
Abstract
Disturbed blood flow (d-flow) induces atherosclerosis by altering the expression of mechanosensitive genes in the arterial endothelium. Previously, we identified >580 mechanosensitive genes in the mouse arterial endothelium, but their role in endothelial inflammation is incompletely understood. From this set, we obtained 84 Drosophila RNAi lines that silences the target gene under the control of upstream activation sequence (UAS) promoter. These lines were crossed with C564-GAL4 flies expressing GFP under the control of drosomycin promoter, an NF-κB target gene and a marker of pathogen-induced inflammation. Silencing of psmd12 or ERN1 decreased infection-induced drosomycin expression, while Bap60 silencing significantly increased the drosomycin expression. Interestingly, knockdown of Bap60 in adult flies using temperature-inducible Bap60 RNAi (C564ts-GAL4-Bap60-RNAi) enhanced drosomycin expression upon Gram-positive bacterial challenge but the basal drosomycin expression remained unchanged compared to the control. In the mammalian system, smarcd3 (mammalian ortholog of Bap60) expression was reduced in the human- and mouse aortic endothelial cells exposed to oscillatory shear in vitro as well as in the d-flow regions of mouse arterial endothelium in vivo. Moreover, siRNA-mediated knockdown of smarcd3 induced endothelial inflammation. In summary, we developed an in vivo Drosophila RNAi screening method to identify flow-sensitive genes that regulate endothelial inflammation.
Similar content being viewed by others

Introduction
Vascular endothelial cells respond to blood flow through mechanosensors, which transduce the mechanical force associated with flow (known as shear stress) into cell signaling events and ultimately to changes in gene expression1,2,3. Disturbed blood flow (d-flow), which occurs in branched and curved arteries, is characterized by complex flow patterns with low magnitude and oscillatory shear stress (OS), whereas stable flow (s-flow) is characterized by high magnitude, unidirectional laminar shear stress (LS) due to blood flow in the straight sections of the vasculature. D-flow and s-flow promote and suppress endothelial inflammation and atherosclerosis, respectively4,5,6,7. The molecular mechanisms underlying the pro-inflammatory effects of d-flow on the endothelium are likely to be regulated by flow-sensitive genes, but the specific mechanisms are still unclear6,8.
Previously, we carried out a transcriptomic study to determine which genes were regulated by blood flow using a partial carotid ligation model of atherosclerosis6. From this study, we identified more than 580 mechanosensitive genes that changed in response to d-flow as compared to s-flow. However, it remains unclear which of these mechanosensitive genes regulate endothelial inflammation, a key proatherogenic response.
In Drosophila, genetic analyses reveal that there are two independent NF-κB signaling pathways, Toll-mediated and immune deficiency (IMD)-mediated pathways9,10. Toll pathway is activated by Gram-positive bacteria or fungi leading to the activation of Dif, a p65-like NF-κB, whereas IMD-pathway is activated by Gram-negative bacteria leading to the activation of Relish, a p105-like NF-κB11. NF-κB-dependent antimicrobial peptide (AMP) gene expression such as Dif-dependent drosomycin and Relish-dependent diptericin is shown to be required for efficient antimicrobial responses12.
To functionally screen candidate mechanosensitive genes that may regulate endothelial inflammation, we have developed an in vivo Toll-mediated NF-κB activation screening assay using Drosophila carrying a fluorescent NF-κB target gene reporter [drosomycin promoter fused to Green Fluorescent Protein (GFP)] along with RNAi targeting the respective mechanosensitive gene under the control of the UAS-GAL4 promoter13,14,15.
Using this in vivo functional screening method, we screened 84 RNAi fly lines and identified novel, flow-sensitive, pro-inflammatory genes including smarcd3 (mammalian ortholog of Bap60) that regulates Toll-mediated NF-κB activation.
Materials and Methods
Animal Study Design
Animals were maintained and cared for in accordance to the National Institutes of Health (NIH) guidelines in our AAALAC-accredited experimental animal facility under a controlled environment (21° ± 2 °C, 50% ± 10% relative humidity and a 12-h light:12-h dark cycle with lights on at 0700 h EST). The Institutional Animal Care and Use Committee (IACUC) at Emory University approved the project protocols and procedures performed in accordance with the established guidelines and regulations consistent with federal assurance. Male C57BL/6 and ApoE−/− mice (Jackson Laboratory) were fed ad libitum with standard chow diet until surgery at 8–9 weeks of age. Mice were anaesthetized with 3.5% isoflurane initially and then 1.5–2% during the entire procedure and underwent partial ligation as we previously described16. Following surgery, analgesic buprenorphine (0.1 mg kg−1) was administered. Following the partial carotid ligation, d-flow in LCA was confirmed by Doppler ultrasonography (Vevo1100, Visualsonics)16,17. The detailed method for endothelial-enriched RNA extraction, and microarray processing and analyses have been previously described in detail in our previous publications6,8,18.
Generation of Drosophila lines carrying RNAi and GFP-NF-κB reporter
We first determined which of the 588 flow-sensitive murine genes previously identified6 have corresponding fly orthologs by comparing amino acid homology between murine and Drosophila genes (http://flybase.org)19. Through this manual comparison, we identified 99 Drosophila genes having significant sequence homology to corresponding murine genes. Of these, we obtained 84 Drosophila RNAi lines expressing corresponding hairpin RNA (Supplementary Table 1). These double-stranded RNAs are processed by Dicer into siRNAs which direct sequence-specific degradation of the target mRNA (UAS-RNAi flies) from Vienna Drosophila Resource Center (VDRC).
To develop fly lines that can report onset of inflammation by NF-κB activation as a surrogate marker, we used C564-GAL4 flies carrying drosomycin-GFP (GFP under control of drosomycin promoter) reporter. Drosomycin is a well-known NF-κB target gene in flies. This allowed us to test the effect of individual RNAi on the Toll-mediated NF-κB activation in the absence or presence of bacterial challenge. To this end, we mated male UAS-RNAi lines to female C564-GAL4 flies expressing drosomycin-GFP. All fly lines were maintained at 25 °C with standard fly medium20.
For validation studies, conditional expression of UAS-RNAi was achieved using temperature-sensitive allele of GAL80 under the control of tubulin promoter (tub-GAL80ts). In these C564-GAL4-Tub-GAL80ts (C564ts-GAL4) flies, GAL80ts (inhibitor of GAL4 activity) is active at 18 °C and thus capable of inhibiting GAL4 activity21. At 29 °C, the GAL80ts becomes inactive allowing RNAi expression. Flies were grown at 18 °C during development and adult flies were shifted to 29 °C for 5 days for the expression of UAS-RNAi before infection experiment.
In vivo GFP screening by fluorescence stereo microscopy
GFP expression in the flies was examined under basal and bacterial infected conditions by fluorescence stereo microscopy. A total of three or more independent cohorts of ~10 adult flies each were examined before and 24 h post-bacterial infection. A representative GFP fluorescence screening set was blindly scored on a scale of 0 (least) to 5 (brightest) by 4 observers for semi-quantitation of fluorescence intensity.
Infection Experiments
Bacterial infection was performed by pricking adult flies in the thorax with a thin needle previously dipped in a concentrated pellet of a Gram-positive bacteria (Micrococcus luteus) culture (OD = 200) or Gram-negative bacteria (Erwinia carotovora 15) as previously described20. Each experiment was performed with approximately 10 flies for each genotype, and the results shown are representative of at least three independent tests. The lethal RNAi lines are shown in Supplementary Table 1 and were not pursued further.
Cell culture and in vitro shear stress system
Human umbilical vein endothelial cells (HUVECs), human aortic endothelial cells (HAECs) and immortalized mouse aortic endothelial cells (iMAECs) were maintained in their respective culture medium as we previously described22,23,24,25. Shear stress was applied to the cells in 10-cm culture plates for 24 hours using a cone-and-plate viscometer that exerts 15 dyn/cm2 of unidirectional flow (laminar shear or LS) and ±5 dyn/cm2 of oscillatory shear stress (OS), respectively, as we previously described26,27. The cell culture plates were secured in place at the bottom by a vacuum pump, and the incubator was maintained at 5% CO2 and 37 °C. Cells with no shear stress (Static) were used as controls. Total RNA was collected by scraping the cells in QIAzol buffer (Qiagen).
Quantitative real-time PCR (qPCR) assays
qPCR was performed to validate the functional screening results in Drosophila. The transcript level of drosomycin was measured in the RNA extracted from RNAi flies in either basal, 6hr or 24hr post-bacterial infection. qPCR assays were also performed using total RNAs obtained from the mouse carotid arterial endothelium and from endothelial cells exposed to shear stress in vitro. The primers used for qPCR analyses are listed in Supplementary Tables 2 and 3.
Immunofluorescence staining
For en face confocal microscopy imaging, the right (RCA) and left carotid arteries (LCA) obtained at 48 hours post-ligation from C57Bl6 mice subjected to partial carotid ligation were used. The carotids were opened longitudinally and fixed on black wax with the endothelial layer facing upwards. The tissues were permeabilized with 0.25% (v/v) Triton X-100 in PBS, washed, blocked for 1.5 h in 10% (v/v) goat serum, and incubated with Rabbit polyclonal anti-smarcd3 antibody (Abcam; ab115917, dilution 1:100) in blocking solution (overnight, 4 °C). After additional washing, the tissues were incubated with DyLight 550-conjugated donkey anti-Rabbit secondary antibody (Abcam; dilution 1:250) in PBS for 2 h at room temperature, and with 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich) in PBS for 20 min. Fluorescence was imaged using a confocal microscope (Carl Zeiss; LSM 510 Meta instrument). Negative control tissues were stained with appropriate IgG isotype control antibodies (Santa Cruz Biotechnology). Nuclei were stained with DAPI.
Monocyte adhesion assay
Human peripheral blood mononuclear leukocytes (THP-1 cells) were cultured in serum-containing RPMI medium (MT10040CM; Fisher) and were labeled in serum-free RPMI with 5 μl/ml 2′7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein (BCECF-AM, B-1170; Molecular Probes) at 37 °C for 30 minutes. HUVECs were transfected with either 150 nM smarcd3-siRNA (Dharmacon RNAi ON-TARGET plus SMART pool) or control siRNA for 24 hours. As a positive control, HUVECs were activated with 3 ng/ml TNFα in serum-free RPMI for 3 hours to induce monocyte adhesion. 5 × 105 THP-1 cells per milliliter (in a total of 6 ml) were incubated with the HUVECs for 30 minutes at 37 °C. Nonadherent monocytes were washed away with HBSS (Corning), and the bound monocytes were fixed with 4% paraformaldehyde for 5 minutes. Bound monocytes were imaged in 8–10 fields per plate using fluorescent and bright-field microscopy, and quantification was done by ImageJ software (NIH)28.
Ex vivo Aorta tissue culture and siRNA transfection
After sacrificing C57BL/6 mice, isolated aortas were longitudinally halved, opened en-face and cultured in a 48-well plate with 150 μl per well of complete medium viz. Dulbecco’s Modified Eagle’s Medium (DMEM, GIBCO, NY) containing 10% (v/v) fetal bovine serum, penicillin/streptomycin, and L-glutamine29. Transient transfections were performed using siRNA for 100 nM smarcd3 (5′- GAGUACAUCAAUGGCGACAAGUAU-3′) or control for 48 h using Lipofectamine RNAiMAX (Invitrogen), following manufacturer’s protocol as described previously8. Post transfection, the en face immunostaining was performed for studying the expression of smarcd3 and vascular cell adhesion molecule (VCAM1). Signal intensities indicating the expression of VCAM1 (Cyan) and Smarcd3 (Red) were quantified by ImageJ software (NIH)28.
Ex vivo endothelium monocyte adhesion assay
The abdominal aortas were isolated from C57BL/6 mice and the surrounding adventitial tissue was gently cleaned. siRNA for smarcd3 or non-targeting controls were transfected as described above for 48 hours. The aortas were opened longitudinally and fixed en face with the endothelial surface-side up using fine needles on a black wax petri dish. A mouse monocyte macrophage cell line J774A.1 (ATCC® TIB-67™) was labeled with Calcein-AM (Molecular probes), and labeled cells (5 × 105) were incubated on the excised aortas that was pinned on the black wax dish for 30 min at 37 °C in a CO2 incubator30. These were then washed twice with phosphate buffered saline (PBS) and the bound J774A.1 cells were counted as above.
Statistical analyses
Statistical analyses were carried out with Graph-Pad Prism (GraphPad Software). All error bars reported are standard error of mean (S.E.M) unless otherwise indicated. Pairwise comparisons were performed using one-way Student’s t-tests. Differences between groups were considered significant at P-values below 0.05.
Results
Selection of Drosophila RNAi lines for functional screening
Previously, we developed a mouse model of d-flow-induced atherosclerosis by the partial carotid ligation surgery6,16,17. Using this mouse model, we identified 588 mechanosensitive genes in the mouse arterial endothelium6. Although it is well-known that d-flow induces endothelial inflammation including NF-κB activation, the role of these mechanosensitive genes is largely unclear. To functionally identify mechanosensitive genes that are involved in NF-κB-dependent inflammation, we developed a Drosophila RNAi-based screening system.
By comparing the Entrez IDs of 588 previously identified mechanosensitive murine gene6 to fly orthologs (http://flybase.org), we first identified and obtained 84 Drosophila RNAi lines carrying Drosophila orthologs of murine mechanosensitive genes (Fig. 1; Supplementary Table 1).

Overall study design and work flow.
A previous mRNA microarray study identified 588 mechanosensitive genes from endothelial cells exposed to disturbed blood flow (d-flow) using the mouse partial ligation model. Of these 588 mechanosensitive genes, we found 84 corresponding Drosophila orthologs. RNAi flies for these genes were obtained and mated with C564-GAL4 flies containing a drosomycin-GFP reporter (drosomycin-GFP RNAi flies). Drosomycin-GFP expression under both basal and Gram-positive bacterial infection was used to screen the RNAi fly line by stereo fluorescence microscopy. IHJ prepared the illustrations of mouse and Drosophila using Adobe Illustrator. SK prepared the illustration for partial carotid ligation model using Microsoft PowerPoint software.
Generation and screening of Drosophila RNAi lines expressing drosomycin-GFP
We tested the effects of 84 Drosophila orthologs of murine mechanosensitive genes on the Toll pathway by using reporter flies carrying drosomycin-GFP. To constitutively induce the expression of a specific RNAi, male UAS-RNAi lines were mated with female C564-GAL4 flies carrying drosomycin-GFP12,13,15. The C564-GAL4 flies that express GAL4 mainly in the fat body (immune tissue in Drosophila) were used for tissue-specific knockdown of the target gene31. The progeny F1 was used to determine NF-κB/Dif-dependent drosomycin expression following Gram-positive bacterial infection condition. We found that expression of six RNAi fly lines in this study showed lethality (Supplementary Table 1), and they were not further studied. Expression of GFP in the remaining 78 RNAi lines was examined by fluorescence stereo microscopy before and after 24 h of Gram-positive bacterial infection as shown by the representative fluorescence images (Supplementary Table 4).
Identification of Bap60, RPN5 and ERN1 as regulators of inflammation in Drosophila
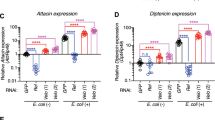
From the above initial in vivo GFP screening using the constitutive RNAi expression system (C564-GAL flies), we identified three genes, RPN5, ERN1, and Bap60, that substantially modified NF-κB activation (Fig. 2). Dif (a Drosophila NF-κB pathway gene indispensable for immunity to Gram-positive bacteria) RNAi fly, which was used as a negative control, did not show activation of NF-κB in response to the bacterial infection, as expected (Fig. 2A). RPN5 RNAi and ERN1 RNAi blocked NF-ΚB activation in response to bacterial infection. Interestingly, Bap60 RNAi fly line showed a significantly higher NF-κB activation in basal condition which did not further increase in response to the bacterial infection (Fig. 2A). These results suggestes that RPN5 and ERN1 may act as NF-κB activators, while Bap60 acts as a NF-κB repressor.

Identification of RNAi lines regulating NF-κB activation in Drosophila.
(A) Stereo fluorescence microscopy was used to monitor the drosomycin-GFP expression in each drosomycin-GFP RNAi fly line under both basal and Gram-positive bacterial infection. As a control, the Dif-RNAi fly was used. GFP expression in three or more independent cohorts comprising ~10 adults each was monitored after 24 h of infection. White Scale Bar = 500μm (B) qPCR assay for drosomycin mRNA levels in non-infection or in response to Gram positive bacterial infection for 24 h. Dif (a Drosophila NF-κB) and Myd88 RNAi flies were used as negative controls. Drosomycin level in the non-infection control was taken arbitrarily as set 1. Values represent the mean ± S.E.M (*P < 0.05) of at least three independent experiments. (C) qPCR assay was carried out for diptericin mRNA level, a NF-κB target gene, in uninfected control and in response to Gram negative bacterial infection for 6 h. Relish and IMD RNAi flies were used as negative controls. Diptericin level in non-infection condition was taken arbitrarily as set 1. Values represent the mean ± S.E.M (*P < 0.05) of at least three independent experiments. (D) qPCR assay for drosomycin mRNA levels in non-infection condition (left) and Gram-positive bacterial infection for 24 h (middle). Respective RNAi was induced by maintaining the C564ts-GAL4 flies at 29 °C for 5 days. Drosomycin levels in the non-infected control flies was taken arbitrarily as 1. Values represent the means ± S.E.M (*P < 0.05) of at least three independent experiments. (right panel) qPCR for diptericin mRNA expression, an NF-κB target gene, in response to Gram-negative bacterial infection for 6 h was carried out using the C565ts-GAL4-driven RNAi flies. Diptericin level in non-infection condition of control flies was taken arbitrarily as 1. Values represent the means ± S.E.M (*P < 0.05) of at least three independent experiments.
Next, we validated these qualitative screening results by determining the expression of drosomycin by qPCR using total RNAs extracted from these flies. The qPCR results using the constitutive RNAi expression system (C564-GAL flies) showed that drosomycin expression was increased by >10-fold by Gram-positive bacterial infection in the control RNAi flies, which was significantly reduced in RPN5 and ERN1 RNAi lines (Fig. 2B). Furthermore, drosomycin expression was increased by >20-fold over the control (Control) in the Bap60 RNAi fly line (Fig. 2B). Importantly, Gram-positive bacterial infection did not further increase the drosomycin expression in Bap60 RNAi flies (Fig. 2B). RNAi flies for Dif and Myd88, two well-known signaling molecules mediating Toll-induced NF-κB activation, were used as controls to test the specificity of drosomycin expression in this screening system (Fig. 2B). Both Dif RNAi and Myd88 RNAi lines showed no significant expression of drosomycin in non-infected and as well as Gram-positive bacterial infection conditions. These results further substantiate the GFP fluorescence imaging results above (Fig. 2A), suggesting the potential role of RPN5 and ERN1 as NF-κB activators and Bap60 as NF-κB repressor.
It is well known that the NF-κB pathway in Drosophila can be activated by Toll-mediated (by Gram-positive bacteria) and IMD-mediated (by Gram-negative bacteria) pathways32. Therefore, we tested whether RPN5, ERN1 and Bap60 can also regulate IMD-mediated NF-κB activation as measured by diptericin expression in response to Gram-negative bacterial infection using the constitutive RNAi expression system. Upon Gram-negative bacterial infection, diptericin expression was increased by >2,000-fold over the non-infected control, which was significantly reduced in RPN5 and ERN1 RNAi lines (Fig. 2C). Interestingly, Bap60 RNAi fly line showed a >1,000-fold increase in the diptericin expression in the non-infected condition which was further increased to >2,000-fold 6 hours after Gram-negative bacterial infection (Fig. 2C). In addition, ERN1 RNAi fly also showed a substantial increase in the diptericin expression in the non-infected condition which also increased by Gram-negative bacterial infection.
Since Relish and IMD are two well-known upstream regulators of diptericin in the IMD-mediated NF-κB pathway in response to Gram-negative bacterial infection, we examined diptericin expression in Relish RNAi and IMD RNAi flies. As expected, both Relish RNAi and IMD RNAi flies showed no significant expression of diptericin in non-infected and Gram-negative bacterial infection conditions (Fig. 2C). These results indicated that knockdown of Bap60 induces NF-κB activation in both the Toll-mediated and IMD-mediated pathways.
Previous studies33,34 indicate that Bap60 mutants may develop melanotic masses which could result in aberrant activation of NF-κB as we observed in our current study. To address this, we tested the role of Bap60 in an inducible RNAi expression system using the temperature-inducible C564-GAL4-Tub-GAL80ts (C564ts-GAL4) flies. Similar to the constitutive RNAi expression, the inducible knockdown of Bap60 in adult flies resulted in an increased drosomycin expression in response to Gram-positive bacterial infection (Fig. 2D). In contrast, unlike the constitutive RNAi flies, temperature-induced knockdown of Bap60 C564ts-GAL4 flies did not change the basal drosomycin expression (Fig. 2D). These results suggest that the increased basal drosomycin expression in the constitutive C564-GAL4-Bap60-RNAi flies might be related to developmental changes resulting in Toll-mediated NF-κB pathway35. However, we failed to observe any obvious signs of melanotic masses in either of the Bap60 RNAi flies in our hands.
Next, we tested whether inducible knockdown of RPN5, ERN1, and Bap60 can also regulate Toll- and IMD-mediated NF-κB activation by measuring drosomycin and diptericin expression, respectively, in Gram-positive and Gram-negative bacterial infection conditions in the C654ts-Gal4 RNAi flies. First, we found that inducible knockdown of Bap60 did not induce drosomycin expression in the basal condition (Fig. 2D), unlike in the constitutive Bap60 knockdown flies (Fig. 2B). However, inducible Bap60 knockdown increased drosomycin expression while reducing diptericin expression compared to the respective infection controls, suggesting that Bap60 plays a differential role in Toll- and IMD-pathways. In contrast, unlike the constitutive ERN1 knockdown, the inducible knockdown of ERN1 showed relatively minor reduction of drosomycin expression and no significant effect on diptericin expression, in response to respective bacterial infection conditions (Fig. 2D). However, inducible knockdown of RPN5 showed a significant inhibitory effect on the Toll- and IMD-mediated NF-κB activation (Fig. 2D) as observed in the constitutive RPN5 RNAi flies (Fig. 2B).
Taken together, by comparing the constitutive and inducible RNAi systems, we have found that RPN5 (activator) and Bap60 (differential regulator) may be mechanosensitive NF-κB regulators while ERN1 is a weak NF-κB regulator.
Smarcd3, a mammalian ortholog of Bap60, is a mechanosensitive protein expressed in mouse aortic endothelium
We next examined the role of RPN5, ERN1 and Bap60 in mouse arterial endothelium. For this, we first examined the mRNA expression of smarcd3, psmd12 (a mammalian ortholog of RPN5) and ERN1, in order to validate the mechanosensitive regulation of these genes in the mouse arterial endothelium. Using our mouse partial carotid ligation model, we induced d-flow in the LCA, while the contralateral RCA continued to experience s-flow16,17. Endothelial-enriched mRNAs obtained from both carotids 48-hour post-ligation showed that expression of smarcd3 in the LCA endothelium was decreased by approximately 50%, while psmd12 was increased by 2.2-fold as compared to the RCAs (Fig. 3A). However, the expression of ERN1 was not statistically different. As a control experiment, we used KLF2 as a well-known flow-sensitive gene6 (Fig. 3A). These findings indicate that smarcd3 and psmd12 are flow-sensitive genes in endothelial cells, whereas the mechanosensitivity of ERN1 could not be confirmed.

Validation of mechanosensitivity of smarcd3 (Bap60 ortholog), psmd12 (RPN5 ortholog) and ERN1 in mouse arterial endothelium.
C57Bl6 mice underwent partial ligation of the LCA and were sacrificed 48 h later. (A) Endothelial-enriched RNAs were isolated from RCA and LCA, and the expression of smarcd3, psmd12 and ERN1 was determined by qPCR (n = 3, *p < 0.05). KLF2 was used as a control. (B) LCAs and RCAs were dissected out 48 h post ligation and were mounted en face. Immunofluorescence confocal microscopy was performed to detect the expression of smarcd3 (red) and DAPI for nuclei (blue) (n = 3). Auto-fluorescence (green) shows internal elastic lamina (IEL). White Scale Bar = 50 μm. (C) Image J was used to perform the semi-quantitative estimation of smarcd3 expression in the RCA vs. LCA by calculating the mean fluorescence intensities from the Red Channel. Data shown as means ± S.E.M from 5 different animals; *p < 0.05. Mean fluorescence intensities from the negative control images (Secondary Antibody only) were used for background subtraction. (D) Human umbilical vein endothelial cells (HUVECs), human arterial endothelial cells (HAECs), and immortalized mouse aortic endothelial cells (iMAECs) were subjected to either laminar shear (LS) or oscillatory shear (OS) for 24 h. RNA was extracted and expression of smarcd3 was determined by qPCR (n = 5, p < 0.05). Static cells were used as controls.
Next, we validated the mechanosensitivity of smarcd3 by a specific antibody, but not psmd12, as the antibody was unavailable. Consistent with our qPCR analyses, we found that Smarcd3 protein expression in the LCA endothelium (where flow is disturbed and endothelium is inflamed) was significantly reduced as compared to the RCA (where flow is stable and endothelium is not inflamed)6,8,17 (Fig. 3B,C). These results demonstrate that smarcd3 expression is significantly reduced in the mouse arterial endothelium exposed to d-flow compared to s-flow. Additional in vitro experiments using the cone-and-plate shear system also confirms that exposure to the pro-inflammatory oscillatory shear stress (OS) for 24 h decreased the expression of smarcd3 in HUVECs, HAECs, and iMAECs compared to the anti-inflammatory laminar shear stress (LS) (Fig. 3D). Taken together, these results demonstrate that smarcd3 expression is reduced by the pro-inflammatory d-flow condition both in vitro and in vivo.
Knockdown of smarcd3 induces endothelial inflammation
To determine whether smarcd3 is involved in endothelial inflammation, we treated HUVECs with smarcd3-siRNA and subjected them to oscillatory shear conditions. As expected and shown previously36,37, oscillatory shear induced VCAM1 and CXCL8 expression while decreasing the KLF2 expression compared to laminar shear (Fig. 4A). Knockdown of smarcd3 further enhanced this oscillatory shear-induced expression of VCAM1 and CXCL8 by 3-fold and 1.9-fold, respectively (Fig. 4A). However, knockdown of smarcd3 did not have any effect on the expression of KLF2 (Fig. 4A). In addition, treatment of smarcd3-siRNA in HUVECs significantly increased monocyte adhesion to the endothelium as compared to the control-siRNA (Fig. 4B,C), confirming the role of smarcd3 in endothelial inflammation.

Knockdown of smarcd3 enhances endothelial activation.
(A) HUVECs were transfected with non-target or smarcd3-siRNA (150 nM) for 2 days and were subjected to either LS or OS for 24 h. RNA was prepared and expression of NF-κB target genes (VCAM1 and CXCL8) was determined by qPCR (mean ± S.E.M, n = 4, *p < 0.05). KLF2 was used as a control. (B) HUVECs were transfected with either non-target or smarcd3-siRNA (150 nM) for 2 days. The number of adhering BCECF-labelled THP1 monocytes (5 × 105) was determined by fluorescence microscopy. As a positive control, some cells were treated with TNFα (3 ng/mL) for 3 hours. White Scale Bar = 200 μm. (C) Graph shows quantification of bound monocytes (mean ± S.E.M, n = 3, *p < 0.05).
We next validated the role of smarcd3 in endothelial inflammation by treating mouse aorta explants ex vivo with smarcd3-siRNA. Treatment of mouse aortic explants with smarcd3-siRNA for 48 hours significantly reduced the expression of smarcd3 in the endothelial layer as compared to the non-targeting control [shown by en-face immunostaining (Fig. 5A,B)], confirming the efficacy of smarcd3-siRNA in our ex vivo system. In this condition, we found that knockdown of smarcd3 resulted in increased VCAM1 expression (Fig. 5A,C) and significantly increased monocyte adhesion (Fig. 5D). These results demonstrate that knockdown of smarcd3 induces endothelial inflammation both in vitro and ex vivo.

Knockdown of smarcd3 induces VCAM1 expression in mouse aortic endothelial cells ex vivo.
(A) Mouse aorta explants were cultured en-face in a 48-well plate. Aortic explants were transfected with siRNAs (100 nM) for smarcd3 (si-Smarcd3) or control for 48 h and expression of smarcd3 and VCAM1 in the endothelium was determined by immunofluorescence staining using antibodies for smarcd3 (red) and VCAM1 (Cyan). Auto-fluorescence (green) shows internal elastic lamina (IEL). White Scale Bar = 50 μm (B,C) Graph shows quantitative estimation of signal intensities for expression of smarcd3 and VCAM1, respectively (mean ± S.E.M, n = 6, *p < 0.05). (D) Aortic explants treated as above were mounted en-face, and the number of BCECF-labeled J774A.1 monocytic cells to the endothelial surface was determined by fluorescence microscopy. Values represent mean ± S.E.M (*P < 0.05; n = 6).
Discussion
Recent technological advances such as microarray and RNA sequencing analyses have enhanced our understanding on a global gene expression pattern during endothelial inflammation and pathogenesis of atherosclerosis. However, it is not easy to individually validate the roles of each gene in the disease progression. Previously, by using a partial carotid ligation model of atherosclerosis, we identified more than 580 mechanosensitive genes that changed in response to d-flow as compared to s-flow6. D-flow is known to promote atherosclerosis by regulating global gene expression changes in the endothelium1. Although one of the key mechanisms by which d-flow causes atherosclerosis is endothelial inflammation4, the functions of these flow-sensitive genes during inflammation remain to be elucidated. Here, we developed an in vivo functional screening system using the constitutive Drosophila RNAi (C564-GAL4 RNAi) flies that express drosomycin-GFP as a surrogate marker of Toll-dependent NF-κB activation. Using this in vivo screening strategy, we screened 84 flow-sensitive genes and identified Bap60, RPN5 and ERN1 as potential regulators of Toll-mediated NF-κB-dependent inflammation pathway. The role of these genes was subsequently validated using the temperature inducible RNAi (C564ts-GAL4 RNAi) flies to rule out the potential secondary effects of these genes during development. From these fly studies, we identified two mechanosensitive NF-κB regulators: RPN5 as an activator of both Toll- and IMD-mediated NF-κB pathways, and Bap60 as repressor and activator of Toll-mediated and IMD-mediated NF-κB pathways, respectively. We also identified ERN1 as a weak activator of the Toll-mediated pathway.
Subsequent studies in mammalian systems validated smarcd3 (mammalian ortholog of Bap60) as a novel mechanosensitive repressor of NF-κB-mediated inflammation in mammalian endothelial cells. To our knowledge, this is the first report where a mammalian mechanosensitive transcriptomic dataset has been functionally screened by taking advantage of Drosophila UAS-RNAi knockdown library.
Here, Bap60/smarcd3 was identified as a novel mechanosensitive molecule involved in the downregulation of NF-κB-dependent inflammation. Smarcd3 is a SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily D member-338. Smarcd3 is part of the large chromatin remodeling complex that is known to regulate gene transcription by modifying the chromatin structure38,39,40,41. As knockdown of smarcd3 significantly enhanced oscillatory shear-induced pro-inflammatory response as measured by VCAM1 and CXCL8 expression (Fig. 4), it is likely that d-flow induces pro-inflammatory gene expression via reducing smarcd3 expression in the endothelial cells.
Recently, Bonnay et al. showed that Drosophila Bap60 interacts with Akirin upon immune challenge, and Bap60-Akirin complex is required for the induction of a subset of IMD target genes, indicating that Bap60 acts as a positive regulator for IMD-pathway42, and is consistent with our result obtained using the inducible Bap60 RNAi flies (Fig. 2D). In contrast, knockdown of Brahma complex component including Bap60 and Brahma is sufficient to enhance the drosomycin reporter activity in S2 cells42,43. Consistent with this, our inducible Bap60 RNAi fly showed increased drosomycin expression upon Gram-positive bacterial infection (Fig. 2D). Together, our results and previous studies show that Bap60 could act as a negative and positive regulator for Toll- and IMD-target gene expression, respectively, in adult flies (Fig. 2C,D). Although, it is not known whether shear stress regulated NF-κB is activated via Toll- and/or IMD-mediated pathways, our mammalian studies suggest that smarcd3 acts as a repressor of NF-κB activation in response to shear stress.
Although our Drosophila screening system enabled us to screen for candidate mechanosensitive genes involved in NF-κB activation, there are some limitations in our experimental approaches. For example, our primary screening was limited to testing the Toll-mediated NF-κB activation pathway, but not the IMD-mediated NF-κB pathway. Furthermore, our primary screening with constitutive C564-GAL4 flies seems to have caused false positive results due to potential secondary effects during development. The use of an inducible GAL4 driver such as C564ts-GAL4 together with a dual reporter system [by the drosomycin-GFP reporter with an IMD-pathway reporter such as diptericin-mCherry44] would have circumvented these technical limitations. Due to lack of proper reagents, the role of RPN5 could not be studied.
In summary, we developed an in vivo Drosophila RNAi screening method to identify flow-sensitive genes that regulate endothelial inflammation. The newly identified mechanosensitive smarcd3 may play a critical role in endothelial inflammation and atherosclerosis. This in vivo functional screening approach may be expanded to identify novel genes that regulate other pathways such as apoptosis and cell proliferation.
Additional Information
How to cite this article: Kumar, S. et al. Functional screening of mammalian mechanosensitive genes using Drosophila RNAi library– Smarcd3/Bap60 is a mechanosensitive pro-inflammatory gene. Sci. Rep. 6, 36461; doi: 10.1038/srep36461 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Chiu, J. J. & Chien, S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiological reviews 91, 327–387, doi: 10.1152/physrev.00047.2009 (2011).
Davies, P. F. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nature clinical practice. Cardiovascular medicine 6, 16–26, doi: 10.1038/ncpcardio1397 (2009).
Tzima, E. et al. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 437, 426–431, doi: 10.1038/nature03952 (2005).
Tarbell, J. M., Shi, Z.-D., Dunn, J. & Jo, H. Fluid Mechanics, Arterial Disease, and Gene Expression. Annual Review of Fluid Mechanics 46, 591–614, doi: 10.1146/annurev-fluid-010313-141309 (2014).
Passerini, A. G. et al. Coexisting proinflammatory and antioxidative endothelial transcription profiles in a disturbed flow region of the adult porcine aorta. Proceedings of the National Academy of Sciences of the United States of America 101, 2482–2487, doi: 101/8/2482 (2004).
Ni, C. W. et al. Discovery of novel mechanosensitive genes in vivo using mouse carotid artery endothelium exposed to disturbed flow. Blood 116, e66–e73, doi: blood-2010-04-278192 10.1182/blood-2010-04-278192 (2010).
Dai, G. et al. Distinct endothelial phenotypes evoked by arterial waveforms derived from atherosclerosis-susceptible and -resistant regions of human vasculature. Proceedings of the National Academy of Sciences of the United States of America 101, 14871–14876, doi: 10.1073/pnas.0406073101 (2004).
Son, D. J. et al. The atypical mechanosensitive microRNA-712 derived from pre-ribosomal RNA induces endothelial inflammation and atherosclerosis. Nature communications 4, 3000, doi: 10.1038/ncomms4000 (2013).
Lemaitre, B., Nicolas, E., Michaut, L., Reichhart, J. M. & Hoffmann, J. A. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 86, 973–983 (1996).
Lemaitre, B. et al. A recessive mutation, immune deficiency (imd), defines two distinct control pathways in the Drosophila host defense. Proceedings of the National Academy of Sciences of the United States of America 92, 9465–9469 (1995).
Lemaitre, B. & Hoffmann, J. The host defense of Drosophila melanogaster. Annu Rev Immunol 25, 697–743, doi: 10.1146/annurev.immunol.25.022106.141615 (2007).
Jang, I. H. et al. A Spatzle-processing enzyme required for toll signaling activation in Drosophila innate immunity. Dev Cell 10, 45–55, doi: 10.1016/j.devcel.2005.11.013 (2006).
DeVorkin, L. & Gorski, S. M. Monitoring Autophagy in Drosophila Using Fluorescent Reporters in the UAS-GAL4 System. Cold Spring Harbor Protocols 2014, pdb.prot080341, doi: 10.1101/pdb.prot080341 (2014).
Duffy, J. B. GAL4 system in Drosophila: a fly geneticist’s Swiss army knife. Genesis 34, 1–15, doi: 10.1002/gene.10150 (2002).
Dietzl, G. et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448, 151–156, doi: 10.1038/nature05954 (2007).
Nam, D. et al. Partial carotid ligation is a model of acutely induced disturbed flow, leading to rapid endothelial dysfunction and atherosclerosis. American journal of physiology 297, H1535–H1543 (2009).
Nam, D. et al. A model of disturbed flow-induced atherosclerosis in mouse carotid artery by partial ligation and a simple method of RNA isolation from carotid endothelium. J Vis Exp, doi: 1861 10.3791/1861 (2010).
Kumar, S., Kim, C. W., Son, D. J., Ni, C. W. & Jo, H. Flow-dependent regulation of genome-wide mRNA and microRNA expression in endothelial cells in vivo. Scientific Data 1, 140039, doi: 10.1038/sdata.2014.39 (2014).
Attrill, H. et al. FlyBase: establishing a Gene Group resource for Drosophila melanogaster. Nucleic Acids Res 44, D786–D792, doi: 10.1093/nar/gkv1046 (2016).
Kambris, Z. et al. Drosophila immunity: a large-scale in vivo RNAi screen identifies five serine proteases required for Toll activation. Curr Biol 16, 808–813, doi: 10.1016/j.cub.2006.03.020 (2006).
McGuire, S. E., Le, P. T., Osborn, A. J., Matsumoto, K. & Davis, R. L. Spatiotemporal rescue of memory dysfunction in Drosophila. Science (New York, N.Y.) 302, 1765–1768, doi: 10.1126/science.1089035 (2003).
Ni, C. W., Kumar, S., Ankeny, C. J. & Jo, H. Development of immortalized mouse aortic endothelial cell lines. Vascular cell 6, 7, doi: 10.1186/2045-824x-6-7 (2014).
Holliday, C. J., Ankeny, R. F., Jo, H. & Nerem, R. M. Discovery of shear- and side-specific mRNAs and miRNAs in human aortic valvular endothelial cells. American journal of physiology 301, H856–H867, doi: 10.1152/ajpheart.00117.2011 (2011).
Tressel, S. L., Huang, R. P., Tomsen, N. & Jo, H. Laminar shear inhibits tubule formation and migration of endothelial cells by an angiopoietin-2 dependent mechanism. Arteriosclerosis, thrombosis, and vascular biology 27, 2150–2156, doi: 10.1161/atvbaha.107.150920 (2007).
Boo, Y. C., Mun, G. I., Tressel, S. L. & Jo, H. Detection of low levels of nitric oxide using an electrochemical sensor. Methods in molecular biology (Clifton, N.J.) 704, 81–89, doi: 10.1007/978-1-61737-964-2_7 (2011).
Ni, C. W., Qiu, H. & Jo, H. MicroRNA-663 upregulated by oscillatory shear stress plays a role in inflammatory response of endothelial cells. American journal of physiology 300, H1762–H1769, doi: ajpheart.00829.2010 10.1152/ajpheart.00829.2010 (2011).
Rezvan, A., Ni, C. W., Alberts-Grill, N. & Jo, H. Animal, in vitro, and ex vivo models of flow-dependent atherosclerosis: role of oxidative stress. Antioxid Redox Signal 15, 1433–1448, doi: 10.1089/ars.2010.3365 (2011).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675 (2012).
Feeley, K. P., Westbrook, D. G., Bray, A. W. & Ballinger, S. W. An ex-vivo model for evaluating bioenergetics in aortic rings. Redox Biol 2C, 1003–1007, doi: 10.1016/j.redox.2014.08.008 (2014).
Kim, C. W. et al. Prevention of abdominal aortic aneurysm by anti-microRNA-712 or anti-microRNA-205 in angiotensin II-infused mice. Arteriosclerosis, thrombosis, and vascular biology 34, 1412–1421, doi: 10.1161/ATVBAHA.113.303134 (2014).
Harrison, D. A., Binari, R., Nahreini, T. S., Gilman, M. & Perrimon, N. Activation of a Drosophila Janus kinase (JAK) causes hematopoietic neoplasia and developmental defects. EMBO J 14, 2857–2865 (1995).
De Gregorio, E., Spellman, P. T., Tzou, P., Rubin, G. M. & Lemaitre, B. The Toll and Imd pathways are the major regulators of the immune response in Drosophila. The EMBO Journal 21, 2568–2579, doi: 10.1093/emboj/21.11.2568 (2002).
Minakhina, S. & Steward, R. Melanotic mutants in Drosophila: pathways and phenotypes. Genetics 174, 253–263, doi: 10.1534/genetics.106.061978 (2006).
Fukuyama, H. et al. Landscape of protein–protein interactions in Drosophila immune deficiency signaling during bacterial challenge. Proceedings of the National Academy of Sciences 110, 10717–10722, doi: 10.1073/pnas.1304380110 (2013).
Zang, Y. et al. Plasma membrane overgrowth causes fibrotic collagen accumulation and immune activation in Drosophila adipocytes. Elife 4, e07187, doi: 10.7554/eLife.07187 (2015).
Orr, A. W. et al. The subendothelial extracellular matrix modulates NF-κB activation by flow: a potential role in atherosclerosis. The Journal of Cell Biology 169, 191–202, doi: 10.1083/jcb.200410073 (2005).
Liang, F., Huang, N., Wang, B., Chen, H. & Wu, L. Shear stress induces interleukin-8 mRNA expression and transcriptional activation in human vascular endothelial cells. Chinese medical journal 115, 1838–1842 (2002).
Jordan, N. V. et al. SWI/SNF chromatin-remodeling factor Smarcd3/Baf60c controls epithelial-mesenchymal transition by inducing Wnt5a signaling. Molecular and cellular biology 33, 3011–3025, doi: 10.1128/mcb.01443-12 (2013).
Coradini, D., Boracchi, P., Oriana, S., Biganzoli, E. & Ambrogi, F. Differential expression of genes involved in the epigenetic regulation of cell identity in normal human mammary cell commitment and differentiation. Chinese journal of cancer 33, 501–510, doi: 10.5732/cjc.014.10066 (2014).
Meng, Z. X., Wang, L., Xiao, Y. & Lin, J. D. The Baf60c/Deptor pathway links skeletal muscle inflammation to glucose homeostasis in obesity. Diabetes 63, 1533–1545, doi: 10.2337/db13-1061 (2014).
Acevedo, N. et al. Age-associated DNA methylation changes in immune genes, histone modifiers and chromatin remodeling factors within 5 years after birth in human blood leukocytes. Clinical epigenetics 7, 34, doi: 10.1186/s13148-015-0064-6 (2015).
Bonnay, F. et al. Akirin specifies NF-kappaB selectivity of Drosophila innate immune response via chromatin remodeling. EMBO J 33, 2349–2362, doi: 10.15252/embj.201488456 (2014).
Kuttenkeuler, D. et al. A large-scale RNAi screen identifies Deaf1 as a regulator of innate immune responses in Drosophila. Journal of innate immunity 2, 181–194, doi: 10.1159/000248649 (2010).
Charroux, B. & Royet, J. Elimination of plasmatocytes by targeted apoptosis reveals their role in multiple aspects of the Drosophila immune response. Proceedings of the National Academy of Sciences of the United States of America 106, 9797–9802, doi: 10.1073/pnas.0903971106 (2009).
Acknowledgements
This work was supported by funding from National Institutes of Health grants HL119798, HL113451, HL095070 and HL124879 to HJ. HJ is John and Jan Portman Professor. This work was supported from National Research Foundation of Korea NRF-2015R1A3A2033475 to W.-J. L.
Author information
Authors and Affiliations
Contributions
Study design: S.K., I.H.J., W.J.L. and H.J. Conceived, and designed the experiments: S.K., I.H.J., W.J.L., C.W.K. and H.J. Performed experiments: S.K., I.H.J., C.W.K. and D.W.K. Analyzed the data: S.K., I.H.J., C.W.K., D.W.K., W.J.L. and H.J. Statistical analysis and paper writing: S.K., I.H.J. and H.J. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Kumar, S., Jang, Ih., Kim, C. et al. Functional screening of mammalian mechanosensitive genes using Drosophila RNAi library– Smarcd3/Bap60 is a mechanosensitive pro-inflammatory gene. Sci Rep 6, 36461 (2016). https://doi.org/10.1038/srep36461
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep36461
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
