
Abstract
Cholera is a devastating diarrhoeal disease caused by certain strains of serogroup O1/O139 Vibrio cholerae. Mobile genetic elements such as genomic islands (GIs) have been pivotal in the evolution of O1/O139 V. cholerae. Perhaps the most important GI involved in cholera disease is the V. cholerae pathogenicity island 1 (VPI-1). This GI contains the toxin-coregulated pilus (TCP) gene cluster that is necessary for colonization of the human intestine as well as being the receptor for infection by the cholera-toxin bearing CTX phage. In this study, we report a GI (designated GIVchS12) from a non-O1/O139 strain of V. cholerae that is present in the same chromosomal location as VPI-1, contains an integrase gene with 94% nucleotide and 100% protein identity to the VPI-1 integrase, and attachment (att) sites 100% identical to those found in VPI-1. However, instead of TCP and the other accessory genes present in VPI-1, GIVchS12 contains a CRISPR-Cas element and a type VI secretion system (T6SS). GIs similar to GIVchS12 were identified in other V. cholerae genomes, also containing CRISPR-Cas elements and/or T6SS’s. This study highlights the diversity of GIs circulating in natural V. cholerae populations and identifies GIs with VPI-1 recombination characteristics as a propagator of CRISPR-Cas and T6SS modules.
Similar content being viewed by others


Introduction
Vibrio cholerae is a species of bacteria commonly found in marine and estuarine waters and the causative agent of the diarrhoeal disease cholera1. Although more than 200 serogroups of the bacterium are known, only two, O1 and O139, are responsible for pandemics of the cholera disease2. Historically, there have been seven pandemics of cholera, with the current seventh pandemic caused by O1 strains of the El Tor biotype and the sixth and presumably the previous five pandemics caused by O1 strains of the classical biotype1. The evolution of pandemic strains has been greatly influenced by lateral gene transfer (LGT), which led to the acquisition of many novel virulence factors3.
Integrative mobile genetic elements (MGEs) such as transposons and genomic islands (GIs) are particularly important in LGT processes, as they help facilitate integration of non-homologous transferred DNA into replicons such as the chromosome or a resident plasmid(s)4. Studies comparing the genomes of V. cholerae strains consistently find MGEs as one of the sources, if not the major source, of genome variation5,6. For example, the seventh pandemic strains contain two GIs called the Vibrio seventh pandemic islands I and II not present in the strains isolated from the previous pandemics7. Most important to pathogenicity of the pandemic strains are genes found on two MGEs, the cholera toxin bacteriophage (CTXϕ) and a GI called the V. cholerae pathogenicity island 1 (VPI-1)8,9. Although these MGEs are common in pandemic strains, examples of CTXϕ and VPI-1 in non-O1/O139 strains have been documented and are indicative of their mobility across natural populations of V. cholerae and close relatives10,11,12. The CTXϕ contains genes encoding a potent enterotoxin that, when secreted in the human intestinal tract, results in significant loss of fluid that can lead to death within 24 hours if left untreated1. VPI-1 contains the genes encoding the toxin-coregulated pilus (TCP; thus, VPI-1 is also known as the TCP pathogenicity island) that is required for adhesion to the intestinal wall as well as genes encoding the accessory colonization factor and the regulatory genes toxT and tcpPH13,14,15. TCP is also the receptor for CTXϕ and thus a prerequisite for infection and subsequent lysogeny in the emergence of toxigenic V. cholerae8.
Another virulence factor associated with frequent LGT is the type VI secretion system (T6SS) of V. cholerae and other Gram-negative bacteria. Bacteria that harbor T6SS produce a membrane-spanning protein complex capable of puncturing eukaryotic or prokaryotic cells and injecting toxic effector proteins into their targets16. In V. cholerae, the presence of T6SS has previously only been reported as a conserved chromosomal element and displays toxic activity against macrophages17. The T6SS is made up of a series of proteins encoded by genes in three different locations of the V. cholerae genome: a main cluster on chromosome 2 and two smaller auxiliary clusters on chromosomes 1 and 217,18. The characteristic proteins of the T6SS are Hcp and VgrG. Hcp is encoded by two alleles on the V. cholerae chromosome and polymerizes to form the nanotube that protrudes from the bacterial cell surface19,20. The tip of Hcp is decorated with VgrG proteins that form a trimer21. VgrG proteins are conserved at their N-termini but carry specialist C-termini with enzymatic activity21. For example, V. cholerae contains three VgrG alleles with two encoding proteins with actin cross-linking (VgrG-1) and peptidoglycan degrading (VgrG-3) activity22,23. Additional effector proteins with diverse enzymatic activities can be added to the VgrG spike for delivery into target cells24.
Acquisition of novel genes through the uptake of MGEs can have obvious beneficial effects for harmless and pathogenic bacteria alike. However, not all LGT is advantageous, and bacteria have evolved a variety of methods to prevent the spread of harmful DNA sequences in their genomes25. One recently discovered defense mechanism against unwanted LGT is the CRISPR-Cas system. CRISPR-Cas modules consist of cas genes and an array of short direct repeats separated by highly variable spacer sequences that correspond to genetic elements such as bacteriophages or MGEs26. Transcription of the CRISPR array with subsequent slicing of the transcript into smaller CRISPR RNAs acts in concert with the Cas proteins to specifically recognize foreign DNA and cleave it26. The system acts as an adaptive immune system for bacteria as it allows for the synthesis and incorporation of new spacers into the array following invasion of a foreign DNA molecule, thus providing immunity to the host cell.
Non-O1/O139 strains of V. cholerae have been hypothesized to be an important source of new MGEs that could relocate into pandemic strains. Here, we report a GI with VPI-1 recombination characteristics that harbors both a CRISPR-Cas module and an auxiliary T6SS locus in a non-O1/O139 strain of V. cholerae from Sydney, Australia. This GI likely provides recipient cells not only with a defense mechanism against maladaptive LGT, but also with a potential competitive advantage over bacteria lacking this GI and perhaps a novel virulence factor. We also show that similar GIs are present in other non-O1/O139 strains from around the globe.
Methods
Bacterial strain, growth conditions, and molecular biology methods
The non-O1/O139 V. cholerae S12 strain was isolated from the Georges River (Sydney, Australia) in 200927 and routinely cultured on Luria-Bertani medium at 37 °C. The whole genome of V. cholerae S12 was paired-end sequenced at the Wellcome Trust Sanger Institute (Hinxton, UK) using Illumina HiSeq 2000 (San Diego, CA, USA) from DNA extracted using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA). For extraction of GIVchS12 circles, a plasmid extraction was carried out on S12 using the PureYield Plasmid Miniprep Kit (Promega). PCR was performed using 2X MangoMix (Bioline, London, UK) that consists of DNA polymerase, dNTPs, Mg2 + and an orange dye with 30 cycles of denaturation at 93 °C for 30 sec, the appropriate annealing temperature for 30 sec and an extension of 72 °C (1 min/kb). All primers were acquired from Integrated DNA Technologies (Coralville, IA, USA) (Table 1) and used at a final concentration of 10 μM. PCR amplicons intended for sequencing were excised from 1% agarose gels and purified using the Wizard SV Gel and PCR Clean-Up Kit (Promega) and sequenced using the Sanger method at Macrogen (Seoul, South Korea).
Bioinformatic analyses
The Illumina HiSeq whole genome sequencing reads for V. cholerae S12 were filtered to remove low quality reads with average read quality less than Q20 and low quality trailing ends with base quality less than Q20 using Prinseq-lite v0.20.428. Reads were then de novo assembled into contiguous sequences (contigs) using Velvet v1.2.1029 and the assemblies were improved by scaffolding using SSPACE v2.030, gap filling using GapFiller v1.1031, reordering of contigs against the V. cholerae N16961 reference genome using Mauve v2.4.032 and removal of contigs shorter than 300 bp. The final improved draft genome assembled into 83 scaffolds from 4,624,354 read pairs with an average read length of 75 bp to give a total genome size of 4,061,577 bp with average depth of coverage of 171 reads.
The genomic region encoding GIVchS12 was identified on contig 000009 by pairwise comparison of the V. cholerae S12 draft genome to the complete reference genome of V. cholerae N16961 using the program Mauve v2.4.032. Analysis of GIVchS12 on contig 000009 identified three assembly gaps between ORFs 13 and 15 due to a putative repeat of ORF14 (annotated as ORF14a and ORF14b). To confirm this repeat, a PCR with primers Gap_F1 and Gap_R1 that anneal outside of this repeat region (see Fig. 1) resulted in an expected ~2.5 kb product (as opposed to a ~1.3–1.5 kb product if only one copy of ORF14 was present). Two of the assembly gaps were closed through sequencing the ends of the amplicon creating the GIVchS12 sequence in GenBank file KU722393. GIVchS12 was annotated using Prokka33 and the automated annotation was manually curated with the aid of BLAST against the non-redundant NCBI protein database (Bethesda, MD, USA) using BLASTP providing putative identification34. The CRISPR-Cas module in GIVchS12 was identified using the online tool CRISPRFinder (http://crispr.i2bc.paris-saclay.fr)35.

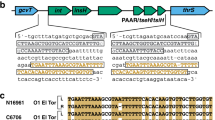
The ~28-kb genomic island, GIVchS12, from Vibrio cholerae S12 containing a CRISPR-Cas module and type VI secretion auxiliary locus.
Schematic representation of GIVchS12 is shown in (A) with the VPI-1 att sites given as hatched boxes and the VPI-1 integrase as a black block arrow. Regions of nucleotide identity to VPI-1 and surrounding regions in V. cholerae N16961 are shown. All ORFs and their orientation are shown as block arrows. Numbers shown in, above, or below the block arrows correlate to the putative identification shown in (B). Primer binding sites are shown by black triangles with their direction of extension indicated by the direction of the triangle.
In order to determine if GIVchS12 or similar islands were present in other V. cholerae, additional V. cholerae genomes were obtained from GenBank. The list of genomes used is provided in Supplementary Material. The genomes were annotated with RAST v2.0 (Rapid Annotation Using Subsystem Technology)36. The GIVchS12 ORFs were compared against the ORFs of the V. cholerae genomes to determine presence/absence by calculating the BLAST score ratio (BSR)37. Significant hits were considered as putative homologues if the BSR values were at least 0.3 (for 30% amino acid identity)38. Furthermore, the whole genomes were aligned using Mugsy v1.2.339, and the core alignment (2,539,853 bp in total length) was extracted from the Mugsy output using Galaxy v16.0440 and Geneious v8.1.841. From this alignment, a maximum likelihood phylogenetic tree was constructed using RAxML v8.1.1742 using the general time reversible (GTR) nucleotide substitution model and gamma distribution pattern. Robustness of branching was estimated with 100 bootstrap replicates. Vibrio metoecus, the closest relative of V. cholerae, was used as an outgroup43.
GenBank accession numbers
The full sequence of the GIVchS12 including flanking sequences and the sequenced attP and attB sites are available in GenBank/ENA/DDBJ and have the accession numbers KU722393, KU722394, and KU722395, respectively. The raw Illumina HiSeq sequencing reads are available under accession number ERR063652 and the improved draft genome assembly of V. cholerae S12 can be accessed at MDST00000000.
Results and Discussion
A novel variant of the Vibrio pathogenicity island
In order to identify regions of interest in the genome of V. cholerae S12, contigs were compared to the closed genome of V. cholerae N16961. A novel GI in the same respective location as VPI-1 (between VC0816 and VC0848 on chromosome 1) was identified on contig 000009. This GI of ~28-kb has been given the designation GIVchS12 and contains an integrase with 94% nucleotide and 100% amino acid identity to the VPI-1 integrase gene and protein, respectively, and characteristic VPI-1 attL and attR sites abutting the GI44. Given GIVchS12’s location, with identical integrase protein and att sites, this GI is likely to have recombination functions identical to VPI-1. Previous studies have observed variations in VPI-1 through PCR analysis of V. cholerae strains or BLASTN analysis of V. cholerae genomes for VPI-1 genes. For the most part, the variations identified represent minor gene gain/loss events or sequence changes to known ORFs in VPI-110,11,12. GIVchS12 is different in that it shares practically no gene content with VPI-1.
A CRISPR-Cas module for self-preservation
Bioinformatic analysis of GIVchS12 identified a CRISPR-Cas module and a T6SS auxiliary locus at the attR and attL ends, respectively (Fig. 1). The CRISPR-Cas module in GIVchS12 contains genes encoding homologues of Cas1, Cas3, and Cas6. Based on the protein sequences and their organization, this CRISPR-Cas system is most likely similar to those of type 1-F26. Several spacers displayed 100% identity to various bacteriophage genomes consistent with the module having a role in acting against invading foreign DNA. The association of a CRISPR-Cas module within a GI is intriguing for two reasons. From an ecological perspective, the mobilization of a CRISPR-Cas system benefits the host not only with an adaptive immune system but also by the instant addition of the immunity that comes with the various spacer sequences it already carries. Thus, a host would immediately gain protection from various bacteriophages and other invading foreign DNA within that ecosystem. Secondly, an intriguing study identified a CRISPR-Cas system within a bacteriophage genome that was able to counteract an inhibitory GI present in the bacterial host genome, thus improving the bacteriophages’ capacity to successfully infect the bacterial host45. As a result, this raises the possibility that the CRISPR-Cas system within GIVchS12 might improve integration efficiency in recipient cells that contain other genetic elements interfering with the GI’s integrity and/or integration. Furthermore, once integrated into the host, the CRISPR-Cas system could prevent the replacement of GIVchS12 by VPI-1 or other GIs competing for the same integration sites. The CRISPR-Cas system found on GIVchS12 could therefore promote direct self-preservation or self-preservation by protecting its host.
A novel T6SS auxiliary locus
Also present on GIVchS12 are genes normally associated with three T6SS loci found in all V. cholerae genomes, known as the main locus and auxiliary loci 1 and 2. The GIVchS12 locus structurally resembles the two T6SS auxiliary loci, with the presence of an hcp gene, a copy of vgrG, a gene encoding a protein with a DUF4123 domain, and putative cargo effectors and immunity proteins further downstream. The lack of proteins making up the T6SS machinery indicates that the proteins on this additional auxiliary locus are dependent on the structural proteins encoded on the main chromosomal T6SS locus for effective translocation into target cells that in S12 is present on contig 00022. The auxiliary loci 1 and 2 are present on contigs 00011 and 00021, respectively, although in contig 00021 the sequence breaks before hcp, presumably due to the difficulty of assembling repeat regions. At the end of contig 00011, the first 392-bp of hcp from auxiliary locus 1 is present before the sequence breaks. In the small contigs of 00079 and 00082 are the first 275-bp and last 108-bp of an hcp homologue, respectively, with hcp from contig 00079 presumably from auxiliary locus 2. The auxiliary 1 and 2 hcp genes in S12 share 99% nucleotide identity with those in V. cholerae N16961 and V52. The hcp from GIVchS12 is clearly different to both the auxiliary 1 and 2 hcp loci sharing 88% nucleotide identity.
The VgrG protein encoded in GIVchS12 is dissimilar to the chromosomal VgrG proteins but, like VgrG-2, lacks a C-terminal effector domain46. The DUF4123 domain found in the protein encoded downstream of the vgrG gene indicates a function as an accessory loading proteins like tap-1 (VC1417 in the auxiliary 1 locus of V. cholerae V52), which is responsible for the loading of cargo effectors with antibacterial activity onto VgrG proteins47,48. Due to the structural similarity of this locus with chromosomal auxiliary loci, it is likely that the hypothetical protein encoded by ORF 11 (Fig. 1) represents such a cargo effector. Antibacterial T6SS effectors are always accompanied by immunity proteins that provide protection against self-intoxication, making it likely that the homologous proteins encoded by ORFs 12, 14a and 14b act as such. ORF 12 is 61% and 62% identical at the nucleotide level to ORFs 14a and 14b, respectively. ORFs 14a and 14b share 89% nucleotide identity. Expression of effectors from the GIVchS12 T6SS locus is likely to increase the range of T6SS-mediated microbial toxicity as evidenced by the divergent VgrG protein and one or more putative other effectors encoded on GIVchS12. It is therefore likely that this increase in effector repertoire gives cells harboring GIVchS12 an advantage over other cells in T6SS-mediated competition. A series of other genes encoding hypothetical proteins are in close proximity and may have a role in either forming the T6SS apparatus or act as effector proteins. For example, ORFs 13 and 15 (also homologues of each other) encode Zn-binding Pro-Ala-Ala-Arg (PAAR) proteins. Zn-binding PAAR proteins form a conical extension on the VgrG tip and also function to attach effector proteins to the spike24. More research is required to gain insight on how V. cholerae S12 regulates expression of the different Hcp and effector proteins and to determine the enzymatic activity of such effector proteins.
A successful and globally distributed genomic island
In order to determine whether GIs similar to GIVchS12 were present in the VPI-1 site of other V. cholerae genomes, we compared GIVchS12 to 28 other V. cholerae strains for which genome sequences are available in public databases. The non-O1/O139 strain from Haiti (2012Env-2) contained a complete GIVchS12-like island with similar CRISPR-Cas and T6SS modules and a complete set of ORFs 16–25 (Fig. 2). RC385 has an almost complete GIVchS12 but lacks ORF14b. A further three GIVchS12-like islands had minor variations. The non-O1/O139 strain 2012Env-32 from Haiti lacks ORF24 and three other ORFs in the CRISPR-Cas module and the non-O1/O139 strain 8–76 and O1 strain A325 from India and Argentina vary in their CRISPR-Cas modules. A325 also lacks ORF14b. Fifteen other GIs were identified containing a CRISPR-Cas module and/or a T6SS, although for many (in the upper most clade; see Fig. 2), we were unable to locate a contig containing VC0816 so it is unclear if the GI continues beyond ORF14a. One GI (in strain VCC19) harboured a T6SS but contained other genes instead of the CRISPR-Cas module and ORFs 16–25. Two other GIs in strains 490–93 and HE-48 contained a divergent CRISPR-Cas modules and other genes instead of ORFs16-25. Given that all these strains have been isolated from different geographic locations (Supplementary Material) including Europe, South America, North America, and Asia, this data indicates that GIs with VPI-1 recombination characteristics are active in disseminating CRISPR-Cas and T6SS modules across the globe.

Presence/absence of the GIVchS12 ORFs in various Vibrio cholerae strains.
The heat map shows the BLAST score ratio (BSR) against the GIVchS12 reference (each column is an ORF). The gradient bar shows the BSR values and their corresponding colours; white indicates the absence of the ORF. Only BSR values of at least 0.3 were included. Strains with similar or complete GIVchS12 are indicated in bold. Striped boxes indicate the presence of a CRISPR-Cas module different from GIVchS12; dotted boxes indicate the presence of other ORFs in those regions different from GIVchS12. Strains with the VPI-1 similar to N16961 are indicated with *. The phylogenetic relationship of the V. cholerae strains is shown on the left of the heat map with Vibrio metoecus OP3H as outgroup. The maximum likelihood phylogenetic tree was constructed from the core alignment of whole genomes (≈2.5 mb) and using the GTR gamma substitution model with 100 bootstrap replicates (indicated on the tree nodes). The scale bar represents nucleotide substitutions per site. Shortened branch lengths, approximately three times the scale bar (0.12), are indicated.
GIVchS12 and associated islands can be divided into sub-clusters or islets, as has been previously done for V. cholerae pathogenicity island 2 (VPI-2)12, consisting of the CRISPR-Cas cluster, the T6SS cluster, and the ORFs 16–25 cluster. Given that multiple GIs (Fig. 2) contain different genes in the ORF 16–25 region and in the case of VCC19, in the CRISPR-Cas region, other islets have clearly been acquired by relatives of GIVchS12. We hypothesize that evolution of GIVchS12 and its relatives proceeded through acquisition of these sub-clusters through homologous recombination processes as supported by the observation that the first 293 bp of the GIVchS12 integrase shares 100% identity to the VPI-1 integrase before dropping to 90% for the remainder of the gene.
A genomic island that readily excises as a circle
To confirm that the GI could excise as a circle, a nested PCR strategy was employed using primers annealing within the GI and facing outward toward the attL and attR ends. First, primers ML130 and ML131 were used in a PCR reaction with template derived from a plasmid preparation of V. cholerae S12. Next, 1 μl from the ML130/ML131 PCR was used as template for a fresh PCR reaction employing primers ML126 and ML127 (relative primer binding sites are shown in Fig. 1A) giving an expected fragment of ~580-bp (Fig. 3). As expected, sequence of the PCR product showed that excision occurs at the att sites abutting the GI producing an attP site identical to what is observed when VPI-1 excises from the genome44.

Cropped DNA agarose gel showing the amplicon of a two-stage nested inverse PCR of the excised GIVchS12 circle (lane 1) and the amplicon of a two-stage PCR of the “empty” chromosomal GIVchS12 site (lane 4).
Controls for each PCR are given in the adjacent lanes that include nested PCR of the negative control from the first-stage PCR and dH2O negative control (lanes 2 and 3 for GIVchS12 circle PCR and lanes 5 and 6 for “empty” chromosomal GIVchS12 site PCR) respectively. Lane M is the DNA marker with relevant band sizes shown on the left of the gel.
PCR of the empty chromosomal site was also conducted using a nested PCR strategy with primers annealing outside the GI and facing in toward the attL and attR ends. Primers ML134 and ML135 were used in a PCR reaction with template derived from a PCR reaction with ML136 and ML137 using genomic DNA as template. The resulting product (Fig. 3) was sequenced and showed precise excision of GIVchS12 leaving an identical attB site as previously seen with excision of VPI-144. VPI-1 uses both a phage-like integrase and a transposase protein called VpiT to facilitate excision of the GI44. Genes encoding these proteins are present within VPI-1.
However, in some V. cholerae pandemic strains, vpiT is in a different location44. VpiT or a homologue of VpiT was not found in GIVchS12 or in the S12 genome.
In conclusion, we report an interesting GI with VPI-1 recombination characteristics housing a CRISPR-Cas element and a T6SS auxiliary locus. This GI is likely to provide its host with a competitive advantage by protecting from bacteriophages as well as adding T6SS-associated bactericidal effectors proteins. Furthermore, this study shows that GIs with VPI-1 recombination characteristics carrying CRISPR-Cas and T6SS modules are circulating in natural V. cholerae populations globally.
Additional Information
How to cite this article: Labbate, M. et al. A genomic island in Vibrio cholerae with VPI-1 site-specific recombination characteristics contains CRISPR-Cas and type VI secretion modules. Sci. Rep. 6, 36891; doi: 10.1038/srep36891 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Kaper, J., Morris, J. Jr & Levine, M. Cholera. Clin Microbiol Rev 8, 48–86 (1995).
Nelson, E. J., Harris, J. B., Glenn Morris, J., Calderwood, S. B. & Camilli, A. Cholera transmission: the host, pathogen and bacteriophage dynamic. Nat Rev Micro 7, 693–702 (2009).
Faruque, S. M. & Mekalanos, J. J. Pathogenicity islands and phages in Vibrio cholerae evolution. Trends in Microbiol 11, 505–510 (2003).
Dobrindt, U., Hochhut, B., Hentschel, U. & Hacker, J. Genomic islands in pathogenic and environmental microorganisms. Nat Rev Microbiol 2, 414–424 (2004).
Chun, J. et al. Comparative genomics reveals mechanisms for short-term and long-term clonal transitions in pandemic Vibrio cholerae. Proc Natl Acad Sci USA 106, 15442–15447 (2009).
Grim, C. J. et al. Genome sequence of hybrid Vibrio cholerae O1 MJ-1236, B-33, and CIRS101 and comparative genomics with V. cholerae. J Bacteriol 192, 3524–3533 (2010).
Dziejman, M. et al. Comparative genomic analysis of Vibrio cholerae: genes that correlate with cholera endemic and pandemic disease. Proc Natl Acad Sci USA 99, 1556–1561 (2002).
Waldor, M. K. & Mekalanos, J. J. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science 272, 1910–1914 (1996).
Karaolis, D. K. R. et al. A Vibrio cholerae pathogenicity island associated with epidemic and pandemic strains. Proc Natl Acad Sci USA 95, 3134–3139 (1998).
Hasan, N. A. et al. Nontoxigenic Vibrio cholerae non-O1/O139 isolate from a case of human gastroenteritis in the U.S. Gulf Coast. J Clin Microbiol 53, 9–13 (2015).
Li, M., Kotetishvili, M., Chen, Y. & Sozhamannan, S. Comparative genomic analyses of the Vibrio Pathogencity Island and cholera toxin prophage regions in nonepidemic serogroup strains of Vibrio cholerae. Appl Environ Microbiol 69, 1728–1738 (2003).
Orata, F. D. et al. The dynamics of genetic interactions between Vibrio metoecus and Vibrio cholerae, two close relatives co-occurring in the environment. Genome Biol Evol 7, 2941–2954 (2015).
Herrington, D. A. et al. Toxin, toxin-coregulated pili, and the toxR regulon are essential for Vibrio cholerae pathogenesis in humans. J Exp Med 168 (1988).
Taylor, R. K., Miller, V. L., Furlong, D. B. & Mekalanos, J. J. Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proc Natl Acad Sci USA 84, 2833–2837 (1987).
Kovach, M. E., Shaffer, M. D. & Peterson, K. M. A putative integrase gene defines the distal end of a large cluster of ToxR-regulated colonization genes in Vibrio cholerae. Microbiology 142, 2165–2174 (1996).
Russell, A. B., Peterson, S. B. & Mougous, J. D. Type VI secretion system effectors: poisons with a purpose. Nat Rev Microbiol 12, 137–148 (2014).
Pukatzki, S. et al. Identification of a conserved bacteria protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc Natl Acad Sci USA 103, 1528–1533 (2006).
Das, S. & Chaudhuri, K. Identification of a unique IAHP (IcmF associated homologous proteins cluster in Vibrio cholerae and other proteobacteria through in silico analysis. In Silico Biol 3, 287–300 (2003).
Williams, S. G., Varcoe, L. T., Attridge, S. R. & Manning, P. A. Vibrio cholerae Hcp, a secreted protein coregulated with HlyA. Infect Immun 64, 283–289 (1996).
Ballister, E. R., Lai, A. H., Zuckermann, R. N., Cheng, Y. & Mougous, J. D. In vitro self-assembly of tailborable nanotubes from a simple protein building block. Proc Natl Acad Sci USA 105, 3733–3738 (2008).
Silverman, J. M., Brunet, Y. R., Cascales, E. & Mougous, J. D. Structure and regulation of the type VI secretion system. Ann Rev Microbiol 66, 453–472 (2012).
Brooks, T. M., Unterweger, D., Bachmann, V., Kostiuk, B. & Pukatzki, S. Lytic activity of the Vibrio cholerae Type VI secretion toxin VgrG-3 is inhibited by the antitoxin TsaB. J Biol Chem 288, 7618–7625 (2013).
Dong, T. G., Ho, B. T., Yoder-Himes, D. R. & Mekalanos, J. J. Identification of T6SS-dependent effector and immunity proteins by Tn-seq in Vibrio cholerae. Proc Natl Acad Sci USA 110, 2623–2628 (2013).
Shneider, M. M. et al. PAAR-repeat proteins sharpen and diversify the type VI secretion system spike. Nature 500, 350–353 (2013).
Croucher, N. J. et al. Horizontal DNA transfer mechanisms of bacteria as weapons of intragenomic conflict. PLoS Biol 14, e1002394, doi: 10.1371/journal.pbio.1002394 (2016).
Makarova, K. S. et al. Evolution and classification of the CRISPR–Cas systems. Nat Rev Microbiol 9, 467–477 (2011).
Islam, A. et al. Indigenous Vibrio cholerae strains from a non-endemic region are pathogenic. Open Biol 3, 120181 (2013).
Schmieder, R. & Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics (2011).
Zerbino, D. R. & Birney, E. Velvet: algorithms for de novo short reads assembly using de Bruijn graphs. Genome Res 18, 821–829 (2008).
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D. & Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579 (2011).
Boetzer, M. & Pirovano, W. Toward almost closed genomes with GapFiller. Genome Biol (2012).
Darling, A. E., Mau, B. & Perna, N. T. progressiveMAUVE: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5, e11147 (2010).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30 2068–2069 (2014).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J Mol Biol 215, 403–410 (1990).
Grissa, I., Vergnaud, G. & Pourcel, C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucl Acids Res 35, W52–W57 (2007).
Aziz, R. K. et al. The RAST server: rapid annotation using subsystems technology. BMC Genomics 9, 75 (2008).
Rasko, D. A., Myers, G. S. A. & Ravel, J. Visualization of comparative genomic analyses by BLAST score ratio. BMC Bioinformatics 6, 1–7, doi: 10.1186/1471-2105-6-2 (2005).
Rost, B. Twilight zone of protein sequence alignments. Protein Engineering 12, 85–94 (1999).
Angiuoli, S. V. & Salzberg, S. L. Mugsy: Fast multiple alignment of closely related whole genomes. Bioinformatics 27, 334–342 (2010).
Goecks, J., Nekrutenko, A. & Taylor, J. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biology 11, 1–13 (2010).
Kearse, M. et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Kirchberger, P. C. et al. Vibrio metoecus sp. nov., a close relative of Vibrio cholerae isolated from coastal brackish ponds and clinical specimens. Int J Syst Evol Microbiol 64, 3208–3214 (2014).
Rajanna, C. et al. The Vibrio pathogenicity island of epidemic Vibrio cholerae forms precise extrachromosomal circular excision products. J Bacteriol 185, 6893–6901 (2003).
Seed, K. D., Lazinski, D. W., Calderwood, S. B. & Camilli, A. A bacteriophage encodes its own CRISPR/Cas adaptive response to evade host innate immunity. Nature 494, 489–491 (2013).
Pukatzki, S., Ma, A. T., Revel, A. T., Sturtevant, D. & Mekalanos, J. J. Type VI secretion system translocates a phage tail spike-like protein into target cells where it cross-links actin. Proc Natl Acad Sci USA 104, 15508–15513 (2007).
Unterweger, D. et al. Chimeric adaptor proteins translocate diverse type VI secretion system effectors in Vibrio cholerae. EMBO 34, 2198–2210 (2015).
Liang, X. et al. Identification of divergent type VI secretion effectors using a conserved chaperone domain. Proc Natl Acad Sci USA 112, 9106–9111 (2015).
Acknowledgements
NRT is supported by the Wellcome Trust, grant #098051 to Wellcome Trust Sanger Institute.
Author information
Authors and Affiliations
Contributions
M.L. conceived the experiments and wrote the manuscript. A.M. and N.R.T. sequenced the V. cholerae S12 genome. N.D.J., W.L.K., and C.A. conducted the experiments. M.L., F.D.O., N.K.P., P.C.K., and Y.B. performed bioinformatic analyses. G.M. and I.G.C. helped supervise the project. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Labbate, M., Orata, F., Petty, N. et al. A genomic island in Vibrio cholerae with VPI-1 site-specific recombination characteristics contains CRISPR-Cas and type VI secretion modules. Sci Rep 6, 36891 (2016). https://doi.org/10.1038/srep36891
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep36891
This article is cited by
-
CRISPR-Cas systems target endogenous genes to impact bacterial physiology and alter mammalian immune responses
Molecular Biomedicine (2022)
-
CRISPR-Cas systems are present predominantly on chromosome and its relationship with MEGs in Vibrio species
Archives of Microbiology (2022)
-
Pandemic Vibrio cholerae shuts down site-specific recombination to retain an interbacterial defence mechanism
Nature Communications (2020)
-
Insights into Klebsiella pneumoniae type VI secretion system transcriptional regulation
BMC Genomics (2019)
-
CRISPR-Cas systems are present predominantly on mobile genetic elements in Vibrio species
BMC Genomics (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
