
Abstract
Genomic gene clusters for the biosynthesis of chemical defence compounds are increasingly identified in plant genomes. We previously reported the independent evolution of biosynthetic gene clusters for cyanogenic glucoside biosynthesis in three plant lineages. Here we report that the gene cluster for the cyanogenic glucoside dhurrin in Sorghum bicolor additionally contains a gene, SbMATE2, encoding a transporter of the multidrug and toxic compound extrusion (MATE) family, which is co-expressed with the biosynthetic genes. The predicted localisation of SbMATE2 to the vacuolar membrane was demonstrated experimentally by transient expression of a SbMATE2-YFP fusion protein and confocal microscopy. Transport studies in Xenopus laevis oocytes demonstrate that SbMATE2 is able to transport dhurrin. In addition, SbMATE2 was able to transport non-endogenous cyanogenic glucosides, but not the anthocyanin cyanidin 3-O-glucoside or the glucosinolate indol-3-yl-methyl glucosinolate. The genomic co-localisation of a transporter gene with the biosynthetic genes producing the transported compound is discussed in relation to the role self-toxicity of chemical defence compounds may play in the formation of gene clusters.
Similar content being viewed by others


Introduction
Plants produce a wide variety of chemical defence compounds that provide protection against herbivores and pathogens. A particular plant species or genus is characterised by the presence of a subset of such defence compounds. Considerable inter- and intraspecific variation is thought to result from various trades-offs, such as between growth and defence, or between competing defence strategies in a varying ecological context1,2. Besides constraints on the allocation of resources, chemical defence also carries the risk of self-toxicity as a metabolic cost. One specific class of chemical defence compounds are the cyanogenic glucosides, which occur widely in the plant kingdom3. These compounds are glucosides of amino acid derived α-hydroxynitriles, and part of a two-component chemical defence system. Hydrolysis of cyanogenic glucosides by a specific β-glucosidase following tissue disruption, for instance by chewing insects, releases the chemically unstable α-hydroxynitrile, which upon dissociation gives rise to the formation of toxic hydrogen cyanide. The cyanogenic glucoside dhurrin is the main chemical defence compound in Sorghum bicolor, an important cereal crop used for food and feed4. Dhurrin is derived from the amino acid tyrosine by the sequential action of two cytochrome P450 enzymes, named CYP79A1 (Sobic.001G012300) and CYP71E1 (Sobic.001G012200), and the glucosylation and stabilisation of the produced α-hydroxynitrile (cyanohydrin) intermediate by the UDP-glucosyltransferase UGT85B1 (Sobic.001G012400)5. As it involves two membrane anchored cytochrome P450 enzymes, dhurrin biosynthesis is thought to take place at the cytosolic surface of the endoplasmic reticulum (ER). The glucosylated compound, which is labile in non-acidic environments due to the ionization of the hydroxyl group on the benzene ring6, is stably stored in the acidic vacuolar compartment but the mechanism of its intracellular transport from the ER to the vacuole is unknown7.
The biosynthetic pathways for cyanogenic glucosides have also been elucidated in dicot plant species such as cassava (Manihot esculenta) and the model legume Lotus japonicus. Gene identification in L. japonicus was helped by the fact that the biosynthetic genes were found to be co-localised in the same genome region, and in this species the second step was catalysed by a member of the CYP736 gene family8. The biosynthetic genes in cassava and sorghum were also found to be organised in a gene cluster, but the three clusters are thought to have evolved independently. This remarkable genomic co-localisation of non-homologous genes encoding biosynthetic enzymes in the same metabolic pathway has also been observed for other classes of plant chemical defence compounds such as terpenoids9,10,11, benzoxazinoids12, and alkaloids13,14. These clusters are proposed to promote the co-inheritance of beneficially interacting alleles and to additionally facilitate the co-expression of the biosynthetic genes by regulation at the chromatin level11,15. An important driver for gene cluster formation and maintenance, via selection for reduced recombination between the interacting genes, is thought to be the fact that incompletely inherited biosynthetic pathways may result in the release of toxic intermediates causing self-toxicity15,16.
Membrane transport is increasingly recognised as an important component of plant specialised metabolism and bioengineering approaches, but the number of characterised transporters remains limited17. Members of the large multidrug and toxic compound extrusion (MATE) gene family are found in both prokaryotes and eukaryotes, and transport a wide range of compounds18. In plants they have been shown to transport xenobiotic compounds, organic acids19, plant hormones, and secondary metabolites such as anthocyanins and other flavonoids20,21,22, and the alkaloid nicotine23,24. Here we report that the biosynthetic gene cluster for dhurrin additionally includes a gene encoding a tonoplast localised MATE transporter for dhurrin uptake, demonstrating that the analysis of plant gene clusters can contribute to transporter identification.
Results and Discussion
The dhurrin gene cluster
Analysis of the genomic region surrounding the dhurrin biosynthetic gene cluster in sorghum, revealed the presence of genes encoding a MATE transporter (Sobic.001G012600) we have named SbMATE2, and a glutathione S-transferase (GST) named SbGST1 (Sobic.001G012500) of the plant specific phi subfamily (Fig. 1a). Additional support for the involvement of these two genes in dhurrin metabolism was their co-expression with the biosynthetic genes, as revealed by searching the MOROKOSHI sorghum transcriptome database containing publically available RNA-seq data25. The genes showing the highest co-expression with CYP79A1, encoding the first enzyme of the dhurrin biosynthetic pathway, were CYP71E1, immediately followed by SbMATE2 (Fig. 1b, Supplementary Table 1). Co-expression with CYP79A1 was additionally observed for the SbGST1 and the UGT85B1 genes, which showed the highest level of co-expression with each other. High relative expression of all genes was observed in shoots of 9-day old seedlings (Fig. 1b, condition 16), which was enhanced by abscisic acid and osmotic stress treatments (conditions 14 and 15, respectively)26,27. Co-expression studies have resulted in transporter identification, as was reported for several MATE vacuolar nicotine transporters in Nicotiana tabacum23,24. Moreover, the co-expression of clustered biosynthetic genes from the same pathway was reported for the synthesis of the triterpene thalianol in Arabidopsis thaliana, and such tight coordinated regulation is suggested to prevent the accumulation of deleterious biosynthetic intermediates11. This toxicity argument also applies to the labile and reactive intermediates of the dhurrin pathway, and to dhurrin itself given its labile and reactive nature at cytoplasmic pH6. The coordinated expression of genes in transport or storage with those of the biosynthesis pathway, is similarly proposed to reduce or prevent the self-toxic effects of the metabolites produced.

The dhurrin gene cluster in sorghum contains SbMATE2 and SbGST1, which are co-expressed with the biosynthetic genes.
(a) genomic organisation of the dhurrin gene cluster. (b) RNA-Seq expression profiles for the dhurrin biosynthetic genes CYP79A1, CYP71E1, and UGT85B1, and for SbGST1 and SbMATE2, and two flanking genes. FPKM values (Fragments Per Kilobase Million) are indicated, for treatment details see Supplementary Table 1 (adapted from the MOROKOSHI transcriptome database).
GSTs are well known for conjugating the tripeptide glutathione to endogenous toxic products and xenobiotic compounds, but also for non-enzymatic roles as carrier proteins for endogenous reactive molecules such as porphyrins and anthocyanins28,29,30. Like in the biosynthesis of cyanogenic glucosides, anthocyanins are produced by cytochrome P450 enzymes on the cytoplasmic surface of the endoplasmic reticulum and it is of particular interest to note that anthocyanin transport to the vacuole requires the action of both a GST and a MATE transporter31. This is for instance demonstrated by the transparent testa mutants in Arabidopsis, where the TT19 gene encodes a GST, and where TT12 encodes a MATE transporter21,32. The precise mechanism of anthocyanin transport is the subject of much debate, may involve vesicle mediated trafficking, and was suggested to be related to the removal of toxic compounds from the cytoplasm33. Although similar mechanisms may have been recruited in the case of dhurrin, a potential role for SbGST1 in dhurrin metabolism remains to be established and could be indirect, such as in dealing with the cellular effects of dhurrin self-toxicity.
SbMATE2 is localised to the vacuolar membrane
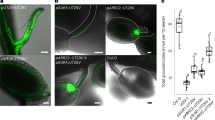
The sequence of the SbMATE2 transporter gene was experimentally verified by cDNA cloning from seedlings and shown to contain two small introns, 135 bp and 97 bp in length respectively, positioned in the C-terminal half of the protein coding region. The transcript encodes a 498 amino acid polypeptide predicted by the Phyre2 web portal for protein modelling to show the topology of the twelve transmembrane helixes typical for prokaryotic and plant MATE transporters34,35,36 (Supplemental Fig. 1). The structural model based on the NorM transporter from Vibrio cholerae, and an additional amino acid sequence alignment that includes mammalian MATE transporters, indicated that SbMATE2 contains conserved amino acids that are part of the cation-binding motif reported for NorM-VC35,37 (Supplemental Fig. 2). The functionality of a predicted N-terminal tonoplast targeting signal was experimentally investigated by transiently expressing a SbMATE2-YFP fusion protein in Nicotiana benthamiana followed by confocal microscopy. Co-expression of SbMATE2-YFP with either one of two aquaporin based organelle specific markers was used to distinguish between the tonoplast and plasma membrane38. SbMATE2-YFP co-localised with the vacuolar membrane marker γ-TIP-CFP, a C-terminal fusion of CFP to full-length γ-TIP, as both signals were observed at positions where the tonoplast was not localised directly adjacent to the cell wall (Fig. 2a). In contrast, the AtPIP2A-CFP marker, consisting of the CFP fused to the plasma membrane aquaporin AtPIP2A, followed the cellular outline precisely (Fig. 2b). Tobacco protoplasts expressing the SbMATE2-YFP construct were used to further exclude a plasma membrane localisation of SbMATE2. Confocal microscopy showed that the SbMATE2-YFP fusion protein was only localised to the vacuolar membrane as it folds around the chloroplasts on the side internal to the cell (Fig. 2c).

Tonoplast localisation of the SbMATE2-YFP fusion protein in epidermal cells and protoplasts of Nicotiana benthamiana.
A SbMATE2-YFP fusion protein under the control of the 35S-CaMV promoter was transiently expressed in N. benthamiana using Agrobacterium infiltration. The SbMATE2-YFP construct was coinfiltrated with expression constructs for either: (a) At.γ-TIP-CFP, a marker for the vacuolar membrane, or (b) AtPIP2A-CFP, a marker for the plasma membrane38. The localisation of the fusion proteins was visualised using confocal microscopy, and the SbMATE-YFP signal co-localised with that of γ-TIP-CIP, showing a tonoplast localisation. Arrows indicate positions where the tonoplast is not adjacent to the plasma membrane. (c) Localisation of SbMATE2-YFP (in yellow) to the vacuolar membrane in isolated N. benthamiana protoplasts. Chloroplasts situated between the vacuolar and plasma membranes are visualised by their autofluorescence (in red).
SbMATE2 transports cyanogenic glucosides
Phylogenetic analysis placed SbMATE2 in what Shitan et al. designated as clade I, consisting of MATE transporters that are functionally characterised (Fig. 3)39. Most of the MATE transporters in this clade function in the accumulation of plant specialised metabolites such as flavonoids and alkaloids, perhaps also a reflection of the experimental interest in transporters for these compounds. The clade includes the seed coat expressed vacuolar anthocyanin transporter AtTT1221,40, MtMATE1 and MtMATE2 from Medicago truncatula transporting flavonoid glycosides (and in the case of MtMATE2 also flavonoid glycoside malonates)20, VvAM1 and VvAM3 from Vitis vinifera transporting acylated anthocyanins22, and the tobacco NtMATE1, NtMATE2, and Nt-JAT2 transporters for the vacuolar sequestration of nicotine in leaves or roots of Nicotiana tabacum23,24.

In a phylogenetic analysis SbMATE2 is part of a clade containing MATE transporters for flavonoids and alkaloids.
A molecular phylogenetic analysis was performed using the Maximum Likelihood method based on the Jones-Taylor-Thornton (JTT) matrix-based model for amino acid sequences. Branch lengths are measured in the number of substitutions per site, and positions containing gaps were eliminated. Bootstrap values (1000x) are indicated at branch points. Analyses were conducted using the MEGA5 software package47. The amino sequences used in the analysis were: SbMATE2 (Sorghum bicolor, Sobic.001G012600), OsPEZ1 (Oryza sativa, Os03g37490), OsPEZ2 (O. sativa, Os03g0572900), OsMATE1 (O. sativa, Os03g08900), TT12 (Arabidopsis thaliana, At3g59030), FFT (A. thaliana, At4g25640), MdMATE1 (Malus domestica, GU64954), MdMATE2 (M. domestica, GU064956), MtMATE1 (Medicago truncatula, FJ858726), MtMATE2 (M. truncatula, HM856605), VvAM1 (Vitis vinifera, Fj264202), VvAM3 (V. vinifera, FJ264203), NtMATE1 (Nicotiana tabacum, AB286961), NtMATE2 (N. tabacum, AB286962), Nt-JAT1 (N. tabacum, AM991692), Nt-JAT2 (N. tabacum, AB922128), and ZmMATE2 (Zea mays, FJ873684). Coloured circles represent the transported compound classes: red = flavonoids, blue = alkaloids, purple = hydroxynitrile glucosides.
The possible role of SbMATE2 in dhurrin transport was studied by export experiments conducted in Xenopus laevis oocytes. Following injection of dhurrin and a 90 min incubation, SbMATE2 expressing oocytes showed an approximate 60% reduction in dhurrin content in comparison with oocytes not expressing SbMATE2, indicating dhurrin transport activity (Fig. 4). The SbMATE2 transporter was additionally able to transport the structurally related aromatic cyanogenic glucosides prunasin and the diglucoside amygdalin, the leucine derived cyanogenic glucoside epiheterodendrin and the non-cyanogenic β-hydroxynitrile glucoside epidermin (Fig. 4). This indicates that SbMATE2 shows a broad tolerance for accepting structurally varying hydroxynitrile glucoside compounds. We additionally tested if the transporter was able to transport cyanidin 3-O-glucoside (C3G), the anthocyanin substrate for AtTT12, and indol-3-yl-methyl glucosinolate (I3M), representing the glucosinolate class of compounds considered to be chemically and biosynthetically related to cyanogenic glucosides, but neither compound was transported in significant levels.

SbMATE2 exports dhurrin and other hydroxynitrile glucosides from X. Laevis oocytes, but not cyanidin 3-O-glucoside. The bars show the relative content of the indicated plant specialised metabolites in SbMATE2 expressing (right bars) and non-expressing control (left bars) oocytes 90 min after injection of the respective compounds into the oocytes (means, ±s.e., statistical significant differences to control oocytes are indicated *p < 0.05). Each of the compound solutions was injected into 25–30 oocytes to an estimated internal concentration of 100 μM. Following incubation, oocytes were analysed in triplicates consisting of 7–10 oocytes. The compounds shown are the cyanogenic glucosides dhurrin, prunasin, amygdalin, epiheterodendrin, the non-cyanogenic β-hydroxynitrile glucoside epidermin, indol-3-yl-methyl glucosinolate (I3M), and the anthocyanin cyanidin 3-O-glucoside (C3G).
Our results demonstrate the presence of a non-biosynthetic component, the SbMATE2 gene encoding a vacuolar transporter for dhurrin, in the gene cluster for a plant chemical defence pathway. Its inclusion in the dhurrin biosynthetic gene cluster is consistent with ideas that selection for reduced recombination between beneficially interacting alleles leads to gene cluster formation15,16,41. Such selection is proposed to result from antagonistic selection pressures, such as the benefits maintaining a functional pathway provides in specific ecological context, e.g. the presence of non-adapted herbivores, against the trade-off costs associated with it1,15. One of the negative cost associated with the production of chemical defence metabolites is the possibility of self-toxicity. Co-inheritance of a co-adapted gene set is thought to provide protection against the self-toxic biochemical nature of many chemical defence compounds or their pathway intermediates15,16. In the case of dhurrin the transport of this pH-dependent unstable cyanogenic glucoside from its cytoplasmic site of production to the acidic vacuole likely contributes to reducing self-toxicity. We previously also reported independently evolved biosynthetic gene clusters for cyanogenic glucosides in cassava and Lotus8. The main cyanogenic compounds produced by these species are linamarin and lotaustralin, respectively, which are more stable than dhurrin. In the most recent version of the cassava genome (Manihot esculenta v6.1), a MATE encoding gene named Manes.12G129000.1, which is not orthologous to SbMATE2, is present at a distance of about 325 kb of the described gene cluster. This physical linkage results in a high level of co-inheritance of Manes.12G129000.1 with the biosynthetic genes, but its physiological role is presently uncharacterised. The incomplete draft of the Lotus japonicus genome does not contain a transporter gene on the sequence contig that contains the biosynthetic gene cluster, but genetics has positioned at least one additional biosynthetic gene in hydroxynitrile glucoside metabolism in the vicinity of the gene cluster8. Eukaryotic biosynthetic gene clusters have been studied more extensively in fungi, and it is of interest to note that the inclusion of transporters is not uncommon in fungal gene clusters. The TRI12 gene in Fusarium sporotrichioides is part of the biosynthetic gene cluster for terpene-derived trichothecene mycotoxins and encodes a trichothecene efflux pump42. Its disruption results in reduced growth and reduced levels of trichothecene production. A clear role in self-protection was reported for the TOXA gene in the fungal pathogen Cochliobolus carbonum, encoding an HC-toxin efflux pump essential for strains producing this toxic cyclic tetrapeptide43. Fungi also contain metabolic gene clusters which provide nutritional benefits under certain ecological conditions. For example, the DAL cluster in Saccharomyces cerevisiae allows the use of allantoin, a degradation product of purines, as a nitrogen source instead of urate, providing an advantage in oxygen-poor natural environments44. Apart from the catabolic genes, the DAL cluster also contains the DAL4 gene encoding an allantoin permease. Given these examples from fungi, it can be expected that the future detailed analysis of genomic regions containing gene clusters for plant specialised metabolites will contribute to the identification of additional non-biosynthetic pathway components such as regulators or transporters.
Methods
Plant material, cDNA isolation and expression constructs
Total RNA was extracted from 3-day old etiolated seedlings of Sorghum bicolor. Following cDNA synthesis, a full length cDNA clone of SbMATE2 was amplified, cloned using the Zero Blunt TOPO PCR Cloning Kit (Invitrogen), and its sequence was verified. For localisation studies, the SbMATE2 protein coding region was amplified from the cDNA clone and fused in frame to YFP using USER cloning as described45. The SbMATE2-YFP construct, under control of the 35S-CaMV promoter, was transformed to Agrobacterium tumefaciens strain AGL1. For oocyte expression the SbMATE2 coding region was cloned downstream of the T7 promoter in the USER compatible Xenopus expression vector pNB1u and linear template for in vitro transcription was generated by PCR. Further details can be found in the Supplementary information.
Transient expression and confocal microscopy
Transient expression in tobacco was performed by Agrobacterium infiltration of Nicotiana benthamiana leaves. Visualisation of the fluorescent protein fusions in epidermal cells or isolated mesophyll protoplasts was carried out using a Leica TCS SP5-II confocal microscope. Excitation/emission wavelengths were 515/525–535 nm for YFP and 435/500–510 nm for CFP. The excitation/emission wavelengths for capturing chlorophyll autofluorescence were 544/660–690 nm.
Oocyte transport assays
Oocytes from Xenopus laevis were obtained from EcoCyte Bioscience (Castrop-Rauxel, Germany). Capped cRNA of SbMATE2 was synthesized using the mMESSAGE mMACHINE® T7 Transcription Kit (ThermoFisher). For expression in oocytes, 25 ng of in vitro produced cRNA for the SbMATE2 transporter was injected into oocytes 4 days prior to performing transport assays essentially as described previously45. Assuming an oocyte volume of ~1 μL, 50 nL of 2 mM compound stock solutions were injected to obtain estimated internal concentrations of 100 μM. Using the same needle each compound was injected into 25–30 oocytes expressing SbMATE2 and 25–30 control (non-expressing) oocytes. Following two washing steps, each batch of 25–30 oocytes was incubated for 90 min in 500 μL Kulori buffer at pH 5. After incubation, all intact oocytes were washed four times in ice-cold Kulori buffer pH 5 and 7–10 oocytes were extracted in triplicate in 50% MeOH as described previously4. Extracts were analysed by LC-MS. Statistical significant differences between the means of SbMATE2 expressing and control oocytes were calculated using a t-test and GraphPad Software (www.graphpad.com).
Chemicals and LC-MS analysis
Amygdalin and cyanidin 3-O-glucoside were obtained from Sigma-Aldrich and indol-3-yl-methyl glucosinolate from Cfm Oskar Tropitzch GmbH. Dhurrin, prunasin, epiheterodendrin and epidermin were chemically synthesized46. LC-MS analysis was performed using a Zorbax SB-C18 column on an Agilent 1100 Series LC coupled to a Bruker HCT-Ultra ion trap mass spectrometer as described previously8. Compounds were localised in extracted ion chromatograms as sodium adduct ions: dhurrin (m/z 334), prunasin (m/z 318), amygdalin (m/z 480), epidermin (m/z 284), epiheterodendrin (m/z 284), cyanidin 3-O-glucoside (m/z 449), indol-3-yl-methyl glucosinolate (m/z 493). Relative quantification was based on peak area using Bruker-DataAnalysis 4.0 (Bruker Daltonik).
Additional Information
How to cite this article: Darbani, B. et al. The biosynthetic gene cluster for the cyanogenic glucoside dhurrin in Sorghum bicolor contains its co-expressed vacuolar MATE transporter. Sci. Rep. 6, 37079; doi: 10.1038/srep37079 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Ballhorn, D. J., Pietrowski, A. & Lieberei, R. Direct trade-off between cyanogenesis and resistance to a fungal pathogen in lima bean (Phaseolus lunatus L.). J. Ecol. 98, 226–236 (2010).
Kempel, A., Schädler, M., Chrobock, T., Fischer, M. & van Kleunen, M. Tradeoffs associated with constitutive and induced plant resistance against herbivory. Proc. Natl. Acad. Sci. USA 108, 5685–5689 (2011).
Gleadow, R. M. & Møller, B. L. Cyanogenic glycosides: synthesis, physiology, and phenotypic plasticity. Annu. Rev. Plant Biol. 65, 155–185 (2014).
Paterson, A. H. et al. The Sorghum bicolor genome and the diversification of grasses. Nature 457, 551–556 (2009).
Jones, P. R., Møller, B. L. & Høj, P. B. The UDP-glucose:p-hydroxymandelonitrile-O-glucosyltransferase that catalyzes the last step in synthesis of the cyanogenic glucoside dhurrin in Sorghum bicolor. J. Biol. Chem. 274, 35482–35491 (1999).
Mao, C.-H. & Anderson, L. Cyanogenesis in Sorghum vulgare. II. Mechanism of the alkaline hydrolysis of dhurrin (p-hydroxymandelonitrile glucoside). J. Org. Chem. 30, 603–607 (1965).
Saunders, J. A. & Conn, E. E. Presence of the cyanogenic glucoside dhurrin in isolated vacuoles from Sorghum. Plant Physiol. 61, 154–157 (1978).
Takos, A. M. et al. Genomic clustering of cyanogenic glucoside biosynthetic genes aids their identification in Lotus japonicus and suggests the repeated evolution of this chemical defence pathway. Plant J. 68, 273–286 (2011).
Wilderman, P. R., Xu, M., Jin, Y., Coates, R. M. & Peters, R. J. Identification of syn-pimara-7,15-diene synthase reveals functional clustering of terpene synthases involved in rice phytoalexin/allelochemical biosynthesis. Plant Physiol. 135, 2098–2105 (2004).
Swaminathan, S., Morrone, D., Wang, Q., Fulton, D. B. & Peters, R. J. CYP76M7 is an ent-cassadiene C11α-hydroxylase defining a second multifunctional diterpenoid biosynthetic gene cluster in rice. Plant Cell 21, 3315–3325 (2009).
Field, B. & Osbourn, A. E. Metabolic diversification – independent assembly of operon-like gene clusters in different plants. Science 320, 543–547 (2008).
Frey, M. et al. Analysis of a chemical plant defense mechanism in grasses. Science 277, 696–699 (1997).
Winzer, T. et al. A Papaver somniferum 10-gene cluster for synthesis of the anticancer alkaloid noscapine. Science 336, 1704–1708 (2012).
Itkin, M. et al. Biosynthesis of antinutritional alkaloids in solanaceous crops is mediated by clustered genes. Science 341, 175–179 (2013).
Takos, A. M. & Rook, F. Why biosynthetic genes for chemical defense compounds cluster. Trends Plant Sci. 17, 383–388 (2012).
McGary, K. L., Slot, J. C. & Rokas, A. Physical linkage of metabolic genes in fungi is an adaptation against the accumulation of toxic intermediate compounds. Proc. Natl. Acad. Sci. USA 110, 11481–11486 (2013).
Nour-Eldin, H. H. & Halkier, B. A. The emerging field of transport engineering of plant specialized metabolites. Curr. Opin. Biotechnol. 24, 263–270 (2013).
Takanashi, K., Shitan, N. & Yazaki, K. The multidrug and toxic compound extrusion (MATE) family in plants. Plant Biotechnol. 31, 417–430 (2014).
Yokosho, K., Yamaji, N. & Ma, J. F. An Al-inducible MATE gene is involved in external detoxification of Al in rice. Plant J. 68, 1061–1069 (2011).
Zhao, J. et al. MATE2 mediates vacuolar sequestration of flavonoid glycosides and glycoside malonates in Medicago truncatula. Plant Cell 23, 1536–1555 (2011).
Debeaujon, I., Peeters, A. J. M., Léon-Kloosterziel, K. M. & Koornneef, M. The TRANSPARENT TESTA12 gene of Arabidopsis encodes a multidrug secondary transporter-like protein required for flavonoid sequestration in vacuoles of the seed coat endothelium. Plant Cell 13, 853–871 (2001).
Gomez, C. et al. Grapevine MATE-type proteins act as vacuolar H+-dependent acylated anthocyanin transporters. Plant Physiol. 150, 402–415 (2009).
Morita, M. et al. Vacuolar transport of nicotine is mediated by a multidrug and toxic compound extrusion (MATE) transporter in Nicotiana tabacum. Proc. Natl. Acad. Sci. USA 106, 2447–2452 (2009).
Shoji, T. et al. Multidrug and toxic compound extrusion-type transporters implicated in vacuolar sequestration of nicotine in tobacco roots. Plant Physiol. 149, 708–718 (2009).
Makita, Y. et al. MOROKOSHI: Transcriptome database in Sorghum bicolor. Plant Cell Physiol. 56, e6 (2015).
Dugas, D. V. et al. Functional annotation of the transcriptome of Sorghum bicolor in response to osmotic stress and abscisic acid. BMC Genomics 12, 514 (2011).
Pasini, L., Bergonti, M., Fracasso, A., Marocco, A. & Amaducci, S. Microarray analysis of differentially expressed mRNAs and miRNAs in young leaves of sorghum under dry-down conditions. J. Plant Physiol. 171, 537–548 (2014).
Lederer, B. & Böger, P. Binding and protection of porphyrins by glutathione S-transferases of Zea mays L. Biochim. Biophys. Acta 1621, 226–233 (2003).
Mueller, L. A., Goodman, C. D., Silady, R. A. & Walbot, V. AN9, a petunia glutathione S-transferase required for anthocyanin sequestration, is a flavonoid-binding protein. Plant Physiol. 123, 1561–1570 (2000).
Dixon, D. P., Skipsey, M. & Edwards, R. Roles for glutathione transferases in plant secondary metabolism. Phytochemistry 71, 338–350 (2010).
Marrs, K. A., Alfenito, M. R., Lloyd, A. M. & Walbot, V. A glutathione S-transferase involved in vacuolar transfer encoded by the maize gene Bronze-2. Nature 375, 397–400 (1995).
Sun, Y., Li, H. & Huang, J.-R. Arabidopsis TT19 functions as a carrier to transport anthocyanin from the cytosol to tonoplasts. Mol. Plant 5, 387–400 (2012).
Gomez, C. et al. In vivo grapevine anthocyanin transport involves vesicle-mediated trafficking and the contribution of anthoMATE transporters and GST. Plant J. 67, 960–970 (2011).
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N. & Sternberg, M. J. E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 (2015).
He, X. et al. Structure of a cation-bound multidrug and toxic compound extrusion transporter. Nature 467, 991–996 (2010).
Zhang, X. et al. Twelve transmembrane helices form the functional core of mammalian MATE1 (Multidrug and Toxin Extruder 1) Protein. J. Biol. Chem. 287, 27971–27982 (2012).
Lu, M. Structures of multidrug and toxic compound extrusion transporters and their mechanistic implications. Channels 10, 88–100 (2016).
Nelson, B. K., Cai, X. & Nebenführ, A. A multicolored set of in vivo organelle markers for co-localization studies in Arabidopsis and other plants. Plant J. 51, 1126–1136 (2007).
Shitan, N. et al. Involvement of the leaf-specific Multidrug and Toxic Compound Extrusion (MATE) transporter Nt-JAT2 in vacuolar sequestration of nicotine in Nicotiana tabacum. PLoS One 9, e108789 (2014).
Marinova, K. et al. The Arabidopsis MATE transporter TT12 acts as a vacuolar flavonoid/H+-antiporter active in proanthocyanidin-accumulating cells of the seed coat. Plant Cell 19, 2023–2038 (2007).
Schwander, T., Libbrecht, R. & Keller, L. Supergenes and complex phenotypes. Curr. Biol. 24, R288–R294 (2014).
Alexander, N. J., McCormick, S. P. & Hohn, T. M. TRI12, a trichothecene efflux pump from Fusarium sporotrichioides: gene isolation and expression in yeast. Mol. Gen. Genet. 261, 977–984 (1999).
Pitkin, J. W., Panaccione, D. G. & Walton, J. D. A putative cyclic peptide efflux pump encoded by the TOXA gene of the plant-pathogenic fungus Cochliobolus carbonum. Microbiology 142, 1557–1565 (1996).
Wong, S. & Wolfe, K. H. Birth of a metabolic gene cluster in yeast by adaptive gene relocation. Nat. Genet. 37, 777–782 (2005).
Nour-Eldin, H. H. et al. NRT/PTR transporters are essential for translocation of glucosinolate defence compounds to seeds. Nature 488, 531–534 (2012).
Møller, B. L., Olsen, C. E. & Motawia, M. S. General and stereocontrolled approach to the chemical synthesis of naturally occurring cyanogenic glucosides. J. Nat. Prod. 79, 1198–1202 (2016).
Tamura, K. et al. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (2011).
Acknowledgements
This work was supported by a post-doctoral grant from the VILLUM Foundation and by its funding of the VILLUM Research Center for Plant Plasticity. H.H.N. acknowledges support from the Danish National Research Foundation to the DynaMo Center.
Author information
Authors and Affiliations
Contributions
B.D., H.H.N., B.L.M. and F.R. designed the study. B.D. performed the localisation studies and transport assays. H.H.N. assisted with transport assays, M.S.M. synthesized chemical compounds, and C.E.O. performed LC-MS. F.R. wrote the manuscript with contributions from the other authors.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Darbani, B., Motawia, M., Olsen, C. et al. The biosynthetic gene cluster for the cyanogenic glucoside dhurrin in Sorghum bicolor contains its co-expressed vacuolar MATE transporter. Sci Rep 6, 37079 (2016). https://doi.org/10.1038/srep37079
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep37079
This article is cited by
-
Flavonoids: a review on biosynthesis and transportation mechanism in plants
Functional & Integrative Genomics (2023)
-
A chromosome-scale genome sequence of sudangrass (Sorghum sudanense) highlights the genome evolution and regulation of dhurrin biosynthesis
Theoretical and Applied Genetics (2023)
-
Regulation of dhurrin pathway gene expression during Sorghum bicolor development
Planta (2021)
-
Transporter proteins in Zymomonas mobilis contribute to the tolerance of lignocellulose-derived phenolic aldehyde inhibitors
Bioprocess and Biosystems Engineering (2021)
-
Influence of domestication on specialized metabolic pathways in fruit crops
Planta (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
