (2003).
Okamoto, K. & Kondo-Okamoto, N. Mitochondria and autophagy: critical interplay between the two homeostats. Biochimica et biophysica acta 1820, 595–600, doi: 10.1016/j.bbagen.2011.08.001 (2012).
Xu, L. G. et al. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Molecular cell 19, 727–740, doi: 10.1016/j.molcel.2005.08.014 (2005).
Guerreiro, L. T. et al. Gene expression profiling specifies chemokine, mitochondrial and lipid metabolism signatures in leprosy. PloS one 8, e64748, doi: 10.1371/journal.pone.0064748 (2013).
Wang, D. et al. Mitochondrial DNA copy number, but not haplogroup, confers a genetic susceptibility to leprosy in Han Chinese from Southwest China. PloS one 7, e38848, doi: 10.1371/journal.pone.0038848 (2012).
Zhang, F. R. et al. Genomewide association study of leprosy. The new england journal of medicine 361, 2609–2618, doi: 10.1056/NEJMoa0903753 (2009).
Wang, D. et al. Association of the LRRK2 genetic polymorphisms with leprosy in Han Chinese from Southwest China. Genes and immunity 16, 112–119, doi: 10.1038/gene.2014.72 (2015).
Marcinek, P. et al. LRRK2 and RIPK2 variants in the NOD 2-mediated signaling pathway are associated with susceptibility to Mycobacterium leprae in Indian populations. PloS one 8, e73103, doi: 10.1371/journal.pone.0073103 (2013).
Fava, V. M. et al. A missense LRRK2 variant is a risk factor for excessive inflammatory responses in leprosy. PLoS neglected tropical diseases 10, e0004412, doi: 10.1371/journal.pntd.0004412 (2016).
Xiang, Y.-L., Zhang, D.-F., Wang, D., Li, Y.-Y. & Yao, Y.-G. Common variants of OPA1 conferring genetic susceptibility to leprosy in Han Chinese from Southwest China. Journal of dermatological science 80, 133–141, doi: 10.1016/j.jdermsci.2015.09.001 (2015).
Jeyaraju, D. V. et al. Phosphorylation and cleavage of presenilin-associated rhomboid-like protein (PARL) promotes changes in mitochondrial morphology. Proceedings of the National Academy of Sciences of the United States of America 103, 18562–18567, doi: 10.1073/pnas.0604983103 (2006).
Civitarese, A. E. et al. Regulation of skeletal muscle oxidative capacity and insulin signaling by the mitochondrial rhomboid protease PARL. Cell metabolism 11, 412–426, doi: 10.1016/j.cmet.2010.04.004 (2010).
Pellegrini, L. & Scorrano, L. A cut short to death: Parl and Opa1 in the regulation of mitochondrial morphology and apoptosis. Cell death and differentiation 14, 1275–1284, doi: 10.1038/sj.cdd.4402145 (2007).
Cipolat, S. et al. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell 126, 163–175, doi: 10.1016/j.cell.2006.06.021 (2006).
Sanjuan Szklarz, L. K. & Scorrano, L. The antiapoptotic OPA1/Parl couple participates in mitochondrial adaptation to heat shock. Biochimica et biophysica acta 1817, 1886–1893, doi: 10.1016/j.bbabio.2012.05.001 (2012).
Curran, J. E. et al. Genetic variation in PARL influences mitochondrial content. Human genetics 127, 183–190, doi: 10.1007/s00439-009-0756-0 (2010).
Wust, R. et al. Mutation analyses and association studies to assess the role of the presenilin-associated rhomboid-like gene in Parkinson’s disease. Neurobiology of aging 39217, e213–e215, doi: 10.1016/j.neurobiolaging.2015.11.025 (2016).
Walder, K. et al. The mitochondrial rhomboid protease PSARL is a new candidate gene for type 2 diabetes. Diabetologia 48, 459–468, doi: 10.1007/s00125-005-1675-9 (2005).
Phasukkijwatana, N. et al. Genome-wide linkage scan and association study of PARL to the expression of LHON families in Thailand. Human genetics 128, 39–49, doi: 10.1007/s00439-010-0821-8 (2010).
Zhang, A.-M., Jia, X., Zhang, Q. & Yao, Y.-G. No association between the SNPs (rs3749446 and rs1402000) in the PARL gene and LHON in Chinese patients with m.11778G>A. Human genetics 128, 465–468, doi: 10.1007/s00439-010-0875-7(2010).
Li, X. et al. Common variants of the PINK1 and PARL genes do not confer genetic susceptibility to schizophrenia in Han Chinese. Molecular genetics and genomics: MGG 290, 585–592, doi: 10.1007/s00438-014-0942-1 (2015).
Gandhi, S. et al. PINK1 protein in normal human brain and Parkinson’s disease. Brain: a journal of neurology 129, 1720–1731, doi: 10.1093/brain/awl114 (2006).
Gautier, C. A., Kitada, T. & Shen, J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proceedings of the National Academy of Sciences of the United States of America 105, 11364–11369, doi: 10.1073/pnas.0802076105 (2008).
Sha, D., Chin, L. S. & Li, L. Phosphorylation of parkin by Parkinson disease-linked kinase PINK1 activates parkin E3 ligase function and NF-kappaB signaling. Human molecular genetics 19, 352–363, doi: 10.1093/hmg/ddp501 (2010).
Jin, S. M. et al. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. The Journal of cell biology 191, 933–942, doi: 10.1083/jcb.201008084 (2010).
Meissner, C., Lorenz, H., Hehn, B. & Lemberg, M. K. Intramembrane protease PARL defines a negative regulator of PINK1- and PARK2/Parkin-dependent mitophagy. Autophagy 11, 1484–1498, doi: 10.1080/15548627.2015.1063763 (2015).
Valente, E. M. et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160, doi: 10.1126/science.1096284 (2004).
Steinlechner, S. et al. Co-occurrence of affective and schizophrenia spectrum disorders with PINK1 mutations. Journal of neurology, neurosurgery, and psychiatry 78, 532–535, doi: 10.1136/jnnp.2006.105676 (2007).
International HapMap Consortium. The International HapMap Project. Nature 426, 789–796, doi: 10.1038/nature02168 (2003).
Boyle, A. P. et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome research 22, 1790–1797, doi: 10.1101/gr.137323.112 (2012).
1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature 526, 68–74, doi: 10.1038/nature15393 (2015).
G. TEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nature genetics 45, 580–585, doi: 10.1038/ng.2653 (2013).
Wu, C., Jin, X., Tsueng, G., Afrasiabi, C. & Su, A. I. BioGPS: building your own mash-up of gene annotations and expression profiles. Nucleic acids research 44, D313–D316, doi: 10.1093/nar/gkv1104 (2016).
Belone Ade, F. et al. Genome-wide screening of mRNA expression in leprosy patients. Frontiers in genetics 6, 334, doi: 10.3389/fgene.2015.00334 (2015).
Warde-Farley, D. et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic acids research 38, W214–W220, doi: 10.1093/nar/gkq537 (2010).
Mira, M. T. et al. Susceptibility to leprosy is associated with PARK2 and PACRG. Nature 427, 636–640, doi: 10.1038/nature02326 (2004).
Chopra, R. et al. PARK2 and proinflammatory/anti-inflammatory cytokine gene interactions contribute to the susceptibility to leprosy: a case-control study of North Indian population. BMJ open 4, e004239, doi: 10.1136/bmjopen-2013-004239 (2014).
Chopra, R. et al. Mapping of PARK2 and PACRG overlapping regulatory region reveals LD structure and functional variants in association with leprosy in unrelated indian population groups. PLoS genetics 9, e1003578, doi: 10.1371/journal.pgen.1003578 (2013).
Li, J. et al. Association study of the single nucleotide polymorphisms of PARK2 and PACRG with leprosy susceptibility in Chinese population. European journal of human genetics: EJHG 20, 488–489, doi: 10.1038/ejhg.2011.190 (2012).
Zhang, D.-F., Wang, D., Li, Y.-Y. & Yao, Y.-G. Integrative analyses of leprosy susceptibility genes indicate a common autoimmune profile. Journal of dermatological science 82, 18–27, doi: 10.1016/j.jdermsci.2016.01.001 (2016).
Misch, E. A., Berrington, W. R., Vary, J. C. Jr. & Hawn, T. R. Leprosy and the human genome. Microbiology and molecular biology reviews 74, 589–620, doi: 10.1128/MMBR.00025-10 (2010).
Alter, A., Grant, A., Abel, L., Alcaïs, A. & Schurr, E. Leprosy as a genetic disease. Mammalian genome 22, 19–31, doi: 10.1007/s00335-010-9287-1 (2011).
Wang, D. et al. Genetic variants of the MAVS, MITA and MFN2 genes are not associated with leprosy in Han Chinese from Southwest China. Infection, Genetics and Evolution, doi: 10.1016/j.meegid.2016.08.021 (2016).
Francesconi, M. & Lehner, B. The effects of genetic variation on gene expression dynamics during development. Nature 505, 208–211, doi: 10.1038/nature12772 (2014).
Williams, R. B., Chan, E. K., Cowley, M. J. & Little, P. F. The influence of genetic variation on gene expression. Genome research 17, 1707–1716, doi: 10.1101/gr.6981507 (2007).
Wang, D. et al. Genetic variants of the MRC1 gene and the IFNG gene are associated with leprosy in Han Chinese from Southwest China. Human genetics 131, 1251–1260, doi: 10.1007/s00439-012-1153-7 (2012).
Zhang, D.-F. et al. PLD3 in alzheimer’s disease: a modest effect as revealed by updated association and expression analyses. Molecular neurobiology 53, 4034–4045, doi: 10.1007/s12035-015-9353-5 (2016).
Ward, L. D. & Kellis, M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic acids research 40, D930–D934, doi: 10.1093/nar/gkr917 (2012).
Gauderman, W. J. Sample size requirements for matched case-control studies of gene-environment interaction. Statistics in medicine 21, 35–50, doi: 10.1002/sim.973 (2002).
Barrett, J. C., Fry, B., Maller, J. & Daly, M. J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265, doi: 10.1093/bioinformatics/bth457 (2005).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. American journal of human genetics 81, 559–575, doi: 10.1086/519795 (2007).
Acknowledgements
We thank Dr. Yu Fan and Miss Xiao Li for technical assistance and Ian Logan for language editing and helpful comments. This study was supported by the National Natural Science Foundation of China (31271346, 81573034) and Yunnan Province (2014FB177). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Y.G.Y. and D.W. designed the study; X.A.L., X.F.Y., H.L. and Y.Y.L. collected the samples and clinical information; D.W., J.Q.F and D.F.Z. performed the experiments; D.W., G.D.L. and D.F.Z. analyzed the data; D.W. and Y.G.Y. wrote the manuscript. All authors approved the submission of this manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wang, D., Zhang, DF., Feng, JQ. et al. Common variants in the PARL and PINK1 genes increase the risk to leprosy in Han Chinese from South China. Sci Rep 6, 37086 (2016). https://doi.org/10.1038/srep37086
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep37086
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
