
Abstract
The genus Dacus is one of the most economically important tephritid fruit flies. The first complete mitochondrial genome (mitogenome) of Dacus species – D. longicornis was sequenced by next-generation sequencing in order to develop the mitogenome data for this genus. The circular 16,253 bp mitogenome is the typical set and arrangement of 37 genes present in the ancestral insect. The mitogenome data of D. longicornis was compared to all the published homologous sequences of other tephritid species. We discovered the subgenera Bactrocera, Daculus and Tetradacus differed from the subgenus Zeugodacus, the genera Dacus, Ceratitis and Procecidochares in the possession of TA instead of TAA stop codon for COI gene. There is a possibility that the TA stop codon in COI is the synapomorphy in Bactrocera group in the genus Bactrocera comparing with other Tephritidae species. Phylogenetic analyses based on the mitogenome data from Tephritidae were inferred by Bayesian and Maximum-likelihood methods, strongly supported the sister relationship between Zeugodacus and Dacus.
Similar content being viewed by others


Introduction
The genus Dacus Fabricius (Diptera: Tephritidae: Dacini) is one of the most economically important fruit flies1. There are about 248 Dacus species most of which show a strong preference for attacking the pods of Asclepiadaceae and Apocynaceae, or the fruits and flowers of Cucurbitaceae1,2. The majority of Dacus species distribute in the African continent, several species are found in the Indian Subcontinent, Southeast Asia, Australia and the Pacific2.
Dacus (Callantra) longicornis Wiedemann has a widespread distribution across southern Asia and Southeast Asia and attacks Cucurbitaceae species3. Very limited studies focused on D. longicornis are available except for taxonomy and records or first record of this species in some countries and areas3,4,5. Molecular data of D. longicornis has not been well studied with only seven records published in GenBank as of May 2016. It is becoming increasingly evident that detailed knowledge of molecular data of D. longicornis is required not only for its population structure and geographical variability studies, but also for a comprehensive phylogeny analysis of the tribe Dacini which consist of two very large genera - Bactrocera Macquart (629 spp.) and Dacus Fabricius (248 spp.) and two small genera - Ichneumonopsis Hardy (one sp.) and Monacrostichus Bezzi (two spp.)2,7,8,9.
The whole mitogenome has become established as one of the most useful markers and has been used for molecular systematic, phylogeography, diagnostics and molecular evolutionary studies10,11,12. By the May of 2016, forty-five complete mitogenomes of 19 Tephritidae species are available in GenBank (Supplementary Table S1), including 16 Bactrocera species which are Bactrocera (Bactrocera) arceae (Hardy & Adachi) (KR233259)8, B. (B.) carambolae Drew & Hancock (EF014414), B. (B.) correcta (Bezzi) (JX456552), B. (B.) dorsalis (Hendel) (DQ845759, DQ917577, B. (B.) papayae Drew & Hancock DQ917578 and B. (B.) philippinensis Drew & Hancock DQ995281; B. (B.) papayae and B. (B.) philippinensis have been proven to be the same species with B. (B.) dorsalis)13,14, B. (B.) latifrons (Hendel) (KT881556)9, B. (B.) melastomatos Drew & Hancock (KT881557)9, B. (B.) tryoni (Froggatt) (HQ130030)15, B. (B.) umbrosa (Fabricius) (KT881558)9, B. (B.) zonata (Saunders) (KP296150)16, B. (Daculus) oleae (Gmelin) (AY210702, AY210703, GU108459 to GU108479)15,17, B. (Tetradacus) minax (Enderlein) (HM776033)18, B. (Zeugodacus) caudata (Fabricius) (KT625491 and KT625492)9, B. (Z.) cucurbitae (Coquillett) (JN635562)19, B. (Z.) diaphora (Hendel) (KT159730)20, B. (Z.) scutellata (Hendel) (KP722192) and B. (Z.) tau (Walker) (KP711431)21; one D. longicornis (KX345846) (from this study); one Ceratitis (Ceratitis) capitata (Wiedemann) (AJ242872)22 and one Procecidochares utilis Stone (KC355248).
Several studies also did phylogenetic analysis of tephritid species based on the published mitogenome sequences8,9,16, however, sixteen of the 19 published species belong to the genus Bactrocera, so that the phylogenetic relationships of higher taxon cannot be explained very well. Recently, the phylogenetic study of Dacini has been attracted more attention especially in the taxonomic status of the subgenus Zeugodacus. Some researchers recommended to raising Zeugodacus to genus level6, and because of the limited molecular data, some researchers also suggested that the exact relationship between Zeugodacus, Dacus and Bactrocera still needs to be properly resolved7.
In this study, we report the first complete mitogenome of Dacus species - D. longicornis, compare the mitogenome data with other tephritid species, and discuss the molecular phylogeny of Dacini in particular.
Results
Sequencing and assembly of Mitogenome
An Illumina library was constructed on the DNA of Dacus longicornis with an average insert size of 480 bp. This library was sequenced on a run of Hiseq2500 and following removal of sequencing adapters, 9,857,102 read-pairs were generated. Approximately 8% of the reads resembled mitochondrial sequences after BLASTn filtering with E ≤ 1e-5. Assemblies constructed with the IDBA-UD assemblers23 resulted in 10 contigs more than 10,000 bp in length of which the longest contig represented complete mitogenome of D. longicornis.
Mitogenome features
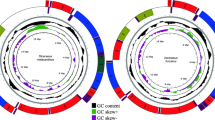
The complete mitogenome of D. longicornis was 16,253 bp in length. It presented the typical set and arrangement of 37 genes found in the ancestral insect mitochondrial genome, including 13 protein-coding genes (PCGs), two ribosomal RNA (rRNA) genes, 22 transfer RNA (tRNA) genes and a control region (A + T-rich region) (Table 1; Fig. 1). Nine PCGs (ND2, COI, COII, COIII, ATP6, ATP8, ND3, ND6 and CYTB), 14 tRNAs (tRNAIle, tRNAMet, tRNATrp, tRNALeu(UUR), tRNALys, tRNAAsp, tRNAGly, tRNAAla, tRNAArg, tRNAAsn, tRNASer(AGN), tRNAGlu, tRNAThr and tRNASer(UCN)) and the control region were located on the major strand (J-strand). Four PCGs (ND5, ND4, ND4L and ND1), eight tRNAs (tRNAGln, tRNACys, tRNATyr, tRNAPhe, tRNAHis, tRNAPro, tRNALeu(CUN) and tRNAVal) and two rRNAs (lrRNA and srRNA) were located on the minor strand (N-strand).

Mitochondrial genome map of Dacus longicornis.
Arrows indicate the orientation of gene transcription. tRNA genes are indicated with their one-letter corresponding amino acids (L1: CUN; L2: UUR; S1: AGN; S2: UCN). The GC content was plotted using a black sliding window, as the deviation from the average GC content of the entire sequence. GC-skew was plotted as the deviation from the average GC-skew of the entire sequence.
Spacing sequences in 14 regions ranged from 1 to 49 bp, the longest located between tRNAArg and tRNAAsn. The overlapping sequences ranged from 1 to 8 bp in 13 regions, the longest was between tRNATrp and tRNACys.
As in other insect mitogenomes24, the nucleotide composition of D. longicornis was all AT biased and positive AT skews and negative GC skews, not only in the whole mitochondrial genome but also in PCGs, rRNAs, tRNAs and the control region (Table 2).
All of the PCGs started with ATN codons (ATG in COII, ATP6, COIII, ND4, ND4L, CYTB and ND1; ATC in ATP8, ND5 and ND6; ATT in ND2; ATA in ND3) except for COI which started with TCG codon. Seven PCGs (COI, COII, ATP8, ATP6, COIII, ND4L and ND6) stopped with TAA codon, three PCGs (ND2, ND3 and ND4) had TAG stop codon, while ND5, CYTB and ND1 had incomplete stop codon T.
Twenty-two typical tRNAs which are usually observed in insect mitogenomes were also found in D. longicornis mitogenome. The size of 22 tRNAs ranged from 64 bp (tRNAHis) to 72 bp (tRNAVal). Most tRNAs could be folded into the cloverleaf structure except for tRNASer(AGN) which lacked the dihydorouridine (DHU) arm (Fig. 2). Twenty-three G-U pairs, four mismatched base U-U pairs and one mismatched base U-C pair were found in D. longicornis mitogenome tRNA secondary structures. The G-U pairs were located in the amino acid acceptor (AA) arm (9 bp), DHU arm (8 bp), anticodon (AC) arm (3 bp) and TψC (T) arm (3 bp). The mismatched base U-U pairs were located in AA arm (2 bp), AC arm (1 bp) and T arm (1 bp). The mismatched base U-C pairs were located in T arm.

Putative secondary structures of tRNAs found in the mitochondrial genome of Dacus longicornis.
The tRNAs are labelled with the abbreviations of their corresponding amino acids. Inferred Watson-Crick bonds are illustrated by lines, whereas GU bonds are illustrated by dots.
The lrRNA was assumed to fill up the blanks between tRNALeu(CUN) and tRNAVal. For the boundary between the srRNA gene and the control region, alignments with homologous sequences in other mitogenomes of Tephritidae were applied to determine the 3′-end of the gene. The lrRNA is 1,331 bp long with an A + T content of 78.5%, and the srRNA is 798 bp long with an A + T content of 74.9%.
The control region (1,343 bp) was flanked by srRNA and tRNAIle and was highly enriched in AT (85.3%). Two 151 bp repeats were found in the control region and one 19 bp poly-T stretch located near the repeats. Furthermore, the region near tRNAIle contained another 22 bp poly-A stretch. Both repeated sequences and poly stretches are common in the control region for most insects25,26, and these motifs may function during processing of the replication and transcription.
Phylogenetic relationship
Four datasets were used in the phylogenetic analysis, there are 14,586 residues in the PCG123RNA matrix (containing nucleotides of 13 PCGs, two rRNAs and 22 tRNAs), 11,148 residues in the PCG123 matrix (containing nucleotides of 13 PCGs), 10,870 residues in the PCG12RNA matrix (containing nucleotides of 13 PCGs but excluding the third codon sites, two rRNAs and 22 tRNAs) and 7,432 residues in the PCG12 matrix (containing nucleotides of 13 PCGs but excluding the third codon sites).
The topology structures conducted from Bayesian and ML analyses were very similar based on these four datasets (Fig. 3). The monophyly of Tephritidae and Dacini tribe were well supported in all trees with posterior probabilities 1.0 and ML bootstraps 100. The genus Bactrocera was not monophyletic but it was different from other Tephritidae mitochondrial genome phylogeny studies which only included Bactrocera speices of Dacini8,9,16,20,21. Members of the subgenera Bactrocera and Zeugodacus formed a distinct clade from the other subgenera Daculus and Tetradacus, respectively. The subgenus Zeugodacus and Callantra were sister groups from our results, which supported the conclusions from several recent studies about phylogenetic relationship of Dacini6,7.

Phylogenetic tree of Tephritidae family based on mitochondrial genomes.
Squares at the nodes are Bayesian posterior probabilities for 1, 2, 5 and 6, ML bootstrap values for 3, 4, 7 and 8. Dataset of PCG123, 1 and 3, PCG123RNA, 2 and 4, PCG12, 5 and 7, PCG12RNA, 6 and 8. Black indicates posterior probabilities = 1.00 or ML bootstrap = 100, gray indicates posterior probabilities ≥ 0.90 or ML bootstrap ≥ 70, white indicates posterior probabilities <0.90 or ML bootstrap < 70, ‘ns’ indicates not support, *indicates posterior probabilities = 1.00 or ML bootstrap = 100 in eight trees.
Discussion
In this study, we are reporting the first complete mitochondrial genome of Dacus species - Dacus longicornis in Dacini tribe of Tephritidae. The size of D. longicornis mitogenome is 16,253 bp, which is the largest one among the other 18 tephritid mitogenomes available with the size ranging from 15,687 bp in B. tau to 16,043 bp in B. minax. The control region of D. longicornis mitogenome is 1,343 bp in length, which is also the longest one in the other published tephritid mitogenomes with the size ranging from 801 bp in B. tau to 1,141 bp in B. minax (Supplementary Table S2).
The A + T contents of the whole mitogenome, PCGs, tRNAs, rRNAs and CR in D. longicornis are 72.33%, 69.4%, 74.81%, 77.17% and 85.26%, average amongst all reported tephritid mitogenomes, which range from 67.28% (B. minax) to 80.83% (P. utilis) in the whole mitogenome, from 64.30% (B. minax) to 78.90% (P. utilis) in PCGs, from 72.31% (B. minax) to 80.61% (P. utilis) in tRNAs, from 73.71% (B. minax) to 85.69% (P. utilis) in rRNAs and from 77.65% (B. minax) to 91.14% (C. capitata) in CR (Supplementary Table S2).
The AT skews and GC skews of D. longicornis in the whole mitogenome, PCGs, tRNAs, rRNAs and CR are 0.101 (from 0.021 in C. capitata to 0.131 in B. minax) and −0.293 (from −0.175 in P. utilis to −0.316 in B. minax), 0.105 (from 0.019 in C. capitata to 0.148 in B. minax) and −0.301 (from −0.170 in P. utilis to −0.319 in B. minax), 0.052 (from 0.005 in P. utilis to 0.055 in B. minax) and −0.126 (from −0.074 in B. cucurbitae to −0.182 in B. minax), 0.087 (from 0.051 in P. utilis to 0.121 in B. minax) and −0.329 (from −0.267 in C. capitata to −0.356 in B. minax), 0.146 (maximum) and −0.354 (minimum), respectively. The CR of D. longicornis shows the most marked AT skews and GC skews compared with the other tephritid mitogenomes (Supplementary Table S2).
Similar to the other tephritid mitogenomes, tRNAs of D. longicornis have three main clusters: (1) tRNAIle – tRNAGln – tRNAMet; (2) tRNATrp – tRNACys – tRNATyr; (3) tRNAAla – tRNAArg – tRNAAsn – tRNASer(AGN) – tRNAGlu – tRNAPhe. The atypical cloverleaf structure of tRNASer(AGN) is similar to this gene in other metazoan mitogenomes27.
Seven PCGs in all Tephritidae species have the same start codons (ATG in ATP6, COII, CYTB, ND4 and ND4L, ATT in ND2, TCG in COI), and five PCGs (ATP6, ATP8, COIII, ND4L and ND6) have the same stop TAA codons (Table 3). It’s worth noting that the subgenera Bactrocera, Daculus and Tetradacus differ from the subgenus Zeugodacus, the genera Dacus, Ceratitis and Procecidochares in the possession of TA instead of TAA stop codon for COI gene. There is a possibility that the TA stop codon in COI is the synapomorphy in Bactrocera group which includes 10 subgenera (Afrodacus, Apodacus, Bactrocera, Bulladacus, Daculus, Gymnodacus, Notodacus, Semicallantra, Tetradacus and Trypetidacus) in the genus Bactrocera28,29 comparing with other Tephritidae species.
Studies on molecular phylogenetic relationship of Dacini fruit flies have been reported by several researchers. Early study supported that Bactrocera and Dacus were each monophyletic based on phylogenetic analysis of 34 tephritid fruit flies including 16 species of Dacini, utilizing 1,391 bp from COII, lrRNA, srRNA, tRNALys and tRNAAsp genes30. Four years later, Segura et al. reported that B. cucurbitae was more closely related to the genus Dacus than to other Bactrocera species using CYTB, ND1 and tRNASer genes from 23 tephritid species, and White also suggested that the subgenus Zeugodacus might be a sister group to the genus Dacus in the same year31,32. In recent four years, Krosch et al. concluded that ‘Zeugodacus’ clade was the sister group to Dacus based on COI, COII, lrRNA and white eye genes from 125 Dacini species6. Virgilio et al. also drew the same conclusion according to two datasets. One dataset included 98 vouchers using COI, ND6, lrRNA, tRNAPro and period genes and the other included 159 vouchers based on COI and lrRNA genes7. Here we added support for this conclusion that Zeugodacus and Dacus are sister groups from mitochondrial genome data.
As for the taxonomic status of the subgenus Zeugodacus, Krosch et al. suggested that taxonomic consideration should be given to raising Zeugodacus to genus level6. Virgilio et al. supported this conclusion, but also proposed that the exact relationship between Zeugodacus, Dacus and Bactrocera still needed to be properly resolved7. Considering that there are 30 recognized subgenera four groups in the genus Bactrocera and eight subgenera in Dacus33, and the subgenera Bactrocera and Zeugodacus have been proven to be not monophyletic34,35, we suggest to accurately resolve the exact relationship of Dacini with more complete taxon sampling and more comprehensive molecular data combining mitochondrial genomes and nuclear genes. Raising Zeugodacus to genus level is also needed to be confirmed further based on more taxonomy, biology and biogeography evidence.
Materials and Methods
Sample collection
The adult specimen of D. longicornis was collected from Yunnan province of China. It was identified based on available taxonomic keys3, and preserved in absolute ethyl alcohol and stored in −20 °C freezer in Chinese Academy of Inspection and Quarantine until use.
Mitogenome sequencing and analysis
The genomic DNA was extracted from one fly’s muscle tissues of the thorax using the DNeasy DNA Extraction kit (QIAGEN) following the manufacturer’s instructions. The concentration of double-stranded DNA (dsDNA) in extraction was assayed on a Qubit fluorometer using a dsDNA high-sensitivity kit (Invitrogen).
An Illumina TruSeq library was generated from the genomic DNA with an average insert size of 480 bp. The library was sequenced on a full run of Illumina Hiseq2500 with 500 cycles and paired-end sequencing (250 bp reads).
A quality assessment of raw FASTQ files for the library was made using FastQCv0.11.4 (www.bioinformatics.babraham.ac.uk/projects/fastqc) prior to the removal of adapter sequences with Trimmomatic v0.30 (ILLUMINACLIP:2:30:10)36. The putative mitochondrial reads were identified in a BLASTn search37 against a custom reference of Tephritidae mitogenomes (E ≤ 1e-5; maximum target sequences 1; DUST filtering disabled). The extracted mitochondrial reads were subjected to whole-genome shot-gun assembly using IDBA-UD23. Assemblies with IDBA-UD used a similarity threshold of 98% and minimum and maximum k values of 80 and 240 bp, respectively. Following assembly, the contig identified as mitogenome was manually checked in Geneious (http://www.geneious.com/) for identical or near-identical overlapping terminal regions and were circularized where possible.
Protein-coding genes (PCGs) and two ribosomal RNA (rRNA) genes were identified by BLAST searches in NCBI (http://www.ncbi.nlm.nih.gov/) and confirmed by alignment with homologous genes from other 18 tephritid species available in GenBank. Transfer RNA (tRNA) genes were identified using the tRNAscan-SE38 and ARWEN39 and checked manually. The circular map of D. longicornis mitogenome sequence was drawn with CGView40. The nucleotide composition and codon usage were analyzed using MEGA 6.041. The composition of skew was measured with the following formula: AT skew = (A − T)/(A + T) and GC skew = (G − C)/(G + C)42. The annotated mitogenome sequence of D. longicornis has been deposited in GenBank with accession number KX345846.
Phylogenetic analyses
To better resolve molecular phylogeny of Dacini especially between Dacus and Zeugodacus, a total of 21 species of Diptera species were used in phylogenetic analysis, including 19 Tephritidae and two outgroup species from Drosophilidae. Detailed information of these species used in this study were listed in Supplementary Table S1.
Sequences of 13 PCGs, two rRNAs and 22 tRNAs were used in phylogenetic analysis. The MAFFT algorithm in the TranslatorX online platform43 under the L-INS-i strategy was utilized to align 13 PCGs based on codon-based multiple alignments and to toggle back to the nucleotide sequences. Before back-translate to nucleotides, poorly aligned sites were removed from the protein alignment using GBlocks within the TranslatorX with default settings. Muscle algorithm implemented in MEGA 6.041 was performed to align the sequences of two rRNAs, ambiguous positions in the rRNAs alignment were filtered by hand. Quality control of the hand alignments44 was performed by comparing with homologous sequences from previously sequenced tephritid mitogenomes to identify 22 tRNAs. Individual genes were concatenated using SequenceMatrix v1.7.845. Four datasets were set up for phylogenetic analysis: (1) nucleotides of 13 PCGs, two rRNAs and 22 tRNAs (P123R) with 14,586 residues, (2) nucleotides of 13 PCGs (P123) with 11,148 residues, (3) nucleotides of 13 PCGs exclude the third codon sites, two rRNAs and 22 tRNAs (P12R) with 10,870 residues and (4) nucleotides of 13 PCGs exclude the third codon sites (P12) with 7,432 residues.
The optimal partition strategy and substitution models for each partition were selected by PartitionFinder v1.1.146. As the software required a user to pre-define partitions, we created input configuration files with 39/42/26/29 (P123/P123R/P12/P12R) pre-defined partitions of the dataset. The “greedy” algorithm were used along with branch lengths estimated as “unlinked” and Bayesian information criterion (BIC)47,48 to search for the best-fit scheme. The best selected partitioning schemes and models of three datasets for ML and BI analyses were listed in Supplementary Table S3.
We performed Bayesian inference (BI) and maximum likelihood (ML) based on the best-fit partitioning schemes recommended by PartitionFinder (Supplementary Table S3). We used MrBayes 3.2.249 to conduct Bayesian analysis. The datasets were conducted with two simultaneous runs of 2 million generations, each with one cold and three heated chains. Samples were drawn every 1,000 Markov chain Monte Carlo (MCMC) steps, with the first 25% discarded as burn-in. The stationarity was considered to be reached and stopped run when the average standard deviation of split frequencies was below 0.01. The ML analysis was conducted with RAxML 8.0.050 with 1,000 bootstrap replicates and using the rapid bootstrap feature (random seed value 12345)51.
Additional Information
Accession Codes: Dacus longicornis mitochondrial genome is available in GenBank database (accession number: KX345846).
How to cite this article: Jiang, F. et al. The first complete mitochondrial genome of Dacus longicornis (Diptera: Tephritidae) using next-generation sequencing and mitochondrial genome phylogeny of Dacini tribe. Sci. Rep. 6, 36426; doi: 10.1038/srep36426 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
White, I. M. & Elson-Harris, M. M. Fruit Flies of Economic Significance: Their Identification and Bionomics (CAB International, Wallingford, 1992).
Drew, R. A. I. Biogeography and speciation in the Dacini (Diptera: Tephritidae: Dacinae). Bishop Museum Bulletin in Entomology 12, 165–178 (2004).
Drew, R. A. I. & Romig, M. Tropical Fruit Flies of South-East Asia (Tephritidae: Dacinae) (CAB International, Wallingford, 2013).
Prabhakar, C. S., Sood, P. & Mehta, P. K. Pictorial keys for predominant Bactrocera and Dacus fruit flies (Diptera: Tephritidae) of north western Himalaya. Arthropods 1(3), 101–111 (2012).
Khan, M. First record of fruit fly, Dacus longicornis Wiedemann (Diptera: Tephritidae) from Bangladesh. Insect Pest Control Newsletter, No. 72, January 2009).
Krosch, M. N. et al. A molecular phylogeny for the Tribe Dacini (Diptera: Tephritidae): Systematic and biogeographic implications. Mol. Phylogenet. Evol. 64, 513–523 (2012).
Virgilio, M., Jordaens, K., Verwimp, C., White, I. M. & De Meyer, M. Higher phylogeny of frugivorous flies (Diptera, Tephritidae, Dacini): Localised partition conflicts and a novel generic classification. Mol. Phylogenet. Evol. 85, 171–179 (2015).
Yong, H. S. et al. Complete mitochondrial genome of Bactrocera arecae (Insecta: Tephritidae) by next-generation sequencing and molecular phylogeny of Dacini tribe. Sci. Rep. 5, 15155 (2015).
Yong, H. S., Song, S. L., Lim, P. E., Eamsobhana, P. & Suana, I. W. Complete mitochondrial genome of three Bactrocera fruit flies of subgenus Bactrocera (Diptera: Tephritidae) and their phylogenetic implications. PLoS ONE 11(2), e0148201 (2016).
Cameron, S. L. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu. Rev. Entomol. 59, 95–117 (2014).
Li, H. et al. Higher-level phylogeny of paraneopteran insects inferred from mitochondrial genome sequences. Sci. Rep. 5, 8527 (2015).
Song, F. et al. Capturing the phylogeny of Holometabola with mitochondrial genome data and Bayesian Site-Heterogeneous MixtureModels. Genome Biol. Evol. 8(5), 1411–1426 (2016).
Yu, D. J., Xu, L., Nardi, F., Li, J. G. & Zhang, R. J. The complete nucleotide sequence of the mitochondrial genome of the oriental fruit fly, Bactrocera dorsalis (Diptera: Tephritidae). Gene 396, 66–74 (2007).
Schutze, M. K. et al. Synonymization of key pest species within the Bactrocera dorsalis complex (Diptera: Tephritidae): taxonomic changes based on a review of 20 years of integrative morphological, molecular, cytogenetic, behavioural and chemoecological data. Syst. Entomol. 40, 456–471 (2015).
Nardi, F. et al. Domestication of olive fly through a multi-regional host shift to cultivated olives: Comparative dating using complete mitochondrial genomes. Mol. Phylogenet. Evol. 57, 678–686 (2010).
Choudhary, J. S., Naaz, N., Prabhakar, C. S., Rao, M. S. & Das, B. The mitochondrial genome of the peach fruit fly, Bactrocera zonata (Saunders) (Diptera: Tephritidae): Complete DNA sequence, genome organization, and phylogenetic analysis with other tephritids using next generation DNA sequencing. Gene 569(2), 191–202 (2015).
Nardi, F., Carapelli, A., Dallai, R. & Frati, F. The mitochondrial genome of the olive fly Bactrocera oleae: two haplotypes from distant geographical locations. Insect Mol. Biol. 12(6), 605–611 (2003).
Zhang, B., Nardi, F., Hull-Sanders, H., Wan, X. W. & Liu, Y. H. The complete nucleotide sequence of the mitochondrial genome of Bactrocera minax (Diptera: Tephritidae). PLoS ONE 9(6), e100558 (2014).
Wu, P. F. et al. The complete mitochondrial genome of the melon fly Bactrocera cucurbitae (Diptera: Tephritidae). Mitochondr. DNA 24(1), 6–7 (2013).
Zhang, K. J. et al. The complete mitochondrial genome of Bactrocera diaphora (Diptera: Tephritidae). Mitochondr. DNA 10.3109/19401736.2015.1089479 (2015).
Tan, M., Zhang, R., Xiang, C. & Zhou, X. The complete mitochondrial genome of the pumpkin fruit fly, Bactrocera tau (Diptera: Tephritidae). Mitochondr. DNA 27(4), 2502–2503 (2016).
Spanos, L., Koutroumbas, G., Kotsyfakis, M. & Louis, C. The mitochondrial genome of the Mediterranean fruit fly, Ceratitis capitata. Insect Mol. Biol. 9(2), 139–144 (2000).
Peng, Y., Leung, H. C. M., Yiu, S. M. & Chin, F. Y. L. IBDA-UD: a de novo assembler for single-cell and metagenomic sequencing data woth highly uneven depth. Bioinformatics 28, 1420–1428 (2012).
Wei, S. J. et al. New views on strand asymmetry in insect mitochondrial genomes. PLoS ONE 5(9), e12708 (2010).
Li, H. et al. Mitochondrial genomes of two barklice, Psococerastis albimaculata and Longivalvus hyalospilus (Psocoptera: Psocomorpha): contrasting rates in mitochondrial gene rearrangement between major lineages of Psocodea. PLoS ONE 8, e61685 (2013).
Song, F. et al. Rearrangement of mitochondrial tRNA genes in flat bugs (Hemiptera Aradidae). Sci. Rep. 6, 25725 (2016).
Jühling, F. et al. Improved systematic tRNA gene annotation allows new insights into the evolution of mitochondrial tRNA structures and into the mechanisms of mitochondrial genome rearrangements. Nucl. Acids Res. 40(7), 2833–2845 (2012).
White, I. M. Morphological features of the Dacini (Dacinae): their significance to behavior and classification. In: Aluja, M. & Norrbom, A. L. (Eds.) Fruit Flies (Tephritidae): Phylogeny and Evolution of Behavior. CRC Press, Boca Raton, 505–533 (1999).
Smith, P. T., Kambhampati, S. & Armstrong, K. A. Phylogenetic relationships among Bactrocera species (Diptera: Tephritidae) inferred from mitochondrial DNA sequences. Mol. Phylogenet. Evol. 26, 8–17 (2003).
Smith, P. T., Mcpheron, B. A. & Kambhampati, S. Phylogenetic analysis of mitochondrial DNA supports the monophyly of Dacini fruit flies (Diptera: Tephritidae). Ann. Entomol. Soc. Am. 95(6), 658–664 (2002).
Segura, M. D., Callejas, C., Fernández, M. P. & Ochando, M. D. New contributions towards the understanding of the phylogenetic relationships among economically important fruit flies (Diptera: Tephritidae). B. Entomol. Res. 96, 279–288 (2006).
White I. M. Taxonomy of the Dacina (Diptera: Tephritidae) of Africa and the Middle East. Afr. Entomol. Memoir 2, 1–156 (2006).
Drew, R. A. I. & Hancock, D. L. Phylogeny of the Tribe Dacini (Dacinae) based on morphological, distributional, and biological data. In: Aluja, M. & Norrbom, A. L. (Eds.), Fruit Flies (Tephritidae): Phylogeny and Evolution of Behavior. CRC Press, Boca Raton, 491–504 (1999).
Nakahara, S. & Muraji, M. Phylogenetic analyses of Bactrocera fruit flies (Diptera: Tephritidae) based on nucleotide sequences of the mitochondrial. Res. Bull. Pl. Prot. Japan 44, 1–12 (2008).
Zhang, B., Liu Y. H., Wu W. X. & Wang, Z. L. Molecular phylogeny of Bactrocera species (Diptera: Tephritidae: Dacini) inferred from mitochondrial sequences of 16S rDNA and COI sequences. Fla. Entomol. 93(3), 369–377 (2010).
Lohse, M. et al. RobiNA: a user-friendly, integrated software solution for RNA-Seqbased transcriptomics. Nucleic Acids. Res. 40(W1), W622–W627 (2012).
Altschu, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic. Acids. Res. 25, 955–964 (1997).
Laslett, D. & Canbäck, B. ARWEN, a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 24, 172–175 (2008).
Grant, J. R. & Stothard, P. The CGViewServer: a comparative genomics tool for circular genomes. Nucleic. Acids. Res. 36, W181–W184 (2008).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA 6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Perna, N. T. & Kocher, T. D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 41, 353–358 (1995).
Abascal, F., Zardoya, R. & Telford, M. J. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic. Acids. Res. 38, W7–W13 (2010).
Cameron, S. L. How to sequence and annotate insect mitochondrial genomes for systematic and comparative genomics research. Syst Entomol. 39, 400–411 (2014).
Vaidya, G., Lohman, D. J. & Meier, R. SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 27, 171–180 (2010).
Lanfear, R., Calcott, B., Ho, S. Y. W. & Guindon, S. PartitionFinder: Combined selection of partitioning schemes and substitution models for phylogenetic analysis. Mol. Biol. Evol. 29, 1695–1701 (2012).
Miller, K. B., Bergsten, J. & Whiting, M. F. Phylogeny and classification of the tribe Hydaticini (Coleoptera: Dytiscidae): partition choice for Bayesian analysis with multiple nuclear and mitochondrial protein-coding genes. Zool. Scr. 38, 591–615 (2009).
Pons, J., Ribera, I., Bertranpetit, J. & Balke, M. Nucleotide substitution rates for the full set of mitochondrial protein-coding genes in Coleoptera. Mol. Phylogenet. Evol. 56, 796–807 (2010).
Ronquist, F. & Huelsenbeck, J. P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 (2003).
Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analysis with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 (2006).
Stamatakis, A., Hoover, P. & Rougemont, J. A rapid bootstrap algorithm for the RAxML Web servers. Syst. Biol. 57, 758–771 (2008).
Acknowledgements
We thank Yuliang Deng from Xishuangbanna Entry-Exit Inspection and Quarantine Bureau for helping the sample collection. The work is supported by the Basic Scientific Research Foundation of Chinese Academy of Inspection and Quarantine (No. 2016JK003) and National Key Projects for Research and Development (2016YFC1200605).
Author information
Authors and Affiliations
Contributions
S.Z., X.P. and F.J. designed the study; F.J. and L.D. collected samples; Y.Y., H.J., Y.Z. and L.D. performed the molecular work; F.J., X.L. and X.P. analysed the data; and F.J., X.L. and X.P. wrote the manuscript with other authors.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Jiang, F., Pan, X., Li, X. et al. The first complete mitochondrial genome of Dacus longicornis (Diptera: Tephritidae) using next-generation sequencing and mitochondrial genome phylogeny of Dacini tribe. Sci Rep 6, 36426 (2016). https://doi.org/10.1038/srep36426
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep36426
This article is cited by
-
De novo assembly and comparative analysis of the complete mitochondrial genome sequence of the pistachio psyllid, Agonoscena pistaciae (Hemiptera: Aphalaridae)
International Journal of Tropical Insect Science (2021)
-
Mitochondrial genome sequencing and phylogeny of Haemagogus albomaculatus, Haemagogus leucocelaenus, Haemagogus spegazzinii, and Haemagogus tropicalis (Diptera: Culicidae)
Scientific Reports (2020)
-
A conserved motif within cox 2 allows broad detection of economically important fruit flies (Diptera: Tephritidae)
Scientific Reports (2018)
-
Characterization and phylogenetic analysis of the complete mitochondrial genome of the medicinal fungus Laetiporus sulphureus
Scientific Reports (2018)
-
Comparative analyses of the mitochondrial genome of the sheep ked Melophagus ovinus (Diptera: Hippoboscidae) from different geographical origins in China
Parasitology Research (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
