
Abstract
Therapeutic manipulation of the gasotransmitter hydrogen sulfide (H2S) has recently been proposed as a novel targeted anticancer approach. Here we show that human lung adenocarcinoma tissue expresses high levels of hydrogen sulfide (H2S) producing enzymes, namely, cystathionine beta-synthase (CBS), cystathionine gamma lyase (CSE) and 3-mercaptopyruvate sulfurtransferase (3-MST), in comparison to adjacent lung tissue. In cultured lung adenocarcinoma but not in normal lung epithelial cells elevated H2S stimulates mitochondrial DNA repair through sulfhydration of EXOG, which, in turn, promotes mitochondrial DNA repair complex assembly, thereby enhancing mitochondrial DNA repair capacity. In addition, inhibition of H2S-producing enzymes suppresses critical bioenergetics parameters in lung adenocarcinoma cells. Together, inhibition of H2S-producing enzymes sensitize lung adenocarcinoma cells to chemotherapeutic agents via induction of mitochondrial dysfunction as shown in in vitro and in vivo models, suggesting a novel mechanism to overcome tumor chemoresistance.
Similar content being viewed by others


Introduction
H2S is produced in mammalian cells by three distinct enzymes, cystathionine beta-synthase (CBS), cystathionine gamma lyase (CSE) and 3-mercaptopyruvate sulfurtransferase (3-MST); during methionine/transsulfuration pathway1,2,3. Deregulation of either H2S production and/or its downstream actions have been implicated in the pathophysiology of several diseases, including cardiovascular disease, shock, inflammation, diabetes, metabolic syndromes and neurodegeneration4,5,6,7,8,9,10,11. In connection with cancer, we have previously showed a marked increase in the expression of CBS in colorectal cancer cells (compared to the surrounding normal mucosal margin), which was also recapitulated in multiple colon cancer cell lines12. ShRNA-mediated silencing, as well as pharmacological inhibition of CBS caused a significant inhibition of the proliferation of colon cancer cells in vitro and in vivo (in tumor-bearing nude mice). Also, silencing or inhibition of CBS suppressed cellular bioenergetics of the colon cancer cells12. The importance of the CBS/H2S in the promotion of cell proliferation and cellular bioenergetics has subsequently been confirmed in ovarian cancer13 and breast cancer14. H2S generated by overexpressed CSE, has been implicated in melanoma15. In addition a rapidly increasing body of literature implicates the endogenously generated H2S to vascular relaxation and angiogenesis, cell proliferation, mitochondrial function, and cell survival - all important factors in cancer biology16,17,18,19,20,21,22. However, the mechanisms by which intra-tumor H2S maintains cancer cell viability are incompletely understood. Here we investigated unique roles of H2S in maintenance of the critical mitochondrial functions involved in chemoresistance of lung adenocarcinoma cells.
Results
Lung adenocarcinoma tumors and cultured cells express high level of H2S-producing enzymes
We compared human lung adenocarcinoma samples to matched adjacent normal lung tissue and detected significantly higher protein levels of all three known H2S-producing enzymes, namely, CBS, CSE and 3-MST (Figs 1A and S1A,B). This was associated with an increased capacity of these tissues to produce H2S, as detected by the H2S-specific fluorescent probe, 7-azido-4-methylcoumarin (AzMC) (Fig. 1B). Increased expression of the various H2S-generating enzymes, and increased H2S production was also noted in multiple cultured lung adenocarcinoma cell lines (A549, H522 and H1944), while lung epithelial cells derived from non-malignant tissue (BEAS 2B) showed significantly lower levels of H2S production (Figs 1C,D and S1B). H2S production was inhibited by aminooxyacetic acid (AOAA), a pharmacological inhibitor of CBS and CSE23 (Fig. 1D). The high levels of endogenous H2S in lung adenocarcinoma cells was further confirmed by fluorescent live cell imaging (Fig. 1E). As expected from the roles of CBS and CSE in the transsulfuration pathway, AOAA induced the accumulation of serine and homocysteine and decreased cystathionine levels in AOAA-treated A549 cells (Fig. S1C). The remainder of H2S production is likely attributed to 3-MST activity (Fig. 1D). Together, these data revealed the high expression of H2S-generating enzymes, and increased production of H2S in lung adenocarcinoma when compared to normal lung tissue and epithelial cells.

Lung adenocarcinoma cells generate enhanced levels of H2S.
(A) The relative protein level of CBS, CSE and 3-MST and (B) production of H2S in human lung adenocarcinoma tumors (T, n = 20) and normal adjacent lung tissues (N, set as 1; n = 20) measured by Western blot (WB) analysis and using H2S-specific fluorescent probe (AzMC). (C) The relative protein level of CBS, CSE and 3-MST analyzed by WB and (D) production of H2S generated by extracts of normal lung epithelial (BEAS 2B, set as 1) and lung adenocarcinoma (A549, H522, H1944) cells in the absence or present of CBS/CSE inhibitor, AOAA (100 μM). (E) The endogenous levels of H2S in BEAS 2B and H1944 cells stained with AzMC. Data are mean ± S.E.M. of at least three independent WB analysis and H2S detection. Representative images of three independent experiments are shown *P < 0.05, **P < 0.01 (based on two-tailed Student’s t-test for pairwise comparison).
Repair of the mitochondrial DNA is regulated by H2S
Mitochondrial DNA (mtDNA) encodes an essential set of proteins to maintain oxidative phosphorylation24. Although, somatic mutations in mtDNA in various cancers have been suggested to play a role in a selective advantage during oncogenesis25,26,27, a recent study showed that cancer cells with severe mtDNA damage lost tumorigenic potential because of a failure of respiratory functions28. Here we compared the DNA integrity in human lung adenocarcinomas with the adjacent lung tissue by the long amplicon PCR assay (LA-qPCR)29. Significantly higher integrity of the mtDNA (less DNA breaks) was detected in human lung adenocarcinoma tumors, when compared to matching lung adjacent tissue (Figs 2A and S2A) with no difference in nuclear DNA (Figs 2B and S2B). Higher number of the mtDNA copies was also detected in adenocarcinoma samples (Fig. S2C). Together, we detected more copies of mtDNA in adenocarcinoma tumors with much better preserve mtDNA integrity (less DNA breaks). However, there were more DNA breaks in the mtDNA but less in the nuclear DNA in cultured lung adenocarcinoma cells (A549 and H1944), when compared to normal BEAS 2B cells (Fig. 2C). To solve this discrepancies, we evaluated the extent of intrinsic reactive oxygen species (ROS) production by measuring the level of mt-specific superoxide using MitoSOX; higher ROS signal intensity was observed in A549 and H1944 cells in comparison to BEAS 2B cells (Fig. 2D). This was further supported by observed higher proton leak, a by-product of oxidative phosphorylation, in A549 and H1944, when compared to BEAS 2B cells (Fig. 2E). To assess the role of enhanced endogenous H2S biosynthesis on DNA integrity, lung cancer-derived and non-tumor derived epithelial cells were treated with increasing concentration of glucose oxidase (GOx; in the presence of glucose in culture media GOx generates low, continuous levels of hydrogen peroxide). The integrity of the mtDNA was better preserved in GOx-treated A549 and H1944 cells when compared to BEAS 2B (Fig. 2F); nuclear DNA in these conditions was largely unaffected (Fig. S2D). We have recently reported that a mitochondria-targeted H2S donor (AP39) protects mtDNA integrity in endothelial cells subjected to oxidative stress16,30. To test whether endogenous H2S production stimulates mtDNA repair in lung adenocarcinoma cells, we monitored the effect of inhibition of CBS/CSE on the integrity of mtDNA and nuclear DNA. The results show that after the pharmacological blockade of tumor H2S production, a time-dependent accumulation of DNA damage in the mtDNA (but not in the nuclear DNA) develops in A549 cells (but not in BEAS 2B cells) (Figs 2G and S2E). In addition, inhibition of H2S biosynthesis sensitized A549 cells to oxidative mtDNA damage (Fig. 2H). SiRNA-mediated depletion of CSE, CBS, and 3-MST or treatment with CSE-specific inhibitor, D,L-propargylglycine (PAG)23, also reduced repair rate of the mtDNA (Figs 2I–K and S2F,G). The oxidative mtDNA damage was prevented by supplementation of A549 cells with mt-specific H2S donor, AP39 (Fig. S2H,I). To rule out possible effect of accumulation of serine and homocysteine in cells treated with AOAA on DNA integrity (Fig. S1C), we incubated A549 cells with serine and homocysteine, and measured DNA integrity after 24 h; no significant changes were detected for both genomes (Fig. S2J). Together, our data show that repair of the mtDNA is enhanced by H2S.

H2S mediates enhanced mtDNA integrity in lung adenocarcinoma cells.
(A,B) Comparison of the mitochondrial and nuclear DNA integrity in human lung adenocarcinoma tumors (T, n = 20) and normal adjacent lung tissue (N, set as 100%; n = 20). (C) Mitochondrial and nuclear DNA integrity in cultured lung epithelial cells (DNA integrity in normal BEAS 2B cells was set as 100%). (D,E) Relative level of oxidative stress measured by MitoSOX Red staining and proton leak-linked to oxygen consumption rate (OCR) profile. (F) Integrity of the mtDNA after 1 h challenge with increasing concentration of glucose oxidase (GOx) (DNA integrity in untreated cells was set as 100%). (G) MtDNA integrity in cells treated with 1 mM AOAA, an inhibitor of CBS and CSE. (H) MtDNA integrity in A549 cells treated with AOAA (1 mM) and GOx for 1 h. (I–K) Restoration of the mtDNA integrity in A549 cells transiently depleted from CBS, CSE or 3-MST (DNA integrity measured immediately after GOx treatment was set as 1). (L) Binary interaction between DNA repair enzymes in the presence or absence of AOAA (300 μM) measured by proximity ligation assay (PLA). Data are mean ± S.E.M of at least three independent analysis of DNA integrity run in technical triplicates. Representative images of three independent experiments are shown *P < 0.05, **P < 0.01 (based on two-tailed Student’s t-test for pairwise comparison).
H2S enhance formation of mitochondrial DNA repair complex
Oxidative stress enhances the formation of DNA repair proficient complexes in the mitochondria31. Here, we monitored the binary interactions of the mt-specific DNA repair enzymes EXOG and DNA Polymerase gamma (PolG) and, for comparison, two selected nuclear-specific DNA repair enzymes (FEN1 and PolB)32,33. The interaction between EXOG with APE1, PolG or Lig3, but not between nuclear-specific DNA repair enzymes FEN1 and PolB, was markedly reduced by H2S biosynthesis inhibition in lung adenocarcinoma (but not in normal epithelial cells) (Fig. 2L). To provide direct evidence of the importance of H2S in observed integrations between mt-specific DNA repair enzymes; the interaction between EXOG with APE1 or Lig3 was restored by pharmacological replacement of mitochondrial H2S in A549 cells treated with AOAA (Fig. S2K). Using mass spectrometry we identified Cys 76 (C76) of EXOG to be post-transnationally modified (sulfhydrated) by oxidized metabolites of H2S (Fig. S3A,B). Next, we accessed the interaction of WT EXOG or mutant EXOG (C76A) with APE1, using a co-immunoprecipitation (IP) approach. These studies showed a detectable interaction of WT EXOG with APE1 that was enhanced by treatment with the H2S donor, NaHS. In contrast, the interaction of mutant EXOG with APE1 was significantly lower and insensitive to regulation by NaHS (Fig. S3C). Together, our data provide evidences about critical role of H2S in formation of mtDNA-specific repair complexes in lung adenocarcinoma cells.
H2S maintain cellular bioenergetics in lung adenocarcinoma cells
Since mtDNA integrity is essential to maintain cellular bioenergetics34, we have evaluated the bioenergetic function of cultured lung cells. As expected, higher basal and maximal mitochondrial respiration was detected in A549 and H1944 cells, when compared to BEAS 2B cells (Fig. 3A,B). Moreover, inhibition of H2S production reduced all major bioenergetic parameters in A549 cells; these effects were reversed by AP39 (Fig. 3C,D). Importantly, BEAS 2B cell bioenergetics were insensitive to inhibitors of H2S production (Fig. 3E). No changes in bioenergetics parameters were detected in A549 cells treated with various concentration of serine and homocysteine (Fig. S3D). From these data, we conclude that lung adenocarcinoma cells are protected from mtDNA damaging agents (e.g., ROS) through the H2S-dependent enhancement of mtDNA repair mechanisms, resulting in cancer cell-specific improvements in mitochondrial electron transport and cellular ATP generation.

H2S is critical for maintaining bioenergetics of lung adenocarcinoma cells.
(A) Changes in profiles of oxygen consumption rate (OCR) of BEAS 2B, A549 and H1944 cells (oligo; oligomycin, an inhibitor of ATP synthase; FCCP; carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone, mitochondrial oxidative phosphorylation uncoupler; AA, antimycin A, ROT, rotenone, inhibitors of complex III and I). (B) Comparison of cellular bioenergetics parameters of BEAS 2B, A549 and H1944 cells based on OCR. (C,D) Time dependent changes in cellular bioenergetics parameters in A549 and BEAS 2B in response to 300 μM AOAA treatment based on OCR. (E) Changes in cellular bioenergetics parameters in A549 treated with 300 μM AOAA, 100 nM AP39 or both for 5 h based on OCR. Data are mean ± S.E.M of at least three independent experiments. *P < 0.05, **P < 0.01 (based on two-tailed Student’s t-test for pairwise comparison).
Inhibition of H2S-producing enzymes sensitize lung adenocarcinoma cells to chemotherapeutic drugs in vitro and in tumor bearing mouse model
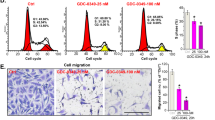
EXOG is essential for mtDNA repair; if its activity is blocked, apoptosis develops32,35, strongly indicates that accumulation of DNA breaks in mtDNA alone induce cell death. We tested the effect of H2S biosynthesis inhibition on the viability of lung cancer cells; A549 and H1944 cells in comparison to BEAS 2B, were significantly less resistant to increasing concentration of AOAA as measured with colony formation assay (Fig. 4A), suggesting essentiality of H2S in survival of lung adenocarcinoma cells. Also proliferation of particularly A549 cells was reduced at 48 h in the presence of AOAA (Fig. 4B). We hypothesized that inhibition of H2S-generating enzymes, would sensitize lung adenocarcinoma cells to chemotherapeutic drugs. Cellular viability determined by the release of LDH (due to necrotic cell death) showed that although AOAA-treatment alone did not result in decreased viability of either BEAS 2B and A549 cells, in combination with camptothecin (CPT, DNA topoisomerase I inhibitor) a concentration-dependent decrease in viability of A549 (but not BEAS 2B) cells was observed (Figs 4C and S4A). Similarly, treatment with another chemotherapeutic drug, oxaliplatin, in combination with AOAA, resulted in decreased cellular viability of A549 (but not BEAS 2B) cells (Fig. S4B). The combination of AOAA and CPT also resulted in time-dependent, synergistic increase in apoptosis (Fig. 4D), mtDNA damage (Fig. 4E) and decrease oxygen consumption linked with ATP synthesis (Fig. 4F) in A549 cells. In order to provide additional evidence that reduced level of endogenous H2S sensitize A549 cells to chemotherapeutic drugs, silencing of CBS also increased LDH release in CPT-treated A549 cells in concentration dependent manner (Fig. 4G).

Inhibition of H2S production sensitize cultured lung adenocarcinoma cells to chemotherapeutic drugs.
(A) Viability of BEAS 2B, A549 and H1944 cells to inhibitor of CSE and CBS (AOAA). (B) Relative proliferation expressed as % of untreated cells after 48 h of 200 μM of AOAA treatment of cultured lung adenocarcinoma cells. (C) Necrotic cell death determined by LDH release of A549 cells treated with AOAA, CPT and AOAA/CPT for 24 h (vehicle treated cells were set as 1). (D) Time dependent changes in amount of apoptotic cells measured by Annexin V staining of A549 cells treated with 300 μM AOAA, 300 nM CPT or combination of both. (E) MtDNA integrity of A549 cells treated for 24 h with 300 μM AOAA, 300 nM CPT or combination of both. (F) Oxygen consumption of A549 cells treated for 24 h with 300 μM AOAA, 300 nM CPT or combination of both. (G) Necrotic cell death determined by LDH release of A549 cells transiently transfected with control or CBS-specific siRNA treated with CPT for 24 h (vehicle treated cells were set as 1). Data are mean ± S.E.M of at least three independent analysis of colony formation assay, LDH release, Annexin V staining and of DNA integrity run in technical triplicates. Representative images of three independent experiments are shown *P < 0.05, **P < 0.01 (based on two-tailed Student’s t-test for pairwise comparison).
Finally, we investigated the effect of inhibition of H2S biosynthesis in a mouse model of A549 xenografts. Although, tumor volume was reduced in mice treated daily with 9 mg/kg AOAA; its growth resumed when AOAA treatment discontinued and animals were switched to vehicle treatment (Fig. S5A). Next, we reduced the dose of AOAA to 3 mg/kg and investigated the effect of combined treatment with irinotecan. We noted the greatest reduction of tumor growth-rate in animals treated with the combination of AOAA and CPT (Figs 5A,B and S5B,C). Similarly, the most pronounced degree of DNA damage was seen in tumors of mice treated with the AOAA/irinotecan combination (Fig. 5C,D). Comprehensive diagnostic blood profile analysis showed no signs of organ injury in mice treated with the AOAA/irinotecan combination (Table S1).

Combination of inhibition of H2S-producing enzymes and chemotherapeutic drugs accelerate decreasing of tumor volume in mouse model.
(A,B) Tumor volumes of mice injected SQ with A549 cells treated with vehicle (PBS), AOAA (3 mg/kg), irinotecan (3 mg/kg) and both (n = 8 for each group). (C,D) DNA integrity measured in tumors of the same animals. (E) Schematic graph of summary of obtained results. Data are mean ± S.E.M. DNA integrity was run in technical duplicates. *P < 0.05 (based on two-tailed Student’s t-test for pairwise comparison).
Discussion
The above data show that lung adenocarcinoma cells upregulate their H2S biosynthesis and use it: (i) to increase the repair capacity of the mtDNA and (ii) to support cellular bioenergetic function; both critical for maintenance of the mitochondrial and cellular homeostasis. Consequently, pharmacological inhibition of H2S production in tumor cells induces mitochondrial dysfunction by reduction of DNA repair capacity in the mitochondria, which sensitizes lung adenocarcinoma cells to chemotherapeutic drugs (Fig. 5E). This approach may form the basis of novel combination approaches in the experimental therapy of lung adenocarcinoma and increase therapeutic index of chemotherapeutic drugs, and reduce side effects.
Mammalian cell has hundreds of mitochondria, each containing approximately 10 copies of 16.5 kb plasmid-like mitochondrial DNA (mtDNA)24. The mutation rate of human mtDNA is 20- to 100-fold higher that in the nuclear DNA36,37. One of the reasons is constant challenge of the mtDNA by oxidants generated as a by-product of mitochondrial oxidative phosphorylation. Also, the repair mechanisms of the mtDNA, in contrast to the nuclear DNA, are limited, due to lack of nucleotide excision repair and lack of back-up for the sole DNA polymerase gamma (PolG) and DNA ligase 3; both essential for the correct repair and replication of the mtDNA33. This means that the mtDNA is more susceptible to genotoxic agents than the nuclear DNA, as demonstrated by our group and other investigators in various cell types16,35,38,39. It is generally accepted that DNA base excision/single strand break repair (BER/SSBR) pathway is the predominant DNA repair mechanism within the mitochondria33. There are only two mitochondria-specific BER/SSBR enzymes identified so far: EXOG and PolG32,33,34, the remaining enzymes are shared by the mitochondria and the nucleus.
Mitochondria research, including studies of maintenance of the mtDNA integrity were for a long time neglected in cancer field since upregulation of glycolysis as a major source of energy was proposed, known as a Warburg effect. However, the functional consequence of decreased mtDNA integrity (accumulation of mtDNA damage) for the survival of the cancer cell has been recently re-defined by several studies. In contrast to the nuclear DNA, where DNA instability is one of the hallmark of cancer cells (increase rate of mutations is used by the cancer cell to evade the body’s antitumor mechanisms, and to develop resistance against anticancer agents); with respect to mtDNA, the priority of the cancer cell is to maintain its integrity (i.e. to reduce the amount of DNA breaks)25,26. It has been recently shown that intact mtDNA is critical for tumorigenic potential28. When the integrity of the mtDNA becomes impaired, mitochondrial cell death pathways become activated, the regulation of mitochondrial protein expression becomes impaired, the cellular bioenergetic homeostasis becomes impaired, and the cell’s viability and survivability is suppressed (reviewed in ref. 40. In fact, mtDNA is so critical for the cancer cell, that if the mtDNA is depleted, the cancer cells develop mechanisms to take it up from the host cell, in order to replenish their cellular pools28. Some authors even call the mtDNA of the cancer cell “a genetic sanctuary”41. Together, various recent studies indicate the importance of the preservation of the mtDNA integrity (meaning proper mtDNA repair); yet the molecular mechanisms of how mtDNA integrity is preserved in cancer cells are poorly defined. Here we are showing for the first time that reduction of the H2S-production in lung adenocarcinoma but not in normal lung epithelial cells results in accumulation of the DNA repair intermediates in the mitochondrial but not nuclear DNA. There are only a limited number of published data connecting H2S with mtDNA transactions. Using a recently developed mitochondrial-specific H2S donor, AP3930, we have shown previously that mitochondria-specific H2S delivery significantly reduces amount of mtDNA damaged induced by oxidative stress in cultured endothelial cells, suggesting a stimulatory role of H2S in process of the mtDNA repair16. Another report demonstrated that cells deficient in H2S production (due to CSE deficiency), have reduced number of mtDNA copies42. Thus - although a correlation between H2S and mtDNA damage/repair has been implicated - the molecular mechanisms of how H2S regulates the integrity of the mtDNA, and how these processes are important for the tumorigenic potential of cancer cells is currently unknown. Here we showed for the first time that H2S directly regulates repair capacity of the mitochondria. Our data indicate that H2S supports formation/stabilize mitochondrial DNA repair complexes and thus increases DNA repair capacity within mitochondria. We have shown earlier an active process of formation of the mitochondrial DNA repair complexes in response to oxidative stress31. Now we identified critical cysteine residue on EXOG modify by H2S which in turn regulates binary interaction with APE1. Both enzymes are critical in DNA end-processing step during BER/SSBR. We showed previously that accumulation of unrepaired DNA intermediates, mostly single strand breaks in the mtDNA induce cell death32.
In light of recently emerging data, the role of H2S in the regulation of cellular bioenergetics has been also completely re-evaluated. In contrast to the earlier observations demonstrating the toxicity of exogenously added H2S as an inhibitor of mitochondrial cytochrome c oxidase (complex IV), recent data indicate that low, physiological concentrations of H2S (produced by endogenous, enzymatic sources) has a stimulatory effect on cellular bioenergetics. Physiologically, H2S acts as an electron donor and a stimulator of bioenergetics43. Hepatoma cells with depleted 3-MST (a constitutive mitochondrial H2S-generating enzyme) exhibit reduced bioenergetics parameters43. We have shown that nanomolar concentrations of the mitochondrial H2S donor, AP39, stimulate cellular bioenergetics in cultured endothelial cells16. These data are consistent with a biphasic biological H2S dose-response, where many effects of H2S at low concentration are beneficial, while at higher concentrations, toxicity develops [reviewed in refs 44 and 45]. However, the role of H2S in regulation of bioenergetics in lung cancer cells has not yet been investigated. Our data suggest that enhanced amounts of H2S in lung adenocarcinoma cells is also used to maintain cellular bioenergetics.
The enhanced H2S biosynthesis demonstrated in the current report selectively supports vital functions of lung adenocarcinoma cells, compared to normal lung epithelial cells. We hypothesized that pharmacological inhibition of H2S-generating enzymes or reducing intracellular level of H2S by specific scavenger would induce mitochondrial dysfunction which in turn would sensitize lung adenocarcinoma cells towards chemotherapeutic drugs. Our in vitro and in vivo data confirm that this is a case. Combination of CBS/CSE inhibitor with commonly used chemotherapeutic agents campothecin or oxaliplatin greatly increases cell death of cultured cells and reduced tumor volume in a mouse model. Aminooxyacetic acid (AOAA) - commonly used inhibitor of CBS and CSE - inhibits multiple PLP-dependent enzymes including several oxidoreductases, transferases, hydrolases or isomerases46. Therefore, there is a great need to develop novel, more specific inhibitors for H2S-producing enzymes, particularly for CSE and CBS, in context of lung adenocarcinoma. Another approach may be to develop mitochondria-specific H2S scavengers in order to reduce mitochondria DNA repair capacity and oxidative phosphorylation to induce mitochondrial dysfunction in cancer cells and to sensitize specifically cancer cells to chemotherapeutic drugs.
Experimental Procedures
Cell culture
The normal lung epithelial cells BEAS 2B (ATCC CRL-9609; Manassas, VA), and lung adenocarcinoma cells A549 (ATCC CCL-185), NCI-H522 (ATCC CRL-5810) and NCI-H1944 (ATCC CRL-5907), were maintained in RPMI 1640 media supplemented with 10% fetal bovine serum and 100 IU/ml penicillin and 100 μg/ml streptomycin at 37 °C in 5% CO2.
Human lung adenocarcinoma tumors and normal adjacent lung tissue
20 pairs of lung adenocarcinoma tumors and normal adjacent lung tissue was obtained from Lung Cancer Biospecimen Resource Network, University of Virginia Health System, Charlottesville, VA. The experimental protocol was approved by the UTMB Institutional Review Board (IRB), and this study was conducted in compliance with ethical and safe research practices involving human tissues.
Animal xenograft model of human lung cancer
All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Texas Medical Branch and were carried out in accordance to the approved guidelines. Athymic nude Balb/c male and female mice (8–10 weeks) purchased from Charles Rivers (Wilmington, MA) were injected subcutaneously in either the right or left dorsum with 5–8 × 106 A549 cells as described12. 25 or 28 days later, mice were randomized and subcutaneous (SQ) injection of phosphate buffered saline (PBS) as a vehicle, AOAA (3 or 9 mg/kg/day), irinotecan (3 mg/kg) or combination of AOAA/irinotecan (3 mg/kg and 3 mg/kg). Tumor dimensions were measured 2 times per week transcutaneously using a caliper and tumor volume was calculated using the formula π/6 (HxWxL).
Detection of H2S and mitochondrial oxidant production in live cells
40,000 of BEAS 2B, A549 and H1944 cells were seeded in Lab-Tek II chamber coverglass system (Nalgen Nunc International) and incubated at 37 °C and 5% CO2 humidified incubator overnight. The cells were loaded with 10 μM of 7-azido-4-methylcoumarin (AzMC, Sigma) for 2 h to detect endogenous H2S or with 5 μM of MitoSOX Red dye (Invitrogen) for 10 min to detect mitochondrial superoxide production. Cells were washed three times with PBS and dye’s specific fluorescence was visualized using Nikon eclipse 80i inverted microscope with Photometric CoolSNAP HQ2 camera and NIS-Elements BR 3.10 software.
Measurement of H2S production in cell homogenates
The cultured BEAS 2B, A549, H522 and H1944 cells were washed twice with PBS and homogenized in NP40 lysing buffer (1% NP40, 150 mM NaCl, 50 mM Tris-Cl, pH 8.0) on ice for 30 minutes. The protein concentration determined with Lowry reagent (Bio-Rad) using BSA as a standard of the extract was adjusted to 10 mg/ml. Cell homogenates (20 μl) were incubated in 96-well plates with 5 μM PLP and 2.5 mM of each L-cysteine and homocysteine, and H2S production was monitored following fluorescent signal of AzMC (10 μM) at 450 nM for 2 h.
Cell proliferation
For assessment of cell proliferation, the xCELLigence system (Roche) was used. Briefly, 8,000 of BEAS 2B, A549 or H1944 cells per well were seeded on a E-plate 96, a specially designed 96-well microtiter plate containing interdigitated microelectrodes to non-invasively monitor the cell proliferation by measuring the relative change in the electrical impedance of the cell monolayer. 24 h after initial seeding, cells were treated with 300 μM of AOAA and proliferation monitored for 48 hours.
Measurement of mitochondrial and nuclear DNA integrity
To measure DNA integrity, we used gene-specific semi-quantitative PCR assays using LongAmp Taq DNA Polymerase (New England BioLabs) as described29. Briefly, total DNA was isolated using DNase Blood and Tissue Kit (QIAGEN). Damage to nuclear DNA was estimated by quantification of the PCR amplification of the 10 kb nuclear-specific DNA fragment using PicoGreen fluorescent dye to detect double-stranded DNA (Quant-iT™ PicoGreen, Molecular Probe). Damage to the mitochondrial DNA was estimated by quantification of the PCR amplification of the 8.9 kb mitochondrial-specific DNA fragment using PicoGreen staining. Obtained data was normalized by the PCR amplification of 211 bp mitochondrial genome-specific fragment for correction of the multiple copies of the mitochondrial genome. Preliminary assays were carried out to ensure the linearity of PCR amplification with respect to the number of cycles and DNA concentration. Note, decreased integrity of the DNA mean increasing amount of DNA breaks detected in amplified region.
Proximity Ligation Assay (PLA)
PLA was performed using Duolink In situ Kit (SIGMA) according to the manufacturer’s recommendations with following a pair of primary antibodies raised in two different species: rabbit (r) EXOG (SIGMA), mouse (m) APE1 (Novus Biologicals), m PolG, m Lig3, r FEN1, m PolB (all from GeneTex). Briefly, cells were fixed and permeabilized with 4% paraformaldehyde, 0.2% Tween-20 in PBS for 20 min at room temperature. After blocking for 1 h at room temperature cells were probed with primary antibodies (at 1:200 dilution) overnight at 4 °C. Signal detection and amplification was performed per manufacturer’s instructions. Images were captured using Nikon eclipse 80i inverted fluorescent microscope with Photometric CoolSNAP HQ2 camera and NIS-Elements BR 3.10 software.
Bioenergetic analysis in cultured cells
The XF24 Extracellular Flux Analyzer (Seahorse Bioscience, Agilent Technologies) was used to measure bioenergetic function. Oxygen consumption rate (OCR) after oligomycin (1.5 μg/ml) was used to assess ATP production rate, OCR after FCCP (0.5 μM) to assess maximal mitochondrial respiratory capacity, OCR after antimycin A (2 μg/ml) and rotenone (2 μM) were used to inhibit the flux of electrons through complex III and I, to detect residual non-mitochondrial oxygen consumption rate, which is considered to be due to cytosolic oxidase enzymes. OCR linked with proton leak was quantified by substation of OCR after antimycin/rotenone from OCR after FCCP. OCR was normalized by μg of protein extract in each well.
LDH assay
Lactate dehydrogenase (LDH) release was used as a cytotoxicity assay, and as a secondary measurement for determination of necrotic cell death. The changes in absorbance were read kinetically at 492 nm for 15 min on a monochromator-based reader (Powerwave HT, Biotek) at 37 °C. LDH activity values are shown as Vmax for kinetic assays in mOD/min.
Colony formation assay
200 cells of BEAS 2B, A549 or H1944 cells were seeded on 6 well plates. After 24 h of incubation at 37 °C in 5% CO2 media was change to containing various concentration of AOAA. Colonies were counted after 10 days using 1% crystal violet in 50% methanol. Each experiment was repeated 3 times with technical triplicates.
Determination of apoptotic cell death
Amount of Annexin V positive cells was determined using PE Annexin V Apoptosis Detection Kit (BD Biosciences) according to manufacture’s recommendation. Briefly, A549 cells were treated with 300 μM AOAA, 300 nM CPT or combination of both for 1, 3, 9 and 24 h. Number of Annexin V positive cells was determined using EasyCyte™ Plus Flow Cytometry System (Guava).
Transient depletion with siRNA
A549 cells were transfected with 40 nM siRNA specific for CBS and 3-MST (#7882, #s194616, Ambion, Life Technologies) and 80 nM siRNA for CSE (#10674, Ambion, Life Technologies) or control siRNA using Lipofectamine 2000 (Invitrogen) per the manufacturer’s protocol. To optimize the siRNA concentration for maximal target depletion, preliminary transfections were carried out with 10–100 nM for each siRNA. The level of depletion was calculated by densitometric analysis of Western blots relatively to loading control of three independent experiments. Cells with 80–90% depletion measured 48 h post-transfection were used in subsequent experiments.
Statistical analysis
Data are shown as means ± standard error of the mean (SEM). Statistical analysis was performed with Prism G, based on two-tailed Student’s t-test for pairwise comparison; *p < 0.05 and **p < 0.01 represent statistically significant differences from control.
Additional Information
How to cite this article: Szczesny, B. et al. Inhibition of hydrogen sulfide biosynthesis sensitizes lung adenocarcinoma to chemotherapeutic drugs by inhibiting mitochondrial DNA repair and suppressing cellular bioenergetics. Sci. Rep. 6, 36125; doi: 10.1038/srep36125 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Szabo, C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov 6, 917–935 (2007).
Kimura, H. Hydrogen sulfide: its production, release and functions. Amino acids 41, 113–121 (2011).
Kabil, O. & Banerjee, R. Enzymology of H2S biogenesis, decay and signaling. Antioxid Redox Signal 20, 770–782 (2014).
Meng, G., Ma, Y., Xie, L., Ferro, A. & Ji, Y. Emerging role of hydrogen sulfide in hypertension and related cardiovascular diseases. Br J Pharmacol 172, 5501–5511 (2014).
di Masi, A. & Ascenzi, P. H2S: a “double face” molecule in health and disease. Bio Factors 39, 186–196 (2013).
Polhemus, D. J. & Lefer, D. J. Emergence of hydrogen sulfide as an endogenous gaseous signaling molecule in cardiovascular disease. Circ Res 114, 730–737 (2014).
Collin, M. & Thiemermann, C. Hydrogen sulfide and sulfite: novel mediators in the pathophysiology of shock and inflammation. Shock 24, 595–606 (2005).
Szabo, C. Roles of hydrogen sulfide in the pathogenesis of diabetes mellitus and its complications. Antioxid Redox Signal 17, 68–80 (2012).
Desai, K. M., Chang, T., Untereiner, A. & Wu, L. Hydrogen sulfide and the metabolic syndrome. Expert Rev Clin Pharmacol 4, 63–73 (2011).
Gong, Q. H. et al. A new hope for neurodegeneration: possible role of hydrogen sulfide. J Alzheimers Dis 24, 173–182 (2011).
Zhang, X. & Bian, J. S. Hydrogen sulfide: a neuromodulator and neuroprotectant in the central nervous system. ACS Chem Neurosci 5, 876–883 (2014).
Szabo, C. et al. Tumor-derived hydrogen sulfide, produced by cystathionine-β-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc Natl Acad Sci USA 110, 12474–12479 (2013).
Bhattacharyya, S. et al. Cystathionine beta-synthase (CBS) contributes to advanced ovarian cancer progression and drug resistance. PLoS one 8, e79167 (2013).
Sen, N. et al. Role of cystathionine β-synthase in human breast cancer. Free Radic Biol Med. 86, 228–38 (2015).
Panza, E. et al. Role of the cystathionine γ lyase/hydrogen sulfide pathway in human melanoma progression. Pigment Cell Melanoma Res 28, 61–72 (2015).
Szczesny, B. et al. AP39, a novel mitochondria-targeted hydrogen sulfide donor, stimulates cellular bioenergetics, exerts cytoprotective effects and protects against the loss of mitochondrial DNA integrity in oxidatively stressed endothelial cells in vitro. Nitric oxide 41, 120–130 (2014).
Baskar, R. & Bian, J. Hydrogen sulfide gas has cell growth regulatory role. Eur J Pharmacol 656, 5–9 (2011).
Módis, K. et al. Effect of S-adenosyl-L-methionine (SAM), an allosteric activator of cystathionine-β-synthase (CBS) on colorectal cancer cell proliferation and bioenergetics in vitro. Nitric oxide 41, 146–156 (2014).
Coletta, C. et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc Natl Acad Sci USA 109, 9161–9166 (2012).
Szabo, C. & Papapetropoulos, A. Hydrogen sulphide and angiogenesis: mechanisms and applications. Br J Pharmacol 164, 853–865 (2011).
Wang, M. J. et al. The hydrogen sulfide donor NaHS promotes angiogenesis in a rat model of hind limb ischemia. Antioxid Redox Signal 12, 1065–1077 (2010).
Szabo, C. Gasotransmitters in cancer: from pathophysiology to experimental therapy. Nat Rev Drug Discov 15, 185–203 (2016).
Asimakopoulou, A. et al. Selectivity of commonly used pharmacological inhibitors for cystathionine β synthase (CBS) and cystathionine γ lyase (CSE). Br J Pharmacol 169, 922–932 (2013).
Anderson, S. et al. Sequence and organization of the human mitochondrial genome. Nature 290, 457–465 (1981).
Maybury, B. D. Mitochondrial DNA damage is uncommon in cancer but can promote aggressive behaviour. Anticancer Res 33, 3543–3552 (2013).
Wallace, D. C. Mitochondria and cancer. Nat Rev Cancer 12, 685–698 (2012).
Larman, T. C. et al. Spectrum of somatic mitochondrial mutations in five cancers. Proc Natl Acad Sci USA 109, 14087–14091 (2012).
Tan, A. S. et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab 21, 81–94 (2015).
Ayala-Torres, S., Chen, Y., Svoboda, T., Rosenblatt, J. & Van Houten, B. Analysis of gene-specific DNA damage and repair using quantitative polymerase chain reaction. Methods 22, 135–147 (2000).
La Trionnaire, S. et al. The synthesis and functional evaluation of a mitochondria-targeted hydrogen sulfide donor, (10-oxo-10-(4-(3-thioxo-3H-1,2-dithiol-5-yl)phenoxy) decyl) triphenylphosphonium bromide (AP39). Med Chem Comm 5, 728–736 (2014).
Szczesny, B., Brunyanszki, A., Olah, G., Mitra, S. & Szabo, C. Opposing roles of mitochondrial and nuclear PARP1 in the regulation of mitochondrial and nuclear DNA integrity: implications for the regulation of mitochondrial function. Nucleic Acids Res 42, 13161–13173 (2014).
Tann, A. W. et al. Apoptosis induced by persistent single-strand breaks in mitochondrial genome: critical role of EXOG (5′-EXO/endonuclease) in their repair. J Biol Chem 286, 31975–31983 (2011).
Alexeyev, M., Shokolenko, I., Wilson, G. & LeDoux, S. The maintenance of mitochondrial DNA integrity–critical analysis and update. Cold Spring Harb Perspect Biol 5, a012641 (2013).
Kazak, L., Reyes, A. & Holt, I. J. Minimizing the damage: repair pathways keep mitochondrial DNA intact. Nat Rev Mol Cell Biol 13, 659–671 (2012).
Furda, A. M., Marrangoni, A. M., Lokshin, A. & Van Houten, B. Oxidants and not alkylating agents induce rapid mtDNA loss and mitochondrial dysfunction. DNA Repair (Amst) 11, 684–692 (2012).
Pesole, G., Gissi, C., De Chirico, A. & Saccone, C. Nucleotide substitution rate of mammalian mitochondrial genomes. J Mol Evol 48, 427–434 (1999).
Mandavilli, B. S., Santos, J. H. & Van Houten, B. Mitochondrial DNA repair and aging. Mutat Res 509, 127–151 (2002).
Szczesny, B. et al. Deficiency in repair of the mitochondrial genome sensitizes proliferating myoblasts to oxidative damage. PLoS One 8, e75201 (2013).
Szczesny, B., Tann, A. W. & Mitra, S. Age- and tissue-specific changes in mitochondrial and nuclear DNA base excision repair activity in mice: Susceptibility of skeletal muscles to oxidative injury. Mech Ageing Dev 131, 330–337 (2010).
Van Houten, B., Hunter, S. E. & Meyer, J. N. Mitochondrial DNA damage induced autophagy, cell death, and disease. Front Biosci 21, 42–54 (2016).
Seoane, M. et al. The mitochondrial genome is a “genetic sanctuary” during the oncogenic process. PLoS One 6, e23327 (2011).
Li, S. & Yang, G. Hydrogen Sulfide Maintains Mitochondrial DNA Replication via Demethylation of TFAM. Antioxid Redox Signal 23, 630–642 (2015).
Módis, K., Asimakopoulou, A., Coletta, C., Papapetropoulos, A. & Szabo, C. Oxidative stress suppresses the cellular bioenergetic effect of the 3-mercaptopyruvate sulfurtransferase/hydrogen sulfide pathway. Biochem Biophys Res Commun 433, 401–407 (2013).
Szabo, C. et al. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br J Pharmacol 171, 2099–2122 (2014).
Módis, K. et al. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part II. Pathophysiological and therapeutic aspects. Br J Pharmacol 171, 2123–2146 (2014).
Toney, M. D. Controlling reaction specificity in pyridoxal phosphate enzymes. Biochim Biophys Acta 1814(11), 1407–18 (2011).
Acknowledgements
This work was supported by grants from the National Institutes of Health (R21ES024648), and American Lung Association (RG-348772) to B.S. This work was also supported by the National Institutes of Health (CA17580302), by a grant from the Cancer Research and Prevention Initiative of Texas (CPRIT DP150074) and by a grant from UTMB’s Institute of Translational Science to C.S. and M.R.H. Financial support from The Hungarian National Science Foundation (OTKA; grant No.: K 109843) for P.N. is also acknowledged. P.N. is a János Bolyai Research Scholar of the Hungarian Academy of Sciences. We thank Dr. Matt Whiteman (University of Exeter, UK) for a kind gift of AP39. Human samples were provided by the Lung Cancer Biospecimen Resource Network which is funded by the Congressionally Directed Medical Research Programs of the Department of Defense.
Author information
Authors and Affiliations
Contributions
Conceptualization, Experimental Design, B.S. and C.S.; Methodology, B.S., J.E.W., M.R.H. and C.S.; Formal analysis, B.S. and M.M.; Investigation, B.S., M.M., J.R.Z. and N.D.; Conceptualization Resources, P.N.; Writing – Original Draft, B.S., M.R.H. and C.S.; Funding Acquisition, B.S., M.R.H. and C.S.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Szczesny, B., Marcatti, M., Zatarain, J. et al. Inhibition of hydrogen sulfide biosynthesis sensitizes lung adenocarcinoma to chemotherapeutic drugs by inhibiting mitochondrial DNA repair and suppressing cellular bioenergetics. Sci Rep 6, 36125 (2016). https://doi.org/10.1038/srep36125
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep36125
This article is cited by
-
Pan-inhibition of the three H2S synthesizing enzymes restrains tumor progression and immunosuppression in breast cancer
Cancer Cell International (2024)
-
The interplay of circulating tumor DNA and chromatin modification, therapeutic resistance, and metastasis
Molecular Cancer (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
