
Abstract
Acute kidney injury (AKI) remains challenging for clinical practice and poses a risk of developing progressive chronic kidney disease (CKD) with no definitive treatment available yet. Tanshinone IIA, an active ingredient of Chinese herbal Salvia miltiorrhiza, has been widely used in Asia for the remarkable organoprotective activities. Its effect on established AKI, however, remains unknown. In mice with folic acid-induced AKI, delayed treatment with Tanshinone IIA, commenced early or late after injury, diminished renal expression of kidney injury markers, reduced apoptosis and improved kidney dysfunction, concomitant with mitigated histologic signs of AKI to CKD transition, including interstitial fibrosis and tubular atrophy, and with an ameliorated inflammatory infiltration in tubulointerstitium and a favored M2-skewed macrophage polarization. Mechanistically, Tanshinone IIA blunted glycogen synthase kinase (GSK)3β overactivity and hyperactivation of its downstream mitogen-activated protein kinases that are centrally implicated in renal fibrogenesis and inflammation. Inhibition of GSK3β is likely a key mechanism mediating the therapeutic activity of Tanshinone IIA, because sodium nitroprusside, a GSK3β activator, largely offset its renoprotective effect. In confirmatory studies, rescue treatment with Tanshinone IIA likewise ameliorated ischemia/reperfusion-induced kidney destruction in mice. Our data suggest that Tanshinone IIA represents a valuable treatment that improves post-AKI kidney salvage via targeting GSK3β.
Similar content being viewed by others


Introduction
Acute kidney injury (AKI), characterized by an abrupt decline in kidney function, is a common and devastating problem in clinical practice. Epidemiologic evidence indicates a growing prevalence of acute kidney disease worldwide, probably associated with increasing incidence of other comorbidities, including diabetes mellitus, hypertension, and caloric nutritional overload1,2. Despite enormous efforts dedicated to the understanding of the underlying pathogenic mechanisms, AKI continues to be a therapeutic challenge with no definitive and specific treatment available yet to rescue kidney injury after AKI. Existing regimens are virtually confined to symptomatic treatments and general supportive cares3, which are, however, of limited utility with unsatisfying therapeutic efficacy, as evidenced by the poor prognosis. Indeed, long-term follow-up studies show that the number of patients with incomplete renal recovery have been underestimated and that AKI per se is an independent risk factor for subsequent transition to chronic kidney disease (CKD) and progression to end stage renal disease4,5,6,7,8. There is therefore a pressing need to develop novel approaches to promote kidney salvage following AKI. To this end, numerous pre-clinical studies have been carried out in the past several decades. However, the majority of these studies were performed in a preventive manner with the intervention imposed prior to kidney insults, thus making these studies clinically irrelevant9,10,11,12. Moreover, as a complex disease, AKI does not have a single cause, but instead involves multiple pathogenic mechanisms13, such as renal tubular damage, inflammation, and fibrogenesis. Consequently, most novel approaches targeting merely a single pathogenic pathway eventually failed in late-stage trials even though they exhibited some promise at early phases. It is imperative to explore multi-pronged approaches for kidney rescue after AKI.
Chinese herbal medicine has being practiced for thousands of years and successfully treated a number of human diseases, including some kidney diseases14,15,16. Although the exact pharmacologic mechanisms remain to be clarified, striking advances have been made in recent years by applying modern pharmacochemistry techniques and methods. Tanshinone IIA, an active ingredient of the traditional Chinese herbal Salvia miltiorrhiza, has been widely used in Asia to treat various diseases for its remarkable organoprotective activities14,17,18,19,20,21,22. Tanshinone IIA has been proven to be a low toxic and safe agent by numerous clinical and experimental studies19,20,21,22. Emerging data suggest that Tanshinone IIA possesses significant anti-inflammatory activities, by which it ameliorates injury in various organ systems, such as the brain, lung and heart23,24,25,26,27,28,29. Notably, two randomized clinical trials are being undertaken to validate its effect on ischemic heart disease30,31. In addition, a prominent beneficial effect of Tanshinone IIA in experimental chronic kidney diseases has also been reported, including remnant kidney disease, diabetic nephropathy, uric acid nephropathy, and hypothermic kidney preservation32,33,34,35,36,37. However, the effect of Tanshinone IIA on AKI has been barely examined. This study aimed to determine the effect of delayed Tanshinone IIA treatment on established AKI in murine models of AKI induced by folic acid or ischemia-reperfusion injury (IRI). We found that Tanshinone IIA therapy initiated at either early or late after injury, was effective in protecting against both acute and chronic kidney damage and dysfunction.
Results
Delayed Tanshinone IIA treatment improves kidney dysfunction incited by folic acid
Following folic acid injury (Fig. 1a), mice rapidly developed AKI, marked by an elevated serum creatinine level that was evident on day 1 and peaked on day 3. This was followed by a partial spontaneous recovery with the serum creatinine level higher than the normal level till day 28 (Fig. 1b). In contrast, Tanshinone IIA therapy, starting 1 day or 5 days after folic acid injury, significantly attenuated the elevation of serum creatinine levels, and completely corrected kidney dysfunction on day 28. In consistency, gross parenchymal changes in the kidney, reflected by reduced kidney to body weight ratios, were evidently observed 28 days after folic acid injection. This effect was considerably abrogated by Tanshinone IIA treatment with a more prominent efficacy exhibited in the early-treated than the late-treated animals (Fig. 1c).

Tanshinone IIA ameliorates folic acid-induced renal dysfunction in mice.
(a) Animal experimental design. Mice were randomized into 5 groups to receive vehicle, folic acid (FA) and/or Tanshinone IIA (TS) given on day 1 [FA+TS(E)] or day 5 [FA+TS(L)] after folic acid injury; (b) Time course changes of serum creatinine levels (mg/dl) measured on days 0, 1, 3, 5, 7, 14 and 28; *P < 0.001, FA+TS(E) vs FA; #P < 0.001 FA+TS(E) vs FA+TS(L); $P < 0.001, FA+TS(L) vs FA; △P = 0.017, FA+TS(E) vs FA; @P = 0.018, FA+TS(L) vs FA; (n = 9). (c) Kidney to body weight ratios were determined on day 28. ◊P < 0.001 vs Control; ★P < 0.001 vs TS; ▲P = 0.004 vs FA; ☆P = 0.024 vs FA; n.s., not significant; (n = 9).
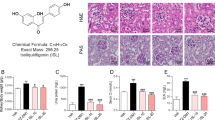
Tanshinone IIA reduces kidney injury markers and ameliorates inflammatory infiltration following folic acid insult
To determine the protective effects of Tanshinone IIA on folic acid injured kidney, kidney sections and urine sample were collected for assessment at 7 days after folic acid injection. Seven days after folic acid injection, mouse kidney demonstrated prominent hypercellularity in the interstitium as revealed by hematoxylin and eosin (H&E) staining (Fig. 2a). Tanshinone IIA treatment markedly attenuated renal inflammatory infiltration with a greater efficacy conferred by the early than the late Tanshinone IIA treatment according to scoring of renal interstitial cell infiltration (Fig. 2b). In parallel, the augmented urine excretion of neutrophil gelatinase-associated lipocalin (NGAL) after folic acid injection was diminished in Tanshinone IIA treated mice with a stronger effect observed in the early treatment group (Fig. 2c). Collectively, these findings suggest that Tanshinone IIA possesses an anti-inflammatory property that prevents the progression of established AKI.

Both early and late Tanshinone IIA treatments ameliorate kidney inflammation and injury induced by folic acid.
(a) Representative micrographs showing hematoxylin-eosin (HE) staining of kidney specimens procured on day 7. Scale bar = 100 μm. (b) Interstitial cell infiltration score assessed based on evaluation of HE staining. ◊P < 0.001 vs Control; ★P < 0.001 vs TS; *P < 0.001 vs FA; #P = 0.016 vs FA; n.s., not significant (n = 9). (c) Time course changes of urinary neutrophil gelatinase associated lipocalin (NGAL) levels. ◊P < 0.001 vs Control; ★P < 0.001 vs TS; *P < 0.001 vs FA; $P < 0.001 vs TS + TS(L); n.s., not significant; (n = 9).
Tanshinone IIA hinders transition of AKI to CKD after folic acid injury
To determine the exact histologic changes in the kidney following folic acid injury and Tanshinone IIA treatment, kidney specimens procured on day 28 were processed for Masson trichrome staining for collagens and peroxidase staining for fibronectin. Shown in Fig. 3a,b, folic acid injury resulted in conspicuous transition of AKI to CKD, marked by tubular atrophy and interstitial fibrosis that is characterized by substantial collagen and fibronectin accumulation in renal interstitium. This effect was significantly mitigated by both early and late treatments with Tanshinone IIA. The morphologic findings were further corroborated by immunoblot analysis of kidney homogenates for collagen I and fibronectin, followed by densitometry analysis of immunoblots (Fig. 3c–e), which demonstrated a greater beneficial efficacy of the early than the late Tanshinone IIA treatment. The seemingly superior efficacy of early versus late Tanshinone IIA treatment is unlikely attributable to the difference in treatment duration. In support of this, additional groups of mice were sacrificed 23 d after early Tanshinone IIA or vehicle treatment (i.e. 24 d after folic acid injury). Histologic signs of kidney destruction, AKI to CKD transition and renal fibrosis, as estimated by morphology based on Masson trichrome staining (Supplemental Figure 1a,b) or by immunoblot analysis of kidney specimens for collagen I and fibronectin (Supplemental Figure 1c~e), were all improved by early Tanshinone IIA treatment to a significantly greater extent when compared with late Tanshinone IIA treatment for the same period.

Tanshinone IIA attenuates folic acid-induced kidney fibrosis.
(a) Representative micrographs showing Masson trichrome staining of kidney specimens procured on day 28; Scale bar = 1 mm. (b) Representative micrographs showing immunoperoxidase staining of kidney specimens procured on day 28 for fibronectin; Scale bar = 500 μm. (c) Representative immunoblots of collagen I and fibronectin in cortical kidney lysates. (d,e) Relative abundance of collagen I (d) and fibronectin (e) in immunoblots expressed as densitometric ratios of collagen I/GAPDH or fibronectin/GAPDH. ◊P < 0.001 vs Control; ★P < 0.001 vs TS; *P < 0.001 vs FA; #P = 0.034 vs FA; $P = 0.026 vs FA+TS(E); @P = 0.001 vs FA; △P = 0.027 vs FA+TS(E); n.s., not significant; (n = 9).
Tanshinone IIA exerts a protective effect directly against the folic acid-elicited kidney damages
One of the major characteristics of the post-AKI pathology is renal tubular epithelial destruction. To determine if Tanshinone IIA treatment directly affects renal tubular injury after folic acid exposure, kidney tissues were procured on day 3 and examined. Scoring of renal tubular injury based on H&E staining (Fig. 4a,b) revealed that early Tanshinone IIA therapy effectively ameliorated the folic acid-elicited renal tubular injuries. This finding was further validated by fluorescent immunohistochemistry staining of kidney sections for kidney injury molecule-1 (KIM-1, Fig. 4c) or immunoblot analysis of kidney homogenates for KIM-1 and NGAL (Fig. 4d~f), well established biomarkers for renal tubular injuries. Renal tubular death, marked by acute tubular necrosis and apoptosis, is a main feature of AKI. To evaluate the possible effect of Tanshinone IIA on renal tubular cell death, kidney specimens were prepared for terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) for apoptotic cells. Shown in Fig. 4g, folic acid insult elicited blatant apoptosis in the kidney with TUNEL staining largely distributed to tubular epithelia. This effect was considerably overridden by Tanshinone IIA treatment with a much stronger effect observed in the early treatment group. Morphologic findings were confirmed by absolute counting of TUNEL positive cells (Fig. 4h). Taken together, these data infer that Tanshinone IIA therapy conveys a direct protective effect on renal tubules.

Early Tanshinone IIA treatment protects against folic acid-induced renal tubular damage.
(a) Representative micrographs showing hematoxylin-eosin (HE) staining of kidney specimens procured on day 3. Scale bar = 500 μm. (b) Kidney tubular injury score assessed based on evaluation of HE staining. ◊P < 0.001 vs Control; ★P < 0.001 vs TS; *P < 0.001 vs FA; n.s., not significant. (c) Representative micrographs showing immunofluorescent staining of kidney specimens procured on day 3 for kidney injury molecule-1(KIM-1). Sections were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Scale bar = 200 μm. (d) Representative immunoblots of KIM-1 and neutrophil gelatinase associated lipocalin (NGAL) in cortical kidney lysates. (e) and (f) Relative abundance of KIM-1 (e) and NGAL (f) in immunoblots expressed as densitometric ratios of KIM-1/GAPDH or NGAL/GAPDH; ◊P < 0.001 vs Control; ★P < 0.001 vs TS; *P < 0.001 vs FA; #P = 0.004 vs FA; n.s., not significant; (n = 9). (g) Representative micrographs showing TUNEL staining of kidney specimens procured on day 3. Scale bar = 200 μm. (h) Absolute counting of TUNEL-positive nuclei per high power field. ◊P < 0.001 vs Control; ★P < 0.001 vs TS; $P = 0.009 vs FA; n.s., not significant; (n = 9).
Tanshinone IIA inhibits early inflammatory response in folic acid-injured kidneys
To understand the mechanism responsible for the extra benefit conferred by early Tanshinone IIA treatment, kidney tissues were collected on day 3 and examined. As compared with kidney specimens from control or Tanshinone IIA alone treated animals, folic acid-injured kidneys exhibited florid infiltrations by inflammatory cells that were positive for leukocyte common antigen (CD45), as determined by fluorescent immunohistochemistry staining or by flow cytometry analysis of isolated single kidney cells (Fig. 5a~c). Early Tanshinone IIA treatment substantially reduced the number of CD45 positive cells in the kidney, denoting an anti-inflammatory activity. Local production of proinflammatory cytokines and chemokines is a prerequisite for recruitment of circulating leukocytes to periphery lesion sites. To further explore if the anti-inflammatory action of Tanshinone IIA possibly stems from a primary effect on the inflamed kidney, kidney specimens were prepared for fluorescent immunohistochemistry staining for tumor necrosis factor-α (TNF-α) and monocyte chemotactic protein-1 (MCP-1), which are cytokines crucial for inflammatory response and leukocyte infiltration. Shown in Fig. 5d, early Tanshinone IIA treatment drastically mitigated the folic acid-elicited renal expression of TNF-α, which was mainly distributed to renal tubules. The morphologic findings were further corroborated by confirmatory immunoblot analysis of kidney homogenates (Fig. 5e) followed by densitometric quantitation (Fig. 5f,g).

Early Tanshinone IIA treatment attenuates folic acid-induced kidney inflammation.
(a) Representative micrographs showing immunofluorescent staining of kidney specimens procured on day 3 for CD45. Sections were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Scale bar = 200 μm. (b) Flow cytometry analysis of isolated single kidney cells for CD45+ cells. SSC, Side-scatter. (c) Quantitation of CD45+ cells in the kidney as measured by flow cytometry. ★P < 0.001 vs TS; *P < 0.001 vs FA; (n = 9). (d) Representative micrographs showing immunofluorescent staining of kidney specimens procured on day 3 for tumor necrosis factor-α (TNF-α) or monocyte chemotactic protein-1 (MCP-1). Sections were counterstained with DAPI. Scale bar = 200 μm. (e) Representative immunoblots of TNF-α and MCP-1 in cortical kidney lysates. f and g, Relative abundance of TNF-α (f) and MCP-1 (g) in immunoblots expressed as densitometric ratios of TNF-α/GAPDH or MCP-1/GAPDH. ◊P < 0.001 vs Control; ★P < 0.001 vs TS; #P = 0.008 vs FA; $P = 0.003 vs FA; n.s., not significant; (n = 9).
Macrophage infiltration in folic acid-injured kidneys is attenuated by Tanshinone IIA therapy
A growing body of evidence suggests that macrophages play a pivotal role in the development and progression of AKI. Kidney specimens collected on day 7 were examined for renal infiltration of macrophages by fluorescent immunohistochemistry staining for F4/80. Shown in Fig. 6a,b, F4/80 positive cells were barely detected in kidney tissues from control or Tanshinone IIA alone treated mice. Following folic acid injury, massive macrophage infiltration was noted in renal interstitium. Delayed treatment with Tanshinone IIA markedly attenuated macrophage accumulation in the folic acid-injured kidney with a greater effect observed in the early treatment group (Fig. 6b). Evidence indicates that macrophage proliferation may contribute to macrophage accumulation in injured tissues. To evaluate macrophage proliferation, dual color fluorescent immunohistochemistry staining for F4/80 and Ki 67 was conducted (Fig. 6c). The number of proliferative macrophages marked by cells positive for both F4/80 and Ki67 in the folic acid-injured kidney was evidently reduced after Tanshinone IIA treatment. These data indicate that Tanshinone IIA treatment impedes macrophage infiltration and proliferation in the folic acid injured kidneys.

Tanshinone IIA mitigates macrophage accumulation in the folic acid injured kidney.
(a) Representative micrographs showing immunofluorescent staining of kidney specimens procured on day 7 for F4/80. Scale bar = 200 μm. (b) Absolute counting of the number of F4/80-positive cell per high-power (HP) field. ◊P < 0.001 vs Control; ★P < 0.001 vs TS; *P < 0.001 vs FA; #P = 0.004 vs FA; $P = 0.007 vs FA+TS(E); n.s., not significant; (c) Representative micrographs showing dual color immunofluorescent staining of kidney specimens procured on day 7 for F4/80 and Ki67. Scale bar = 100 μm. White arrows indicate the dually positive cells in the sections. (d) The percentage of F4/80+Ki67+ cells among all F4/80+ cells. ★P < 0.001 vs TS; *P < 0.001 vs FA; @P = 0.004 vs FA; n.s., not significant; (n = 9).
Tanshinone IIA treatment promotes M2 macrophage polarization in the folic acid injured kidney
Macrophages in diseased kidneys exist in different subtypes, such as M1 and M2, which play distinct or even opposite roles in renal injury and repair. Following folic acid injury, the majority of the infiltrating F4/80 positive macrophages were negative for mannose receptor (MR) on dual color fluorescent immunohistochemistry staining and flow cytometry analysis of isolated single kidney cell suspensions (Fig. 7a,b), consistent with an M1 phenotype. Tanshinone IIA treatment markedly increased the number of F4/80+MR+ M2 subtype of macrophages in the folic acid injured kidney with a greater effect observed in the early treatment group (Fig. 7a–c). In agreement, immunoblot analysis of kidney homogenates for M2 macrophage markers, like MR or arginase I, suggests that M2 polarization was considerably amplified after Tanshinone IIA treatment with a stronger effect observed in the early treatment group (Fig. 7d~f). These results imply that Tanshinone IIA treatment favors an M2-skewed macrophage polarization in the folic acid injured kidneys.

Tanshinone IIA promotes M2 macrophages polarization in the folic acid injured kidney.
(a) Representative micrographs showing immunofluorescent staining of kidney specimens procured on day 7 for F4/80 and mannose receptor (MR). Sections were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Scale bar = 50 μm. White arrows indicate the dually positive cells in the sections. (b) Flow cytometry analysis of isolated single kidney cells for cells positive for MR and F4/80; cells were gated on F4/80+ macrophages. (c) The percentage of F4/80+MR+ cells among all F4/80+ cells as determined by flow cytometry. *P < 0.001 vs FA; #P < 0.001 vs FA+TS(E); (n = 9). (d) Representative immunoblots of Arginase I, MR, and F4/80 in cortical kidney lysates prepared on day 7. (e) and (f) Relative abundance of Arginase I (e) and MR (f) in immunoblots expressed as densitometric ratios of Arginase I/F4/80 or MR/F4/80. *P < 0.001 vs FA; $P = 0.041 vs FA+TS(E); @P = 0.002 vs FA; △P = 0.029 vs FA+TS(E); (n = 9).
Tanshinone IIA modulates glycogen synthase kinase (GSK) 3β-MAPK signaling pathway activity in the folic acid injured kidney
Burgeoning evidence indicates that GSK3β is a point of convergence of multiple pathways involved in acute and chronic kidney injuries38. GSK3β has been known to dictate the activity of mitogen-activated protein kinases (MAPK)39,40,41, which are implicated in modulating renal inflammation and fibrogenesis42,43,44,45,46,47. Indeed, folic acid injury drastically induced GSK3β overactivity, reflected by the diminished inhibitory phosphorylation of GSK3β in the kidney, and triggered hyperphosphorylation and hyperactivation of p38 (Fig. 8a,c), extracellular regulated protein kinase (ERK, Fig. 8a,d) and c-Jun N-terminal kinase (JNK, Fig. 8a,e), as shown by immunoblot analysis of kidney homogenates. The augmented activity of GSK3β and diverse MAPK pathways was strikingly blunted following Tanshinone IIA treatment with a stronger effect detected in the early treatment group, concomitant with the anti-inflammatory and anti-fibrotic action of Tanshinone IIA observed above.

Tanshinone IIA mitigates GSK3β overactivity and hyperactivation of its downstream mitogen-activated protein kinases (MAPK) in folic acid-injured kidneys.
(a) Representative immunoblots of GSK3β and phosphorylated GSK3β-Ser9(p-GSK3β); p38 and phosphorylated p38 (p-p38); extracellular regulated protein kinase (ERK) and phosphorylated ERK (p-ERK); c-Jun N-terminal kinase (JNK) and phosphorylated JNK (p-JNK) in cortical kidney lysates prepared from indicated animal groups on day 7. (b) Relative abundance of p-GSK3β in immunoblots expressed as densitometric ratios of p-GSK3β/GSK3β. ◊P < 0.01 vs Control; ★P < 0.01 vs TS; %P = 0.041 vs Control; &P = 0.027 vs TS; *P < 0.001 vs FA; ●P = 0.004 vs FA; P = 0.014 vs FA+TS(E). (c) Relative abundance of p-p38 in immunoblots expressed as densitometric ratios of p-p38/p38; ◊P < 0.01 vs Control; ★P < 0.01 vs TS; *P < 0.001 vs FA; #P = 0.048 vs FA; $P = 0.031 vs FA+TS(E). (d) Relative abundance of p-ERK in immunoblots expressed as densitometric ratios of p-ERK/ERK. ◊P < 0.01 vs Control; ★P < 0.01 vs TS; *P < 0.001 vs FA; @P = 0.029 vs FA; △P = 0.033 vs FA+TS(E). (e) Relative abundance of p-JNK in immunoblots expressed as densitometric ratios of p-JNK/JNK. ◊P < 0.01 vs Control; ★P < 0.01 vs TS; *P < 0.001 vs FA; ▲P = 0.007 vs FA; ☆P = 0.011 vs FA+TS(E); n.s., not significant; (n = 9).
Inhibition of GSK3β is a key signaling mechanism mediating the kidney protective effect of Tanshinone IIA
GSK3β has been shown to play a crucial role in mediating kidney injury38. To determine if inhibition of GSK3β possibly represents a key mechanism conveying the beneficial activity of Tanshinone IIA after AKI, additional mice received Tanshinone IIA therapy in the presence of daily treatment with vehicle or sodium nitroprusside, a GSK3β activator48, starting 1 day or 5 days after folic acid injury. Kidney specimens were procured 7 days after folic acid injury for immunoblot analysis (Fig. 9a~e) and histologic evaluation (Fig. 9f,g). Shown in Fig. 9a~e, both early and late Tanshinone IIA therapy restored inhibitory phosphorylation of GSK3β and overrode hyperphosphorylation of the downstream MAPK kinases, including p38, ERK and JNK. These cellular signaling effects were markedly abrogated by simultaneous sodium nitroprusside treatment. In parallel, histological signs of kidney destruction (Fig. 9f), including vacuolization of proximal tubular epithelium, luminal ectasia, tubular epithelial simplification and atrophy, and inflammatory infiltration in tubulointerstitium were improved after early or late Tanshinone IIA therapy, as corroborated by semi-quantitative evaluation of tubular injury scores (Fig. 9g). The beneficial effect of Tanshinone IIA was markedly offset by sodium nitroprusside co-treatment, entailing that inhibition of GSK3β is essential for the kidney protective activity of Tanshinone IIA.

Inhibition of GSK3β is essential for the beneficial effect of Tanshinone IIA in folic acid-injured kidney.
Mice received Tanshinone IIA therapy in the presence of daily treatment with vehicle or sodium nitroprusside (SNP, 200 μg/kg), a GSK3β activator, starting 1 day or 5 days after folic acid injury. Kidney specimens were procured day 7 after folic acid injury. (a) Representative immunoblots of GSK3β and phosphorylated GSK3β-Ser9(p-GSK3β); p38 and phosphorylated p38 (p-p38); extracellular regulated protein kinase (ERK) and phosphorylated ERK (p-ERK); c-Jun N-terminal kinase (JNK) and phosphorylated JNK (p-JNK) in cortical kidney lysates prepared on day 7. (b) Relative abundance of p-GSK3β in immunoblots expressed as densitometric ratios of p-GSK3β/GSK3β. ◊P < 0.001 vs FA+TS(E); ★P < 0.001 vs FA+TS(L). (c) Relative abundance of p-p38 in immunoblots expressed as densitometric ratios of p-p38/p38. @P < 0.001 vs FA+TS(E); #P = 0.027 vs FA+TS(L). (d) Relative abundance of p-ERK in immunoblots expressed as densitometric ratios of p-ERK/ERK. $P < 0.001 vs FA+TS(E); &P = 0.009 vs FA+TS(L). (e) Relative abundance of p-JNK in immunoblots expressed as densitometric ratios of p-JNK/JNK. ▲P < 0.001 vs FA+TS(E); △P = 0.004 vs FA+TS(L). (f) Representative micrographs showing hematoxylin-eosin (HE) staining of kidney specimens procured on day 7. Scale bar = 200 μm. (g) Kidney tubular injury score assessed based on evaluation of H&E staining. P < 0.001 vs FA+TS(E); ☆P < 0.001 vs FA+TS(L); (n = 9).
Delayed administration of Tanshinone IIA attenuates kidney destruction following IRI
To further ascertain if the beneficial effect of delayed Tanshinone IIA therapy could be generalized to other types of AKI, an additional murine model of AKI induced by IRI was employed (Fig. 10a). Shown in Fig. 10b, Tanshinone IIA treatment given 24 hours after IRI markedly improved kidney function, reflected by the lowered levels of serum creatinine. This was accompanied with amelioration of renal histologic injuries, as revealed by scoring of tubular injury based on H&E staining and by fluorescent immunohistochemistry staining of NGAL in conjunction with morphometric analysis (Fig. 10c~f). Moreover, histologic signs of renal inflammation, including neutrophil infiltration and renal expression of MCP-1, in IR-injured kidneys were significantly attenuated after Tanshinone IIA therapy (Fig. 10g~j).

Tanshinone IIA attenuates ischemia-reperfusion injury induced renal dysfunction and kidney damage and inflammation.
(a) Animal experimental design. Mice were randomized into four groups to receive bilateral kidney ischemia-reperfusion (IR) injury and treatment with vehicle or Tanshinone IIA (TS) given 24 hours after ischemia. (b) Time course changes of serum creatinine levels measured at 0, 24 and 48 hours. ◊P < 0.001 vs Control; ★P < 0.001 vs TS; *P = 0.009 vs IR. (c) Representative micrographs showing hematoxylin-eosin (HE) staining of kidney specimens procured at 48 h. Scale bar = 200 μm. (d) Renal tubular injury score assessed based on evaluation of HE staining. ◊P < 0.001 vs Control; ★P < 0.001 vs TS; #P = 0.008 vs IR. (e) Representative micrographs showing immunofluorescent staining of kidney specimens procured at 48 h for neutrophil gelatinase associated lipocalin (NGAL). Scale bar = 200 μm. (f) Semi-quantification of NGAL staining by morphometric analysis. ◊P < 0.001 vs Control; ★P < 0.001 vs TS; #P = 0.011 vs IR. (g) Representative micrographs showing immunoperoxidase staining of kidney specimens procured at 48 h for neutrophils; Scale bar = 100 μm. (h) Absolute counting of the number of neutrophils positive for myeloperoxidase per high-power (HP) field. ◊P < 0.001 vs Control; ★P < 0.001 vs TS; #P = 0.002 vs IR. (i) Representative micrographs showing immunoperoxidase staining of kidney specimens procured at 48 h for monocyte chmotactic protein-1 (MCP-1); Scale bar = 500 μm. (j) Semi-quantification of MCP-1 expression estimated by morphometric analysis. ◊P < 0.01 vs Control; ★P < 0.01 vs TS; @P = 0.031 vs IR. n.s., not significant; (n = 6).
Discussion
In this study, we found that delayed Tanshinone IIA therapy, commenced early or late after AKI, could effectively attenuate kidney injury elicited by folic acid or IRI and hinder AKI to CKD transition. This beneficial effect of Tanshinone IIA is likely associated with a direct protection against renal tubular damage and an anti-inflammatory activity. To the best of our knowledge, this study is the first to demonstrate the protective effect of delayed Tanshinone IIA therapy on established AKI and the ensuing transition to CKD.
The mechanism responsible for the protective effect of Tanshinone IIA on renal tubular damage is not clear, but our data suggest that it might be associated with a direct anti-apoptotic action. However, the effect of Tanshinone IIA on cellular apoptosis has been highly controversial with both proapoptotic and antiapoptotic activities reported before. For instance, in cardiomyocytes injured with doxorubicin or angiotensin II, Tanshinone IIA was able to confer a potent anti-apoptotic effect49,50. Similarly, in cultured endothelial cells, Tanshinone IIA was capable of counteracting the proapoptotic effect of hydrogen peroxide51. In contrast, Tanshinone IIA has been shown to repress cell growth through inducing apoptosis in multiple cancer cells, including leukemia cells, osteosarcoma cells, and prostate cancer cells52,53,54. This disparity might be due to distinct responses of different cell types, but is more likely associated with difference in Tanshinone IIA doses. Indeed, the doses of Tanshinone IIA applied in studies showing proapoptotic activities53 were almost 10 times higher than those used in studies demonstrating cytoprotective effects49. This seemingly bimodal effect of Tanshinone IIA has been leveraged to kill cancer cells at high doses, but to protect parenchymal cells at much lower doses. The dose of Tanshinone IIA used in this study was very low (50 mg/kg/d) and had been reported by other studies to be safe23,55. No noticeable adverse effect was noted in our animals, according to the observation of body weight, daily behavior and activity and to the analysis of other organ specimens. In this study, Tanshinone IIA treatment lessened renal tubular damage and apoptosis following AKI. Whether this effect was exerted via a renal tubular cell autonomous mechanism warrants in-depth exploration.
In contrast to the arguable effect on apoptosis, Tanshinone IIA has been reproducibly demonstrated to possess a potent anti-inflammatory activity in animal models of diverse diseases, including chronic kidney disease, ischemic brain injury, endotoxin-associated acute lung injury, ischemic heart disease and atherosclerosis19,23,55,56. However, it is largely unknown whether Tanshinone IIA is also beneficial in AKI or whether Tanshinone IIA is able to alter inflammatory injuries in AKI. Inflammation per se is involved in the pathogenesis of AKI and plays a key role in the following transition to CKD. Inflammatory cell infiltration, a hallmark of inflammatory response, is not only closely associated with the severity of AKI, but also a crucial determinant of the long-term outcome of the injured kidney. Data from animal experiments showed that neutrophil infiltration can be detected as early as just few hours after kidney injury. Although the role of neutrophil infiltration in AKI remains to be defined, recent evidence suggests that blocking the recruitment of neutrophil into the kidney can effectively protect the IRI incited kidney injury57. In consistency, in the present study, we found that delayed Tanshinone IIA treatment after initial insult could significantly decrease the numbers of leukocytes in the folic acid injured kidneys and the amount of neutrophils in IR injured kidneys, in parallel with amelioration of kidney dysfunction and renal histologic injury. This finding is in accordance with some previous studies demonstrating that Tanshinone IIA could impede neutrophils infiltration into lipopolysaccharide (LPS)-injured lung or the brain following ischemia injury23,29. In conjunction with previous findings, it seems that Tanshinone IIA may ameliorate established AKI in part by suppressing inflammatory infiltration at the acute phase.
Macrophage is a key player in the development and progression of AKI58. They usually infiltrate into the injured kidney several days after the initial insult. Emerging data prove that the numbers of infiltrating macrophages at the early phase of injury is associated with the severity of AKI59. In consistency, early depletion of macrophages significantly attenuated the renal injury in various models of AKI. In our study, both early and late treatments with Tanshinone IIA after AKI could significantly reduce the accumulation of macrophage in folic acid injured kidney. The underlying mechanism is not clear. One possibility is that Tanshinone IIA may be able to inhibit in situ macrophage proliferation, as shown by our results. Another possibility could be that Tanshinone IIA directly hampers macrophage recruitment into the kidney. In support of this, Tanshinone IIA treatment inhibited the expression of chemokines like MCP-1 in renal parenchyma after folic acid injury or IRI60. Different macrophage subtypes are known to play distinct or even opposite roles in tissue injury and repair59,61. It is well known that M1 macrophages possess pro-inflammatory activities by releasing cytokines that damage surrounding tissue, whereas M2 macrophages exert anti-inflammatory effects by releasing cytokines that promote tissue recovery61,62,63. In the present study, Tanshinone IIA was found to favor an M2-skewed macrophage polarization in folic acid injured kidneys, suggestive of a proreparative effect. This finding is consistent with a previous study showing that Tanshinone IIA could inhibit the expression of inducible nitric oxide synthase in LPS-stimulated macrophages in vitro64,65. However, whether the effect of Tanshinone IIA on macrophage polarization in the injured kidney is attributable to a direct effect on macrophages per se or secondary to the improved local tissue microenvironments merits further investigation.
The molecular basis of progression of AKI is complex. Accumulating evidence points to GSK3β as a point of convergence for a number of nephropathic pathways downstream diverse renal injurious signals. GSK3β has been known to dictate the activity of MAPK kinases39,40,41, including p38, ERK and JNK, which have been implicated in renal inflammation and fibrogenesis in progressive AKI38. MAPKs are implicated in NF-κB activation and production of proinflammatory cytokines and chemokines in the kidney upon injury42,43,44. In addition, MAPKs play a critical role in AKI to CKD transition or renal fibrogenesis, characterized by tubular atrophy and interstitial fibrogenesis45,46,47. Furthermore, activation of MAPKs is associated with renal fibroblast proliferation and activation46, a hallmark of kidney fibrogenesis. In our study, Tanshinone IIA treatment was able to effectively mitigate GSK3β overactivity and subsequent hyperactivation of MAPK pathways in the injured kidney. Conversely, sodium nitroprusside, a GSK3b activator48, offset the effect of Tanshinone IIA on the GSK3β/MAPK kinases signaling pathways and abolished its renoprotective activity, suggesting that the fundamental mechanism responsible for the beneficial effect of Tanshinone IIA in AKI may lies in its regulation of the signaling activity of GSK3β/MAPK kinases pathway.
In aggregate, our study demonstrated that delayed treatment with Tanshinone IIA in established AKI can effectively ameliorate progressive kidney destruction through protecting against kidney damage and inhibiting inflammation. Our findings suggest that Tanshinone IIA represents a novel and pragmatic therapeutic strategy that rescues the kidney from AKI via a multi-pronged mechanism. Future clinical trials are certainly merited to examine its safety and effectiveness in AKI patients.
Materials and Methods
The Animal Care and Use Committee at Nanjing University approved the animal studies, which conformed to the United States Department of Agriculture regulations and the National Institutes of Health guidelines for humane care and use of laboratory animals. C57BL/6 male mice aged 6 to 8 weeks and weighing 20 to 22 grams were obtained from Nanjing University Model Animal Research Center and used for this study. Six to nine mice were randomly assigned to each group for each observed time point.
Animal Experimental Design
Murine Models of folic acid-induced AKI
Mice were randomized to receive an intraperitoneal injection of folic acid (250 mg/kg, Sigma-Aldrich, St. Louis, MO, USA) dissolved in 0.3 mM sodium bicarbonate (FA) or equal volume of vehicle (0.3 mM sodium bicarbonate) on day 0. Starting day 1, mice were randomly assigned to receive daily Tanshinone IIA (50 mg/kg/d, Jiangsu Carefree Group Co., Nanjing, China) in corn oil [TS(E)] according to the doses safely used in animal models in published studies22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37 or an equal volume of corn oil by gavage. For late Tanshinone IIA treatment [TS(L)] treatment, some folic acid-injured mice were fed daily with Tanshinone IIA by gavage starting day 5. Some Tanshinone IIA-treated mice were co-treated intraperitoneally with 200 μg/kg/d sodium nitroprusside (Sigma) or its vehicle daily. Animals were euthanized on post injury days 3, 7 and 28, followed by organ harvest for further investigation (Fig. 1a). Additional mice were killed on day 24 after injury. On indicated days, blood and urine samples were collected.
Murine Models of Renal IRI
Mice were anesthetized by ketamine (75 mg/kg) and dexdomitor (50 mg/kg). A flank incision was made on both left and right sides. The bilateral renal arteries and veins were isolated from the surrounding tissue by blunt dissection and then occluded with a nontraumatic vascular clamp for 30 min at 37 °C. In the sham group, renal pedicles were isolated but no clamps were applied. At the end of the ischemic period, the vascular clamps were removed and the kidneys were observed for 5 minutes to document reflow. At 24 h after the surgery, all mice started to receive daily Tanshinone IIA (50 mg/kg) in corn oil or an equal volume of corn oil by gavage. Blood and urine samples were collected at 24 and 48 hours and all animals were euthanized 48 h after IRI (Fig. 10a).
Blood and urine biochemistry analyses
Serum creatinine levels were measured using commercial assay kits according to the manufacturer’s instructions (K625–100, Biovision, Milpitas, CA, USA). Urinary NGAL concentration was determined by a commercially available ELISA test kit (Abcam, Cambridge, MA, USA).
Tissue preparation and single kidney cell isolation
Mice were sacrificed and perfused with ice-cold PBS via the left ventricle for 2 minutes. The right kidney was prepared for histologic examination. The left kidney was collected and enzymatically digested for flow cytometry analysis. In brief, kidneys were minced and incubated with collagenase IV (10 mg/mL, Sigma-Aldrich) and DNase I (200 U/mL, Sigma) in Hanks buffer for 20 minutes at 37 °C using a gentleMACS Dissociator (Miltenyi Biotec Inc, San Diego, CA, USA). Digestion was stopped by addition of fetal bovine serum. Samples of tissue suspensions were passed through a 100-μm cell strainer (Thermo Fisher Scientific, Waltham, MA, USA) and centrifuged at 300 g for 5 minutes at 4 °C to collect the pellet, which was then incubated with ACK Lysing Buffer (0.15 M NH4Cl, 10 mM KHCO3, and 0.1 mM Na2EDTA) to remove red blood cells. The samples were centrifuged again and pellet was then resuspended with PBS containing 1% BSA and 0.1% sodium azide (Sigma-Aldrich) and passed through a 40-μm cell strainer. The isolated single kidney cells were processed for flow cytometry.
Flow cytometry
The isolated single kidney cells were analyzed by flow cytometry (FACSArray; BD Biosciences, San Jose, CA USA) for CD45, F4/80 and MR expression. The following antibodies conjugated with fluorochromes were used: PE-labeled anti-CD45 (BD Biosciences), PE-labeled anti-F4/80 (eBioscience, San Diego, CA, USA), FITC-labeled anti-MR (BD Biosciences). Antibodies and their isotype controls were used according to the manufacturer’s recommendations. Data were collected on a FACSArray flow cytometer (BD Biosciences) and analyzed using FlowJo software (TreeStar, Ashland, OR, USA).
Renal morphology assessment
Formalin fixed paraffin embedded kidney specimens were prepared into 3-μm–thick sections. Sections were subjected to H&E staining to determine gross histological change or Masson trichrome staining to evaluate collagen deposition. One observer performed semi-quantitative morphometric analysis in a blinded manner. Tubular injury scores and interstitial cell infiltration scores were accessed based on HE staining and fibrosis scores were assessed based on Masson trichrome staining by using semi-quantitative measurements according to the proportion relative to the total section area and classified as follows: 0 (nil), 1 (<25%), 2 (25–50%), 3 (50–75%), and 4 (>75% of tubulointerstitial fields). Each score was determined by evaluating five random fields per section and three sections per kidney.
Peroxidase immunohistochemistry
Paraffin sections were deparaffinized and rehydrated. After antigen retrieval, sections were incubated overnight with antibodies against fibronectin (ab2413, Abcam) at 1:100 dilution, MCP-1 (ab25124, Abcam) at 1:200 dilution, or anti-neutrophil antibody (ab2557, Abcam) at 1:200 dilution at 4 °C. The ImmPRESS peroxidase polymer detection kit (Vector Laboratories, Burlingame, CA, USA) was used to conjugate secondary antibody for 30 minutes at 37 °C. Slides were finally developed with DAB substrate (Vector Laboratories). Computerized morphometric analysis of MCP-1staining was performed by using the Image Pro-Plus software (Media-Cybernetics, Silver Spring, MD). The final value was determined based on measuring five random fields per section and three sections per kidney.
Fluorescence immunohistochemistry staining
Frozen sections were fixed and incubated overnight at 4 °C with antibodies against CD45 (sc-25590, Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 1:150 dilution, F4/80 (ab6640, Abcam) at 1:200 dilution, MR (AF2535, R&D Systems, Minneapolis, MN, USA) at 1:150 dilution, MCP-1 (ab25124, Abcam) at 1:150 dilution, NGAL (ab63929, Abcam) at 1:200 dilution, TNF-α (ab6671, Abcam) at 1:250 dilution, Ki-67 (AB9260, EMD Millipore, Darmstadt, Germany) at 1:400 dilution and KIM-1 (AF1817, R&D Systems) at 1:150 dilution. FITC-conjugated secondary antibodies (Invitrogen, CA, USA) against various species were incubated for 1 hour at room temperature. Finally, sections were counterstained with 4′,6-diamidino-2-phenylindole (H-1200, Vector Laboratories), and visualized using a fluorescence microscope. As a negative control, the primary antibodies were replaced by preimmune IgG from the same species; no staining occurred. The positively stained cells in the sections were counted in five random fields per section and three sections per kidney. The staining of NGAL was quantitated by computerized morphometric analysis of five random fields per section and three sections per kidney using Image Pro-Plus software (Media-Cybernetics, Silver Spring, MD).
Western Blotting Analysis
Cortical kidney lysates with equal amounts of total protein (50 μg/ml) were fractionated by 7.5–15% SDS-polyacrylamide gels under reducing conditions and analyzed by immunoblot. The antibodies against fibronectin, collagen I (ab34710, Abcam), MR, arginase I (ab64693, Abcam), NGAL, KIM-1, TNF-α, p38 (sc-535, Santa Cruz Biotechnology), MCP-1, phosphorylated-p38 (p-p38, 9211, Cell Signaling Technology, Danvers, MA, USA), ERK1/2 (sc-292838, Santa Cruz Biotechnology), GSK3β (sc-53931, Santa Cruz Biotechnology), phosphorylated GSK 3β-Ser 9 (p-GSK3β-Ser 9, sc-11757, Santa Cruz Biotechnology), phosphorylated ERK (p-ERK 1/2, 9101, Cell Signaling Technology), JNK (sc-572, Santa Cruz Biotechnology), phosphorylated JNK (p-JNK, ab124956, Abcam) and GAPDH (sc-48166, Santa Cruz Biotechnology) were used as primary antibodies.
TUNEL staining
TUNEL staining was performed on frozen sections with a DeadEnd Fluorometric TUNEL System kit (DeadEnd Fluorometric TUNEL System kit (G3250, Promega, USA) according to the manufacturer’s instructions. The number of TUNEL-positive nuclei per field was evaluated in five fields per section and three sections per kidney.
Statistical Analysis
Data are expressed as mean ± standard error. One-way analysis of variance was applied for multiple comparisons of values, followed by the post hoc Student-Newman-Keuls test. A value of p < 0.05 was considered statistically significant, and all tests were two-tailed. All statistical analyses were performed with the SPSS software (version 17.0 for Windows: SPSS Institute, Chicago, IL, USA).
Additional Information
How to cite this article: Jiang, C. et al. Rescue therapy with Tanshinone IIA hinders transition of acute kidney injury to chronic kidney disease via targeting GSK3β. Sci. Rep. 6, 36698; doi: 10.1038/srep36698 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Li, P. K., Burdmann, E. A., Mehta, R. L. & World Kidney Day Steering, C. Acute kidney injury: global health alert. Kidney international 83, 372–376, doi: 10.1038/ki.2012.427 (2013).
Rewa, O. & Bagshaw, S. M. Acute kidney injury-epidemiology, outcomes and economics. Nature reviews. Nephrology 10, 193–207, doi: 10.1038/nrneph.2013.282 (2014).
Murugan, R. & Kellum, J. A. Acute kidney injury: what’s the prognosis? Nature reviews. Nephrology 7, 209–217, doi: 10.1038/nrneph.2011.13 (2011).
Venkatachalam, M. A. et al. Acute kidney injury: a springboard for progression in chronic kidney disease. American journal of physiology. Renal physiology 298, F1078–F1094, doi: 10.1152/ajprenal.00017.2010 (2010).
Chawla, L. S. & Kimmel, P. L. Acute kidney injury and chronic kidney disease: an integrated clinical syndrome. Kidney international 82, 516–524, doi: 10.1038/ki.2012.208 (2012).
Xue, J. L. et al. Incidence and mortality of acute renal failure in Medicare beneficiaries, 1992 to 2001. Journal of the American Society of Nephrology: JASN 17, 1135–1142, doi: 10.1681/ASN.2005060668 (2006).
Mammen, C. et al. Long-term risk of CKD in children surviving episodes of acute kidney injury in the intensive care unit: a prospective cohort study. American journal of kidney diseases: the official journal of the National Kidney Foundation 59, 523–530, doi: 10.1053/j.ajkd.2011.10.048 (2012).
Ishani, A. et al. Acute kidney injury increases risk of ESRD among elderly. Journal of the American Society of Nephrology: JASN 20, 223–228, doi: 10.1681/ASN.2007080837 (2009).
Kinsey, G. R. et al. Regulatory T cells suppress innate immunity in kidney ischemia-reperfusion injury. Journal of the American Society of Nephrology: JASN 20, 1744–1753, doi: 10.1681/ASN.2008111160 (2009).
Castoldi, A. et al. TLR2, TLR4 and the MYD88 signaling pathway are crucial for neutrophil migration in acute kidney injury induced by sepsis. PloS one 7, e37584, doi: 10.1371/journal.pone.0037584 (2012).
Ferenbach, D. A. et al. Macrophage/monocyte depletion by clodronate, but not diphtheria toxin, improves renal ischemia/reperfusion injury in mice. Kidney international 82, 928–933, doi: 10.1038/ki.2012.207 (2012).
Dong, X. et al. Resident dendritic cells are the predominant TNF-secreting cell in early renal ischemia-reperfusion injury. Kidney international 71, 619–628, doi: 10.1038/sj.ki.5002132 (2007).
Bao, H. et al. Delayed administration of a single dose of lithium promotes recovery from AKI. Journal of the American Society of Nephrology: JASN 25, 488–500, doi: 10.1681/asn.2013040350 (2014).
Gao, S. et al. Cardiovascular actions and therapeutic potential of tanshinone IIA. Atherosclerosis 220, 3–10, doi: 10.1016/j.atherosclerosis.2011.06.041 (2012).
Jiang, B. et al. Tanshinone IIA sodium sulfonate protects against cardiotoxicity induced by doxorubicin in vitro and in vivo. Food and chemical toxicology: an international journal published for the British Industrial Biological Research Association 47, 1538–1544, doi: 10.1016/j.fct.2009.03.038 (2009).
Xu, S. & Liu, P. Tanshinone II-A: new perspectives for old remedies. Expert opinion on therapeutic patents 23, 149–153, doi: 10.1517/13543776.2013.743995 (2013).
Jia, Y., Huang, F., Zhang, S. & Leung, S. W. Is danshen (Salvia miltiorrhiza) dripping pill more effective than isosorbide dinitrate in treating angina pectoris? A systematic review of randomized controlled trials. International journal of cardiology 157, 330–340, doi: 10.1016/j.ijcard.2010.12.073 (2012).
Zhou, L., Zuo, Z. & Chow, M. S. Danshen: an overview of its chemistry, pharmacology, pharmacokinetics, and clinical use. Journal of clinical pharmacology 45, 1345–1359, doi: 10.1177/0091270005282630 (2005).
Zhang, H., Long, M., Wu, Z., Han, X. & Yu, Y. Sodium tanshinone IIA silate as an add-on therapy in patients with unstable angina pectoris. Journal of thoracic disease 6, 1794–1799, doi: 10.3978/j.issn.2072-1439.2014.12.37 (2014).
Shen, W. et al. Effects of tanshinone on hyperandrogenism and the quality of life in women with polycystic ovary syndrome: protocol of a double-blind, placebo-controlled, randomised trial. BMJ open 3, e003646, doi: 10.1136/bmjopen-2013-003646 (2013).
Wang, J. et al. Promising therapeutic effects of sodium tanshinone IIA sulfonate towards pulmonary arterial hypertension in patients. Journal of thoracic disease 5, 169–172, doi: 10.3978/j.issn.2072-1439.2013.02.04 (2013).
Adams, J. D., Wang, R., Yang, J. & Lien, E. J. Preclinical and clinical examinations of Salvia miltiorrhiza and its tanshinones in ischemic conditions. Chinese medicine 1, 3, doi: 10.1186/1749-8546-1-3 (2006).
Xu, M. et al. Tanshinone IIA therapeutically reduces LPS-induced acute lung injury by inhibiting inflammation and apoptosis in mice. Acta pharmacologica Sinica 36, 179–187, doi: 10.1038/aps.2014.112 (2015).
Wang, D. T. et al. Tanshinone IIA attenuates renal fibrosis and inflammation via altering expression of TGF-beta/Smad and NF-kappaB signaling pathway in 5/6 nephrectomized rats. International immunopharmacology 26, 4–12, doi: 10.1016/j.intimp.2015.02.027 (2015).
Li, W., Zhang, Y., Xing, C. & Zhang, M. Tanshinone IIA represses inflammatory response and reduces radiculopathic pain by inhibiting IRAK-1 and NF-kappaB/p38/JNK signaling. International immunopharmacology 28, 382–389, doi: 10.1016/j.intimp.2015.06.032 (2015).
Hu, Q. et al. Sodium tanshinone IIA sulfonate ameliorates ischemia-induced myocardial inflammation and lipid accumulation in Beagle dogs through NLRP3 inflammasome. International journal of cardiology 196, 183–192, doi: 10.1016/j.ijcard.2015.05.152 (2015).
Hu, H. et al. Protective effects of tanshinone IIA on myocardial ischemia reperfusion injury by reducing oxidative stress, HMGB1 expression, and inflammatory reaction. Pharmaceutical biology 53, 1752–1758, doi: 10.3109/13880209.2015.1005753 (2015).
Zhang, K. et al. Tanshinone IIA inhibits lipopolysaccharide-induced MUC1 overexpression in alveolar epithelial cells. American journal of physiology. Cell physiology 306, C59–C65, doi: 10.1152/ajpcell.00070.2013 (2014).
Yin, X. et al. Tanshinone IIA attenuates the inflammatory response and apoptosis after traumatic injury of the spinal cord in adult rats. PloS one 7, e38381, doi: 10.1371/journal.pone.0038381 (2012).
Shang, Q., Wang, H., Li, S. & Xu, H. The Effect of Sodium Tanshinone IIA Sulfate and Simvastatin on Elevated Serum Levels of Inflammatory Markers in Patients with Coronary Heart Disease: A Study Protocol for a Randomized Controlled Trial. Evidence-based complementary and alternative medicine: eCAM 2013, 756519, doi: 10.1155/2013/756519 (2013).
Mao, S. et al. Sodium tanshinone IIA sulfonate for reduction of periprocedural myocardial injury during percutaneous coronary intervention (STAMP trial): rationale and design. International journal of cardiology 182, 329–333, doi: 10.1016/j.ijcard.2014.12.166 (2015).
Ahn, Y. M. et al. Renoprotective effect of Tanshinone IIA, an active component of Salvia miltiorrhiza, on rats with chronic kidney disease. Phytotherapy research: PTR 24, 1886–1892, doi: 10.1002/ptr.3347 (2010).
Wu, X. et al. Tanshinone IIA prevents uric acid nephropathy in rats through NF-kappaB inhibition. Planta medica 78, 866–873, doi: 10.1055/s-0031-1298487 (2012).
Kim, S. K., Jung, K. H. & Lee, B. C. Protective effect of Tanshinone IIA on the early stage of experimental diabetic nephropathy. Biological & pharmaceutical bulletin 32, 220–224 (2009).
Liu, X. et al. Proteomic assessment of tanshinone IIA sodium sulfonate on doxorubicin induced nephropathy. The American journal of Chinese medicine 39, 395–409, doi: 10.1142/S0192415X11008907 (2011).
Zhang, X., He, D., Xu, L. & Ling, S. Protective effect of tanshinone IIA on rat kidneys during hypothermic preservation. Molecular medicine reports 5, 405–409, doi: 10.3892/mmr.2011.639 (2012).
Tang, J., Zhan, C. & Zhou, J. Effects of tanshinone IIA on transforming growth factor beta1-Smads signal pathway in renal interstitial fibroblasts of rats. Journal of Huazhong University of Science and Technology. Medical sciences = Hua zhong ke ji da xue xue bao. Yi xue Ying De wen ban = Huazhong keji daxue xuebao. Yixue Yingdewen ban 28, 539–542, doi: 10.1007/s11596-008-0511-0 (2008).
Liu, Z. & Gong, R. Remote ischemic preconditioning for kidney protection: GSK3beta-centric insights into the mechanism of action. American journal of kidney diseases: the official journal of the National Kidney Foundation 66, 846–856, doi: 10.1053/j.ajkd.2015.06.026 (2015).
Radinger, M., Kuehn, H. S., Kim, M. S., Metcalfe, D. D. & Gilfillan, A. M. Glycogen synthase kinase 3beta activation is a prerequisite signal for cytokine production and chemotaxis in human mast cells. J Immunol 184, 564–572, doi: 10.4049/jimmunol.0902931 (2010).
Takada, Y., Fang, X., Jamaluddin, M. S., Boyd, D. D. & Aggarwal, B. B. Genetic deletion of glycogen synthase kinase-3beta abrogates activation of IkappaBalpha kinase, JNK, Akt, and p44/p42 MAPK but potentiates apoptosis induced by tumor necrosis factor. J Biol Chem 279, 39541–39554, doi: 10.1074/jbc.M403449200 (2004).
Ma, C. et al. Interaction between ERK and GSK3beta mediates basic fibroblast growth factor-induced apoptosis in SK-N-MC neuroblastoma cells. J Biol Chem. 283, 9248–9256, doi: 10.1074/jbc.M707316200 (2008).
Sakai, N. et al. p38 MAPK phosphorylation and NF-kappa B activation in human crescentic glomerulonephritis. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association 17, 998–1004 (2002).
Pan, Y. et al. Targeting JNK by a new curcumin analog to inhibit NF-kB-mediated expression of cell adhesion molecules attenuates renal macrophage infiltration and injury in diabetic mice. PloS one 8, e79084, doi: 10.1371/journal.pone.0079084 (2013).
Cassidy, H. et al. The role of MAPK in drug-induced kidney injury. J Signal Transduct 2012, 463617, doi: 10.1155/2012/463617 (2012).
Wang, D. et al. Inhibition of p38 MAPK attenuates renal atrophy and fibrosis in a murine renal artery stenosis model. American journal of physiology. Renal physiology 304, F938–F947, doi: 10.1152/ajprenal.00706.2012 (2013).
Pat, B. et al. Activation of ERK in renal fibrosis after unilateral ureteral obstruction: modulation by antioxidants. Kidney international 67, 931–943, doi: 10.1111/j.1523-1755.2005.00157.x (2005).
Ma, F. Y. et al. A pathogenic role for c-Jun amino-terminal kinase signaling in renal fibrosis and tubular cell apoptosis. Journal of the American Society of Nephrology: JASN 18, 472–484, doi: 10.1681/ASN.2006060604 (2007).
Mariappan, M. M. et al. Activation of glycogen synthase kinase 3beta ameliorates diabetes-induced kidney injury. J Biol Chem 289, 35363–35375, doi: 10.1074/jbc.M114.587840 (2014).
Hong, H. J. et al. Tanshinone IIA prevents doxorubicin-induced cardiomyocyte apoptosis through Akt-dependent pathway. International journal of cardiology 157, 174–179, doi: 10.1016/j.ijcard.2010.12.012 (2012).
Hong, H. J., Liu, J. C., Cheng, T. H. & Chan, P. Tanshinone IIA attenuates angiotensin II-induced apoptosis via Akt pathway in neonatal rat cardiomyocytes. Acta pharmacologica Sinica 31, 1569–1575, doi: 10.1038/aps.2010.176 (2010).
Chan, P. et al. Tanshinone IIA Attenuates H(2)O(2) -induced injury in human umbilical vein endothelial cells. The American journal of Chinese medicine 40, 1307–1319, doi: 10.1142/S0192415X12500966 (2012).
Zhang, K., Li, J., Meng, W., Xing, H. & Yang, Y. Tanshinone IIA inhibits acute promyelocytic leukemia cell proliferation and induces their apoptosis in vivo. Blood cells, molecules & diseases 56, 46–52, doi: 10.1016/j.bcmd.2015.10.007 (2016).
Zhang, Y. et al. Tanshinone IIA induces apoptosis and inhibits the proliferation, migration, and invasion of the osteosarcoma MG-63 cell line in vitro. Anti-cancer drugs 23, 212–219, doi: 10.1097/CAD.0b013e32834e5592 (2012).
Li, C., Han, X., Zhang, H., Wu, J. & Li, B. The interplay between autophagy and apoptosis induced by tanshinone IIA in prostate cancer cells. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine, doi: 10.1007/s13277-015-4602-9 (2015).
Chen, Y. et al. Neuroprotection of tanshinone IIA against cerebral ischemia/reperfusion injury through inhibition of macrophage migration inhibitory factor in rats. PloS one 7, e40165, doi: 10.1371/journal.pone.0040165 (2012).
Xu, S. et al. Effectiveness of combination therapy of atorvastatin and non lipid-modifying tanshinone IIA from Danshen in a mouse model of atherosclerosis. International journal of cardiology 174, 878–880, doi: 10.1016/j.ijcard.2014.04.226 (2014).
Kelly, K. J. et al. Intercellular adhesion molecule-1-deficient mice are protected against ischemic renal injury. The Journal of clinical investigation 97, 1056–1063, doi: 10.1172/JCI118498 (1996).
Jo, S. K., Sung, S. A., Cho, W. Y., Go, K. J. & Kim, H. K. Macrophages contribute to the initiation of ischaemic acute renal failure in rats. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association 21, 1231–1239, doi: 10.1093/ndt/gfk047 (2006).
Cao, Q., Wang, Y. & Harris, D. C. Pathogenic and protective role of macrophages in kidney disease. American journal of physiology. Renal physiology 305, F3–11, doi: 10.1152/ajprenal.00122.2013 (2013).
Ren, Z. H., Tong, Y. H., Xu, W., Ma, J. & Chen, Y. Tanshinone II A attenuates inflammatory responses of rats with myocardial infarction by reducing MCP-1 expression. Phytomedicine: international journal of phytotherapy and phytopharmacology 17, 212–218, doi: 10.1016/j.phymed.2009.08.010 (2010).
Lee, S. et al. Distinct macrophage phenotypes contribute to kidney injury and repair. Journal of the American Society of Nephrology: JASN 22, 317–326, doi: 10.1681/ASN.2009060615 (2011).
Murray, P. J. & Wynn, T. A. Protective and pathogenic functions of macrophage subsets. Nature reviews. Immunology 11, 723–737, doi: 10.1038/nri3073 (2011).
Wang, Y. et al. Ex vivo programmed macrophages ameliorate experimental chronic inflammatory renal disease. Kidney international 72, 290–299, doi: 10.1038/sj.ki.5002275 (2007).
Fan, G. W. et al. The anti-inflammatory activities of Tanshinone IIA, an active component of TCM, are mediated by estrogen receptor activation and inhibition of iNOS. The Journal of steroid biochemistry and molecular biology 113, 275–280, doi: 10.1016/j.jsbmb.2009.01.011 (2009).
Chen, T. H., Hsu, Y. T., Chen, C. H., Kao, S. H. & Lee, H. M. Tanshinone IIA from Salvia miltiorrhiza induces heme oxygenase-1 expression and inhibits lipopolysaccharide-induced nitric oxide expression in RAW 264.7 cells. Mitochondrion 7, 101–105, doi: 10.1016/j.mito.2006.11.018 (2007).
Acknowledgements
This work was supported in part by the Key Science and Technology Development Program of Nanjing Municipal Government (YKK11093) and the U.S. National Institutes of Health grant DK092485. The funders had no role in the design and conduct of this study, collection and interpretation of the data, or preparation and approval of the manuscript.
Author information
Authors and Affiliations
Contributions
C.J. and R.G. conceived the study, and developed the study design and protocol with contributions from all authors. C.J., W.Z., X.Y., Q.S. and B.X. performed the experiments. C.J. extracted the data and undertook the analysis, supervised by M.Z. and R.G., and overseen by all authors. W.Z. prepared the figures. All authors contributed to the data interpretation. C.J. and R.G. drafted the paper and all authors contributed to revisions and approved the final version.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Jiang, C., Zhu, W., Yan, X. et al. Rescue therapy with Tanshinone IIA hinders transition of acute kidney injury to chronic kidney disease via targeting GSK3β. Sci Rep 6, 36698 (2016). https://doi.org/10.1038/srep36698
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep36698
This article is cited by
-
Tanshinone IIA changed the amniotic fluid volume and regulated expression of AQP1 and AQP3 in amniotic epithelium cells: a promising drug treating abnormal amniotic fluid volume
Molecular Medicine (2023)
-
Natural products: potential drugs for the treatment of renal fibrosis
Chinese Medicine (2022)
-
Therapeutic Effect of Trigonella foenum-graecum l Seeds Extract on Folic Acid-Induced Acute Kidney Injury
Proceedings of the National Academy of Sciences, India Section B: Biological Sciences (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
