
Abstract
The bacterium Microbacterium aurum strain B8.A degrades granular starches, using the multi-domain MaAmyA α-amylase to initiate granule degradation through pore formation. This paper reports the characterization of the M. aurum B8.A MaAmyB enzyme, a second starch-acting enzyme with multiple FNIII and CBM25 domains. MaAmyB was characterized as an α-glucan 1,4-α-maltohexaosidase with the ability to subsequently hydrolyze maltohexaose to maltose through the release of glucose. MaAmyB also displays exo-activity with a double blocked PNPG7 substrate, releasing PNP. In M. aurum B8.A, MaAmyB may contribute to degradation of starch granules by rapidly hydrolyzing the helical and linear starch chains that become exposed after pore formation by MaAmyA. Bioinformatics analysis showed that MaAmyB represents a novel GH13 subfamily, designated GH13_42, currently with 165 members, all in Gram-positive soil dwelling bacteria, mostly Streptomyces. All members have an unusually large catalytic domain (AB-regions), due to three insertions compared to established α-amylases, and an aberrant C-region, which has only 30% identity to established GH13 C-regions. Most GH13_42 members have three N-terminal domains (2 CBM25 and 1 FNIII). This is unusual as starch binding domains are commonly found at the C-termini of α-amylases. The evolution of the multi-domain M. aurum B8.A MaAmyA and MaAmyB enzymes is discussed.
Similar content being viewed by others


Introduction
Many micro-organisms are able to use starch as a carbon and energy source. They generally employ α-amylase enzymes for extracellular hydrolysis of amylose and amylopectin1. α-Amylases represent endo-acting glycoside hydrolases that hydrolyze the (1–4)α-D-glucosidic linkages between glucose residues and rapidly degrade long polymers into shorter oligosaccharides. Degradation into glucose requires the subsequent action of exo-acting enzymes such as α-glucosidase or glucoamylase. Most α-amylases belong to the Glycoside Hydrolase family 13 (GH13) (www.CAZy.org)2. Based on sequence similarity, family GH13 enzymes are currently classified into 41 subfamilies2,3. So far α-amylases have been found in 15 of these subfamilies (GH13_1, 5, 6, 7, 10, 14, 15, 19, 24, 27, 28, 32, 36, 37, 39, 41)4,5. Not all GH13 α-amylases listed in the CAZy database have been classified into one of the currently established subfamilies2,3,4. α-Amylases have a catalytic domain that usually consists of 3 regions. Region-A contains the common (β/α)8 barrel and all the catalytic residues6. Region-B is a loop lacking a clearly defined topology, located between β3 and α3 of region-A, containing calcium binding sites6. Due to the close interactions between regions-A and -B they are usually identified as one (catalytic) domain in domain databases such as the Conserved Domain Database (CDD)7. Region-C is located at the C-terminal end of regions-AB. It consists of β-sheets only, including a Greek key motif4,6,8. Although it is required for amylase activity, the function of region-C is not yet fully understood9,10. In some cases region-C has been linked to raw starch binding11,12.
α-Amylase enzymes acting on raw starch commonly have additional carbohydrate binding modules (CBM) at their C-terminal ends2,13,14. Starch binding domains (SBD) are a subgroup of CBMs which bind to starches14. Only about 10% of all GH13 enzymes contain additional domains such as SBDs. Most do not have more than two of them, although some α-amylases contain several additional domains (6 or more) resulting in large complex enzymes2,15. We recently described MaAmyA, a large α-amylase (148 kDa) with two CBM25 and four FNIII domains, plus a novel CBM74 domain, that is able to form large pores in granular starch (Fig. 1)15,16. MaAmyA has been isolated from Microbacterium aurum B8.A, a granular starch degrading bacterium that was obtained from a potato waste water treatment plant17. Interestingly, the pores created by MaAmyA were all of similar size while the M. aurum culture fluid produced a range of pore sizes16. In addition, when staining for starch degradation activity on polyacrylamide gels, a 130 kD protein band was detected in the M. aurum culture fluid that could not be related to MaAmyA16. A total of 14 GH13 members were annotated in the recently obtained genome sequence of M. aurum B8.A (V. Valk, R. M. van der Kaaij, and L. Dijkhuizen, manuscript in preparation), one of which is located directly downstream of MaAmyA (designated MaAmyB). These findings, together with the reported synergy between α-amylases and glucoamylases which significantly increased the degradation rate of raw starches18,19,20,21, resulted in the suggestion that M. aurum B8.A may employ one or more additional enzymes to assist MaAmyA in raw starch degradation.

Detailed domain organization of MaAmyA and MaAmyB, compared to the general domain organization of GH13_42 members; numbers indicate the first and last aa of the domain or conserved insert.
Domains are indicated as follows: white: signal sequence; red: AB-regions of the catalytic domain; pale yellow, C-region; blue: FNIII domain; green: CBM25 domain; pink: CBM74, a novel CBM domain15; orange yellow: aberrant C-region. Conserved insertions in the AB-regions of GH13_42 (including MaAmyB) are indicated as follows: light blue box: region-B; grey box: region-A insert 1; black box: region-A insert 2.
Here we report the characterization of MaAmyB, a large and multi-domain enzyme with exo-acting starch hydrolyzing activity (Fig. 1). MaAmyB is located directly downstream of MaAmyA in the genome of M. aurum B8.A (V. Valk, R. M. van der Kaaij and L. Dijkhuizen, unpublished data). Unlike the exo-acting glucoamylases, which belong to the GH15 family, MaAmyB belongs to the GH13 family and is the first characterized member of a newly defined GH13 subfamily (GH13_42). The 165 enzyme members of this subfamily share a conserved but aberrant multi-domain organization with additional domains N-terminal of their unusually large catalytic domain.
Results
Gene identification
In previous work we characterized MaAmyA, a 148 kDa raw starch degrading and pore-forming α-amylase (glycoside hydrolase 13, subfamily 32: GH13_32) that was isolated from M. aurum B8.A (Fig. 1)16. The gene encoding MaAmyA was first isolated from a DNA library clone with an 11.6 kb insert. This insert contained a second large but C-terminally incomplete open reading frame (ORF) of 3,810 nt. The full 3,834 nt ORF was originally obtained through genome walking. The sequence was confirmed when the full genome of M. aurum B8.A was obtained (V. Valk, R. M. van der Kaaij and L. Dijkhuizen, manuscript in preparation). For this study the gene was obtained from genomic DNA of M. aurum B8.A using specific primers and designated amyB. It encoded a large multi-domain (putative) α-amylase of 1,287 amino acids, including a signal sequence of 53 aa, with a predicted mass of 135 kDa, designated MaAmyB. The genome sequence also confirmed that the gene encoding MaAmyB is located directly downstream of the gene encoding MaAmyA.
MaAmyB contains the A-, B-, and C-regions (but see specific details below) typical for the α-amylase superfamily22, as well as all catalytic residues and conserved regions generally present in family GH13 α-amylases23. MaAmyB does not belong to any established GH13 subfamily although a large number of sequences similar to MaAmyB was identified in databases (Table 1). We decided to heterologously express amyB and to study the activity and possible function of the produced M. aurum B8.A MaAmyB protein.
Expression and purification of MaAmyB
MaAmyB was successfully expressed in E. coli as a full length protein with both N- and C-terminal His-Tags. During purification it became apparent that neither of the two His-Tags present was functional. Most likely they are not accessible in the folded protein structure. The MaAmyB enzyme was purified on basis of its efficient binding to wheat starch granules, followed by washing with standard assay buffer and elution with maltose. Purification was followed by SDS-PAGE analysis (Fig. 2). The MaAmyB starch hydrolyzing activity was confirmed through in gel iodine activity staining of the refolded MaAmyB protein incubated with soluble starch (Fig. 2B). A clear zone of activity with a visible band was apparent for MaAmyB while no activity was observed in the empty vector control sample, confirming the absence of E. coli starch degrading enzymes. The activity of MaAmyB was 62 ± 2 U∙μg−1 (CNP assay). MaAmyB activity was only observed in the presence of 10 mM of CaCl2.

SDS-PAGE analysis and activity staining of MaAmyB.
(A) Regular SDS-PAGE analysis. (B) In gel staining for starch hydrolyzing activity. Arrows indicate the position of MaAmyB. After SDS-PAGE the gels were washed with standard assay buffer, then incubated for 2 h at 37 °C in standard assay buffer containing 0.5% soluble starch and activity was visualized by staining with Lugol’s Iodine solution. The band seen in A corresponds to the main activity band in B. Both bands run at the predicted molar mass of 135 kDa for MaAmyB. The empty vector control did not show a band at 135 kDa (A) and lacked any other activity band (B). For each sample 10 μg protein was loaded. B = MaAmyB eluted from starch granules; EV = Empty vector control eluted from starch granules; Blank = Blank control eluted from starch granules; M = marker proteins.
MaAmyB activity on different substrates
MaAmyB was incubated with maltose (G2), maltoheptaose (G7) soluble and granular starch as substrates. The results showed that MaAmyB is inactive on G2, but active on G7 as well as on soluble and granular starches. Incubation of MaAmyB with soluble starch resulted in initial accumulation of maltohexaose (G6), which in time was further hydrolyzed to smaller maltooligosaccharides, resulting in accumulation of mostly maltotriose, maltose and glucose after 6 h (Fig. 3). Incubation with G7 yielded similar products, while incubation of granular starch with MaAmyB did not result in accumulation of maltohexaose but in direct release of glucose, maltose and maltotriose (see Supplementary Fig. S1).

TLC analysis of MaAmyB products obtained in time by incubation of the enzyme (155 U, CNP assay) with soluble potato starch up to 6 h.
EV = empty vector control incubated for 6 h. M = markers, size consisting of G1–G7 (glucose; maltose; maltotriose; maltotetraose; maltopentaose; maltohexaose; maltoheptaose).
MaAmyB activity was also determined using blocked PNPG7 as a substrate (Fig. 4). This substrate is generally used in a commercial kit for assaying α-amylase activity (Megazyme), based on the release of PNP. Normally the PNP is released in a two-step reaction, first involving endo-acting α-amylase activity to split the blocked substrate. In the second step, an exo-acting α-glucosidase activity (provided in large excess in the commercial kit) removes the remaining glucose units attached to the PNP group. When using limiting amounts of the exo-acting α-glucosidase the overall reaction shows an acceleration in time, due to the sequential action of the endo-acting α-amylase (PNPG7 cleavage) and the α-glucosidase (PNP release) (Fig. 4, blue triangles). To our surprise, MaAmyB alone was able to degrade blocked PNPG7 and showed immediate PNP release, resulting in straight lines of absorbance versus time (Fig. 4). Negative controls containing E. coli extracts prepared from cells with an empty vector (EV), or combined with either endo-acting α-amylase (MaAmyA2) or exo-acting α-glucosidase, showed no PNP release (Fig. 4). The combined data shows that MaAmyB acts as an α-glucan 1,4-α-maltohexaosidase, an amylase releasing a maltohexaose from soluble starch, also cleaving the blocked PNPG7 substrate, and thus is not affected by the 4,6-linked-O-benzylidine group at the non-reducing end of this substrate.

PNP release (absorbance at 405 nm) from the blocked PNPG7 substrate upon incubation with 6 U MaAmyB enzyme preparations and various control samples, (done in triplicate ± SD).
Results obtained for MaAmyB and the positive control (EV + MaAmyA2 α-amylase + α-glucosidase) are significantly different (T-test for 0–500 sec, p-value: 0.028).  = MaAmyB;
= MaAmyB;  = empty vector (EV) control;
= empty vector (EV) control;  = Empty vector (EV) + MaAmyA2 α-amylase;
= Empty vector (EV) + MaAmyA2 α-amylase;  = EV + α-glucosidase;
= EV + α-glucosidase;  = EV + MaAmyA2 α-amylase + α-glucosidase.
= EV + MaAmyA2 α-amylase + α-glucosidase.
Identification of MaAmyB as a member of a novel GH13 subfamily
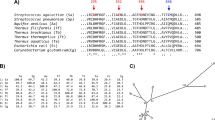
A search of the NCBI non redundant protein sequences database with the catalytic domain (regions-AB) of MaAmyB yielded a total of 164 sequences with at least 54% identity (February 2016). None of these putative starch hydrolases has been purified and characterized biochemically. Only AmlC of Streptomyces lividans TK24 has been identified as an α-amylase based on analysis in crude cell extracts (Yin et al. 1998). In the CAZy database, none of these MaAmyB homologs has been assigned to a specific GH13 subfamily. A phylogenetic tree containing all MaAmyB homologs, as well as selected enzymes from defined GH13 subfamilies, was prepared based on the partial AB-regions of the catalytic domain as identified by dbCAN (GH13.hmm in dbCAN), aa 690–1,121 for MaAmyB (Fig. 5). All MaAmyB homologs cluster together as a new group, which we propose to designate as the new GH13 subfamily 42 (GH13_42).

Phylogenetic tree of MaAmyB plus its 164 homologs, and a selection of members from the defined GH13 subfamilies that are either associated with EC3.2.1.1 or EC3.2.1.982 (see Supplementary Table S1).
The tree is based on the alignment of the catalytic AB-regions (GH13.hmm) as obtained from dbCAN. Domain organization shown is based on a combination of CDD and dbCAN data. The inserts in the A-region, the B-region and aberrant C-region as indicated are based on BLAST results of the identified regions from MaAmyB. Subfamily information was obtained from CAZy. Aberrant C-region information was obtained from Supplementary Fig. S2. The number 70 shows the location of a cluster of 70 putative α-amylases that share the aberrant C-region with the MaAmyB homologs but do not belong to subfamily GH13_42 (designated as GH13_VV) (see Supplementary Figs S2 and S3). At numbers 24 and 63, clades with these numbers of sequences similar to the nearby shown sequences have been collapsed to improve the readability of the tree. CBM74 is a novel CBM domain in MaAmyA15.
MaAmyB homologs were found predominantly in strains of the genera Streptomyces (115x), Paenibacillus (7x), Kitasatospora (7x), Micromonospora (6x), Cystobacter (4x). Up to three homologs were found in strains of 17 other genera. All GH13_42 homologs thus are found in soil-dwelling Gram-positive bacteria.
GH13_42 domain organization: enlarged AB-regions and an aberrant C-region
Most of the identified GH13_42 members share a similar domain organization starting with 2 CBM25 domains, 1 FNIII domain and the catalytic domain (AB-regions) (Fig. 5), which contains all catalytic residues and the 7 conserved regions generally found in the α-amylase superfamily (Fig. 6)1,22,23. The only exceptions are formed by two sequences separated by a single stop codon which are actually forming one single enzyme (CCK25026.1 and CCK25027.1), as well as one incomplete sequence (GenBank WP 03109885.1). Some homologs have an additional FNIII domain between the CBM25 domains. MaAmyB differs from its homologs in that it has 4 FNIII domains (Fig. 5), of which the N-terminal 3 are highly similar to each other (>97% identity), while the 4th FNIII domain has lower similarity (~55% identity) (Fig. 7).

Alignment of the AB-regions of MaAmyB and 3 additional GH13_42 members (CAB06816, BAJ27020.1, BAL91723.1), Pfam CL0058 and CDD CD11339 from the databases and 4 characterized α-amylases: Pig pancreatic α-amylase (PPA) (P00690.3), Taka amylase A from Aspergillus oryzae (TAKA) (P0C1B4.1), Bacillus licheniformis α-amylase (BLA) (P06278.1) and MaAmyA (AKG25402.1).
The B-region is indicated in purple, the two insertions in the A-region of GH13_42 are indicated in grey. The α-helices of the (β/α)8 barrel are indicated in green, β-sheets in red. Conserved Ca2+ binding aspartate residues are indicated in dark grey23. Other conserved residues are indicated in blue23. The 7 conserved regions of α-amylases are indicated with dashed orange lines and roman numbers23. Amino acid numbering as in MaAmyB.

(A) Protein domain organization of MaAmyA and MaAmyB and 4 related proteins. All proteins are shown with the N-terminus on the left hand side. The dotted line box indicates a stretch of domains that is highly similar between MaAmyA and MaAmyB (>99% identity), suggesting a relatively recent duplication event. Equal fill patterns indicate domains with high similarity. Solid filled domains do not have similarity to any other domain shown. AmlB (CAB06622.1) and BAA22082.1 are included as representatives of GH13_32 with high similarity to MaAmyA. AmlC (CAB06816.1) is included as a representative of GH13_42 with high similarity to MaAmyB. BAB86376.1 is included as a representative of a group of chitinases containing FNIII domains with high similarity to the 3 C-terminal FNIII domains of MaAmyA and all FNIII domains of MaAmyB. CBM74 is a novel CBM domain15. (B) Schematic representation showing the orientations of the genes encoding the M. aurum B8.A (MaAmyA, MaAmyB) and S. lividans TK24 (AmlB, AmlC) α-amylases.
In domain databases, α-amylases are listed as proteins with 2 domains: one domain represents the AB-regions, while the C-region is considered as a second and separate domain. The AB-regions of most GH13 α-amylases are represented by clan CL0058 in the Pfam conserved domain database24. In comparison to CL0058, the AB-regions of GH13_42 (534 aa for MaAmyB) are about 100 aa longer. The AB-regions of MaAmyB and 3 additional GH13_42 α-amylases were aligned with CL0058 from Pfam and 3 well-characterized α-amylases (Pig pancreatic α-amylase, Taka amylase A from Aspergillus oryzae and Bacillus licheniformis α-amylase) (Fig. 6). After manual tuning of the alignment using secondary structure information, 3 insertions could be identified in all GH13_42 members compared to CL0058 (Table 1). Two insertions were identified in the A-region. The first one is a 19 aa insertion between the 5th β-sheet and 5th α-helix (Fig. 6), which is uniquely found in all GH13_42 homologs (E value < 1∙10−5). The 2nd, a 37 aa insertion between the 6th α-helix and the 7th β-sheet, is predicted to form an α-helix located in close proximity to the C-region (Fig. 6). This insert is not only found in all 165 GH13_42 homologs but also in 41 additional putative α-amylases that share the aberrant C-region (see below and discussion) (E value < 1∙10−4). In addition, the B-region is 54 aa longer, compared to the 74 aa B-region from the CL0058 clan. This enlarged B-region is strongly conserved and uniquely found in GH13_42 homologs (Table 1).
The region C-terminal of the AB-regions in GH13_42 is not recognized by CDD as a classical C-region. Using Phyre2, which is based on protein (3D) structure prediction comparison, we observed a strong structural similarity (Phyre2 confidence of 97% or higher) between this C-terminal region in MaAmyB and several C-regions of α-amylases (such as the C-region of Bacillus subtilis α-amylase PDB1ua7, aa 348–425), although sequence identity was less than 30%. This low identity explains why the aberrant C-region of MaAmyB was not recognized by CDD. When used as a query sequence in a BLAST search, the aberrant C-region of MaAmyB (aa 1,194–1,259) returned 234 hits including all 164 GH13_42 members (query cover >95% and E-value < 1∙10−13), thus showing full conservation in the GH13_42 subfamily. The other 70 hits make up an additional cluster of putative α-amylases (designated as GH13_VV) which are not part of GH13_42 (see discussion). The aberrant C-region is 66 aa long, similar in length to known α-amylase C-regions. Phyre2 structure prediction for the aberrant C-region of MaAmyB showed an all β-sheet fold including a Greek key motif, as is typical for α-amylase C-regions.
Evolutionary origin of MaAmyB
Figure 7 shows the domain organization of the M. aurum B8.A MaAmyA and MaAmyB proteins, and four closely related relatives. As representatives of the novel GH13_42 subfamily, MaAmyB of M. aurum B8.A and AmlC of S. lividans share the three inserts in the AB-regions (Figs 1 and 6) and the aberrant C-region (see Supplementary Fig. S2). The CBM25 and FNIII domains of MaAmyB however differ from those found in other members of GH13_42. The two CBM25 domains and 3 of the 4 FNIII domains of MaAmyB are almost identical (99% identity at the nucleotide level) to the domains found in the M. aurum B8.A MaAmyA (Fig. 7A). To study the origin of these duplicated domains, additional phylogenetic trees were constructed based on alignment of the CBM25 and FNIII domains (see Supplementary Figs S4 and S5). This revealed that the CBM25 domains of MaAmyB do not cluster with the same domains in the GH13_42 subfamily (26–45% identity) (represented by AmlC in Fig. 7A), but with CBM25 domains attached to 4 GH13_32 amylases, including MaAmyA (42–49% identity) (see Supplementary Fig. S4) (represented by BAA22082.1 in Fig. 7). Phylogenetic analysis also showed that the FNIII domains of MaAmyB and the 3 C-terminal FNIII domains of MaAmyA, do not cluster with the GH13_42 subfamily FNIII domains (18–38 identity, 32–59% similarity) but instead with FNIII domains found in chitinases (44–62% identity, 59–71% similarity) (represented by BAB86376.1 in Fig. 7A). The N-terminal FNIII domain of MaAmyA, however, clusters with FNIII domains of the GH13_42 subfamily (40–60% identity, 59–80% similarity) (see Supplementary Fig. S5). These results, combined with the fact that enzymes with a domain organization similar to those of MaAmyA and MaAmyB currently are not found in databases, suggest that the MaAmyA and MaAmyB enzymes originated after horizontal gene transfer and (intensive) gene (re)shuffling in M. aurum B8.A.
Interestingly, 15 of the 115 Streptomyces strains with a GH13_42 have a MaAmyA homolog located directly upstream of the GH13_42 homolog in their genomes. In these Streptomyces strains the GH13_42 is located on the complementary DNA strand, while the GH13_32 homolog is on the regular strand (Fig. 7B); the M. aurum amyA and amyB genes are both on the complementary strand. The Streptomyces MaAmyA homologs only possess a single CBM20 domain, lacking all of the CBM25, CBM74 and FNIII domains found in MaAmyA (Fig. 7A).
Discussion
This paper reports the characterization of MaAmyB, a large and multi-domain amylase enzyme of M. aurum B8.A with hydrolytic activity on both granular and soluble starches, and on a blocked PNP-maltoheptaose. The highly unusual primary structure of this enzyme, with enlarged AB-regions as well as an aberrant C-region, was also identified in 164 other uncharacterized GH13 enzymes. Given the high similarity between these enzymes, we defined the new GH13_42 subfamily. MaAmyB is the only GH13_42 member with 4 FNIII domains, and with 2 CBM25 domains that do not cluster with the CBM25 domains in other GH13_42 members (see Supplementary Figs S4 and S5). Previously we reported that also the M. aurum B8.A MaAmyA α-amylase enzyme is unusual, the only known GH13_32 member with FNIII domains, and with a CBM74 domain that represents a novel SBD15,16. This raises questions about the evolutionary origins of these M. aurum B8.A MaAmyA and MaAmyB enzymes.
All currently know GH13_42 members share a general domain organization with 2 CBM25 domains and 1 FNIII domain N-terminal of the catalytic domain (Fig. 1). This strong conservation is remarkable as usually domain organizations vary within subfamily members2. Recently we have shown that CBM25 domains in MaAmyA are required for granular starch activity, but have no effect on soluble starch activity16. The role of FNIII domains in amylases has not been studied in detail. In MaAmyA truncation studies we did not see any effect for FNIII domains16. The role of FNIII domains in carbohydrate acting enzymes in general has not been studied in detail. Studies that addressed the roles of FNIII domains in such enzymes are inconclusive25,26,27,28,29,30,31,32,33,34,35,36,37. FNIII domains have been suggested to act as flexible linkers25,29,36 which corresponds to our findings for MaAmyA. CBM25 domains are usually located C-terminally of the catalytic domain, but N-terminally in GH13_42 proteins. We hypothesize that the FNIII domain functions as a flexible linker which enables the CBM25 domains in GH13_42 proteins to function efficiently at the N-terminus. With FNIII domains representing a linker to attach the CBM25 domains, and CBM25 domains enabeling enzymes to degrade granular substrates, we hypothesize that GH13_42 enzymes are involved in the degradation of granular starches.
The M. aurum B8.A MaAmyA and MaAmyB enzymes appear to have originated from at least three different enzymes: a GH13_32 α-amylase, a GH13_42 α-amylase and a chitinase (Fig. 7). Based on the sequence identities at the DNA level, we speculate that a single FNIII domain was duplicated twice resulting in three FNIII domains with high identities (95–98% at the nucleotide level). Together with 2 CBM25 domains, the 3 FNIII domains were duplicated between MaAmyA and MaAmyB. The high identity of the duplicated region (99% at the nucleotide level) suggests that this happened relatively recently. It is likely that in the environment from which M. aurum B8.A was isolated (a potato wastewater treatment facility), an efficient system for potato starch granule degradation is beneficial. We hypothesize that this evolutionary pressure caused horizontal gene transfer and gene (re)shuffling resulting in formation of MaAmyA and MaAmyB. This hypothesis is supported by the absence of enzymes with similar extensive domain organizations as MaAmyA and MaAmyB in the current databases, including 27 Microbacterium genome sequences38, (see Supplementary Figs S2, S4 and S5)15,16.
Members of the novel subfamily GH13_42 are characterized by enlarged AB-regions and an aberrant C-region (Figs 1 and 6). Compared to known α-amylases, the subfamily GH13_42 proteins have three strongly conserved insertions in the AB-regions (Table 1). The largest insert (54 aa) is found in the B-region which is known to be involved in calcium binding6. The aberrant C-region of GH13_42 proteins is strongly conserved among the subfamily members (Table 1). While it shares less than 30% sequence identity with the common C-region in GH13 α-amylases, it shows a strong structural similarity. Therefore it is likely that this domain functions as an α-amylase C-region. Additional bioinformatics analysis of 70 aberrant C-regions that were identified in family GH13 members that did not belong to the GH13_42 subfamily revealed that they are also part of putative starch hydrolases with enlarged AB-regions, though different from GH13_42. None of these 70 putative α-amylases have been characterized biochemically. A phylogenetic tree based on all 234 identified aberrant C-regions and some well know α-amylases shows that the aberrant C-regions cluster as a separate group away from other C-regions (see Supplementary Fig. S2). In addition, the 234 aberrant C-regions do not show clear clusters other than those expected based on the different species/genus harboring the genes encoding these proteins (see Supplementary Fig. S2). Despite the close sequence relationship between the aberrant C-regions, the additional 70 putative α-amylases do not belong to GH13_42 in view of the low sequence similarities in the AB-regions (<19% similarity); in a phylogenetic tree based on the AB-regions, they cluster together as an additional group, designated as GH13_VV (Fig. 5 and Supplementary Fig. S3). In these trees the various GH13_42 homologs cluster across multiple species, which is an indication for horizontal gene transfer of the AB-regions. Interestingly most of the GH13_VV members also have highly conserved, enlarged AB-regions with 3 inserts at similar places as in GH13_42. Region-A insert 2 is partly conserved between GH13_VV and GH13_42 (see Supplementary Table S3). Region-B and region-A insert 1 are uniquely conserved within the GH13_VV cluster (see Supplementary Table S3). Unlike GH13_42 not all regions are fully conserved in all GH13_VV members.
So far the aberrant C-region has only been identified in starch hydrolases with enlarged AB-regions that also have additional domains, like CBMs. In all GH13_42 enzymes and most GH13_VV members, additional domains are located N-terminal of the catalytic domain, while generally in GH13 such domains are located at the C-terminus of the enzyme2. The absence of domains attached to the aberrant C-region may be relevant (for its fold and function).
Structure prediction using the Phyre2 protein fold recognition server shows that region-A insert 2 in GH13_42 members forms an α-helix which lies in close proximity to the Greek key motif formed by the C-region (see Supplementary Fig. S6). Both the A-region inserts as well as the B- and C-regions in MaAmyB are predicted to be on the same side of the (β/α)8-barrel, although with low reliability (see Supplementary Fig. S6). This may indicate a functional relationship between the different enzyme parts as well as between the enlarged AB-regions and the C-region. We speculate that the C-region has a specific function in GH13_42, as may also be deduced from its concurrence with the enlarged AB-regions. Additional data preferably including the 3D structure of a GH13_42 homolog is needed to reveal the exact mechanism of interaction between the different domains, and the function of the C-region in GH13_42 enzymes.
Only one MaAmyB homolog, AmlC from S. lividans TK24, has been described previously39. AmlC is listed in CAZy as an α-amylase (EC 3.2.1.1), based on an activity analysis of crude extracts, which does not discriminate between α-amylases and exo-acting amylases. MaAmyB is able to release PNP from blocked PNPG7 without requiring any additional enzymes (Fig. 4). According to literature, the PNP from blocked PNPG7 can only be released through successive activity of an α-amylase and an exo-acting glycoside hydrolase enzyme, such as an α-glucosidase40. This is also demonstrated in one of the included controls, with MaAmyA2 α-amylase + α-glucosidase: due to the low exo-amylase activity used, an acceleration in the PNP release during the first 750 sec was observed (Fig. 4). Furthermore, α-amylases are not active on maltotriose or smaller substrates and release maltose as smallest product41. In this study, we show that MaAmyB can release glucose from small substrates (Fig. 3). It produces a series of oligosaccharides ranging from glucose to maltohexaose when soluble starch is used as a substrate. After 4 h the initially produced maltohexaose (G6) starts to decrease followed by degradation of maltopentaose (G5) and maltotetraose (G4). This pattern corresponds to that of an exo-acting enzyme with a preference for releasing G6 from starch, but which is also capable of producing glucose, like α-glucan 1,4-α-maltohexaosidases (EC 3.2.1.98)42,43,44,45,46,47. The activity of an α-glucan 1,4-α-maltohexaosidase on a blocked PNPG7 substrate has not been described in literature. Only the maltotetraose-forming amylase (EC 3.2.1.60) from Pseudomonas saccharophila48,49 is described in a GRAS notice50, as able to degrade blocked PNPG7 despite the blocking group, and to release maltotetraose (G4). It is likely that the maltohexaose releasing MaAmyB acts in a similar way (Figs 3 and 4). Contamination of MaAmyB by a low concentration of an E. coli α-amylase is unlikely as control experiments showed a clear increase in activity over time when an α-amylase and α-glucosidase were used (Fig. 4). In addition the empty vector control experiments did not show any activity even after prolonged incubation and addition of either an α-amylase or α-glucosidase. We therefore conclude that MaAmyB is an α-glucan 1,4-α-maltohexaosidase (EC 3.2.1.98).
The finding that MaAmyB acts as an exo-amylase allows for speculation about its function. Synergistic action between an α-amylase and glucoamylase in granular starch degradation has already been described18,19,20,21. MaAmyB is located in the proximity of an α-amylase (MaAmyA) on the M. aurum B8.A genome, and MaAmyA introduced a different pore pattern in starch granules than the M. aurum B8.A culture fluid16. MaAmyB also is active on granular starch (see Supplementary Fig. S1). Therefore we hypothesize that the exo-acting MaAmyB enzyme contributes to degradation of starch granules by rapidly hydrolyzing the helical and linear starch chains that become exposed after pore formation by MaAmyA. In addition, bioinformatics analysis revealed that in at least 15 Streptomyces species a (putative) GH13_32 α-amylase is located near a GH13_42 homolog. This suggests that the synergy between GH13_32 and GH13_42 members is more widely spread.
We conclude that MaAmyB is an α-glucan 1,4-α-maltohexaosidase, the first characterized member of the novel GH13_42 subfamily, which functions in the degradation of granular starch. It is likely that the enlarged AB-regions and aberrant C-region contribute to this specific activity. Further research into the structure/function relationship of these or similar enzymes with their N-terminal CBM domains, enlarged AB-regions and aberrant C-region, is required to elucidate the roles of their different protein domains.
Materials and Methods
Bacterial strains, media, and plasmids
M. aurum strain B8.A was isolated from a waste water treatment plant of a potato starch processing factory. Isolation and growth conditions have been described previously17. The M. aurum strain B8.A strain was deposited (deposit number LMG S-26033) in the BCCM/LMG culture collection of the University of Gent, Belgium17. Escherichia coli TOP10 and C43 were cultivated at 37 °C overnight in LB with orbital shaking (220 rpm). When required, ampicillin or kanamycin was added to a final concentration of 100 or 50 μg·ml−1, respectively. pBAD-VV16, a modified pBAD/Myc-His B (Novagen, Madison, WI, USA), was used as expression vector for the amyB gene constructs in E. coli C43(DE3).
Gene identification
The full amyB gene was amplified from the M. aurum B8.A genome (V. Valk, R. M. van der Kaaij and L. Dijkhuizen, manuscript in preparation) Fwd and Rev primers: CGTCTTCGACCTCCATATGCGAAGGAGCAGGGTCTTGCGAGAAAGCACAC and GCCGGCACAGAGTGCTTGAGGTAGGCGCTGCCGCTACCGATCTTG. Chromosomal DNA was isolated from a Microbacterium aurum B8.A culture grown at 37 °C overnight in LB with orbital shaking (220 rpm) using the GenElute Bacterial Genomic DNA Kit (Sigma-Aldrich, Zwijndrecht, the Netherlands). The product was cloned into the pBAD-VV vector16 using NdeI and DraIII restriction sites. All products were confirmed through sequencing (GATC-Biotech, Konstanz, Germany). The amyB constructs were prepared with both N- and C-terminal His-Tags from the pBAD-VV vector.
Protein expression
Recombinant E. coli strains were grown as 500 ml cultures in 3 l flasks, with 100 μg·ml−1 ampicillin and 0.05% (final concentration) arabinose (Sigma-Aldrich, Zwijndrecht, the Netherlands) for 6 h at 30 °C (220 rpm) and then for 40 h at 18 °C (220 rpm). Cells were collected by centrifugation at 4,250 g for 20 min at 4 °C (Thermo Lynx 4000). Pellets were resuspended in 50 mM Tris-HCl buffer pH 6.8 containing 10 mM CaCl2. Protease inhibitors (Roche Mini EDTA-free Protease Inhibitor, Sigma-Aldrich, Zwijndrecht, the Netherlands) were added and cells were broken by sonication (15 sec at 10,000 Ω, 30 sec cooling, 7x). Cell debris and intact cells were removed by centrifugation at 15,000 g for 20 min at 4 °C. Resulting cell free extracts were immediately used for purification of MaAmyB.
Standard Assay buffer
A 50 mM Tris-HCl buffer pH 6.8 containing 10 mM CaCl2 was used as standard assay buffer for enzyme incubations. Unless indicated otherwise all incubations were performed at 37 °C.
Purification of MaAmyB protein through binding to starch granules
Granular wheat starch (Sigma-Aldrich, Zwijndrecht, the Netherlands catalog no. S5127) was sterilized through gamma irradiation (Synergy Health, Ede, the Netherlands). The sterile granules were washed two times with standard assay buffer. After the second wash, granules were collected through centrifugation and the liquid removed. Then, 5 gram freshly washed (wet) granules was added to 20 ml of freshly prepared cell free extracts of the E. coli cultures expressing MaAmyB, or the E. coli empty vector culture as control. The tubes were mixed until a homogeneous suspension was obtained followed by incubating overnight at 4 °C on a roller bench (VWR, Amsterdam, the Netherlands). The suspension was centrifuged at 4,000 g for 5 min and the supernatant removed. The starch granules were washed 2 times with 40 ml standard assay buffer for 30 min, and the supernatant removed through centrifugation at 4,000 g for 5 min. To elute MaAmyB protein, 5 ml of 10% maltose in assay buffer was added, mixed for 30 min and the supernatant collected through centrifugation at 4,000 g for 5 min. The maltose was removed and the sample concentrated over 50 kD cut off Millipore spin filters and washed with standard assay buffer. Samples were concentrated to 1 ml and stored at 4 °C in standard assay buffer.
Activity staining on SDS-PAGE
SDS-PAGE analysis was used to determine protein masses. After refolding the location of the MaAmyB protein could be visualized in the gels by staining for its starch-acting activity. SDS-PAGE51 was performed using precast THX gels (Bio Rad, Veenendaal, the Netherlands). After loading and running, gels were washed 3 times for 5 min in Milli Q water to remove SDS and allow protein refolding, then incubated for 2 h at 37 °C in standard assay buffer containing 0.5% soluble potato starch (Sigma-Aldrich, Zwijndrecht, the Netherlands)52. After incubation the gels were stained with Lugol’s iodine (2.5% I2/5% KI) to visualize protein bands with α-amylase activity. After imaging, gels were partly destained by washing in Milli Q water and then stained with Bio-Safe™ Coomassie Stain (Bio Rad, Veenendaal, the Netherlands) to visualize proteins. The Fermentas PageRuler prestained marker was included in each gel.
Activity assay with CNPG3
The activity of MaAmyB and other enzymes was determined and standardized through incubation of the enzyme with 2-Chloro-4-nitrophenyl-α-D-maltotrioside (CNPG3). This compound has a 2-Chloro-4-nitrophenyl (CNP) group coupled to maltotriose (G3) at its reducing end. The assay is based on the detection of the released CNP group from CNPG3 by absorbance reading at 405 nm. The enzyme solution or mixture (15 μl) containing approximately 6.25 μg∙ml−1 MaAmyB protein to be tested was prepared in 96-well microtiter plates. Pre-warmed substrate (100 μl 2 mM CNPG3) was added and the reaction was followed through absorbance reading at 405 nm for 20 min in a microtiter plate reader (Spectramax plus, Molecular Devices, Sunnyvale CA, USA) set at 37 °C. One unit is defined as the amount of enzyme required to release 1 μmol CNP min−1 under standard assay conditions. This Unit definition is used throughout this paper.
Activity assay with Blocked PNPG7 substrate
MaAmyB activity was tested by incubation of the enzyme with blocked 4-nitrophenyl-α-maltoheptaoside (blocked PNPG7) (Megazyme, Wicklow, Ierland). This compound has a 4-nitrophenyl (PNP) group coupled to maltoheptaose (G7) at its reducing end while the non-reducing end is blocked by an 4,6-linked-O-benzylidine group. Due to this block, release of PNP generally requires both endo-acting activity (α-amylase) and subsequently exo-acting activity (α-glucosidase). In the present work, blocked PNPG7 was tested as substrate by only adding the MaAmyB enzyme. The assay, based on the detection of the released PNP group from the blocked PNPG7 substrate by absorbance reading at 405 nm, was performed as described for the CNP assay using 6 U (based on the CNP assay) MaAmyB. Negative controls included an empty vector (EV) control without additional enzymes, EV with added α-glucosidase (Megazyme, Wicklow, Ierland), EV with added α-amylase (MaAmyA2, a truncation of MaAmyA from M. aurum B8.A with all starch binding domains removed16. A positive control consisted of EV with both α-glucosidase and α-amylase added. MaAmyA2 and the α-glucosidase were diluted in assay buffer until the activity on CNPG3 was 1.5–2 times the activity of MaAmyB on this substrate.
Activity assays with other substrates
The activity of MaAmyB was tested on maltose (G2), maltoheptaose (G7), soluble potato starch (Sigma Aldrich, Zwijndrecht, the Netherlands, catalog no. S2004) and granular wheat starch (Sigma Aldrich, Zwijndrecht, the Netherlands, catalog no. S5127). The 900 μl substrate solution (or suspension in case of the granular wheat starch), containing 10 mg∙ml−1 of the substrate to be tested in standard assay buffer, was prepared and pre-heated at 37 °C in a heating block. Then 100 μl enzyme preparation containing 25 μg MaAmyB (155 U), or a control, was added. The solution was immediately mixed and the first 100 μl sample was taken and frozen (−20 °C) (t = 0 h). Additional samples were taken after 1, 2, 3, 4, 5, and 6 h for soluble substrates, and 0, 24, 48, 72, 96, and 120 h for granular substrates. Samples of 1 μl were analyzed on TLC (Merck, Darmstadt, Germany) and run overnight using a butanol, ethanol, water (5:5:3 v/v/v) running buffer, and stained using 40% sulfuric acid and 5% Orcinol (9,10-dihydrophenanthrene) in methanol53,54.
Bioinformatics tools
All BLAST55 searches were performed with NCBI BLASTP using standard settings. The MaAmyB catalytic domain (aa 650–1,183) (regions-AB, Fig. 1) was used as query in BLAST searches for related sequences, using standard settings but with the maximum target sequences increased to 1,000 (instead of the default 100). Conserved domains were detected using both the NCBI conserved domain finder7 with forced live search, without low-complexity filter, using the conserved domain database (CDD), and dbCAN56 with standard settings. Signal sequences were predicted with SignalP4.157 using standard settings. Catalytic AB-regions in the related GH13 proteins were identified using the full length sequences based on dbCAN domain information. The C-region in the related GH13 proteins was defined based on the BLAST search results obtained using the aberrant C-region of MaAmyB (aa 1,194–1,259) (Fig. 1). CBM25 domain sequences and FNIII domain sequences were extracted from all proteins listed in the 02–02–2016 release of CAZy2 (after their sequences were obtained from GenBank), based on CDD information. The extracted domain sequences were used for construction of alignments with Mega6.058 using its build-in muscle alignment with standard settings. Alignments were checked for the positions of conserved residues and manually tuned when needed. Phylogenetic trees were made with Mega6.0 using the Maximum Likelihood method with gaps/missing data treatment set on “partial deletion” instead of “full deletion”. Trees were visualized with Interactive Tree Of Life v259. The protein domain annotations shown in the trees are based on combined data from CDD and dbCAN. Information about GH13 subfamilies was obtained from the CAZy database2. An initial tree with a diverse selection of all GH13 subfamilies was constructed to select the closest related subfamilies for display in the final tree. The selection included multiple GH13 members from each of the 40 defined subfamilies, which varied in domain organization and origin. The final tree based on the catalytic domain regions-AB included MaAmyB and all homologs found in the NCBI non redundant protein sequences database, as well as a selection of members that were most closely related based on the initial tree. Protein structure predictions were done with the Phyre2 protein fold recognition server using standard settings60.
Nucleotide sequence accession number
The sequence of amyB, isolated from M. aurum B8.A has been deposited into GenBank database under accession no. KX447523.1.
Additional Information
How to cite this article: Valk, V. et al. Characterization of the starch-acting MaAmyB enzyme from Microbacterium aurum B8.A representing the novel subfamily GH13_42 with an unusual, multi-domain organization. Sci. Rep. 6, 36100; doi: 10.1038/srep36100 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
van der Maarel, M. J. E. C., van der Veen, B., Uitdehaag, J. C. M., Leemhuis, H. & Dijkhuizen, L. Properties and applications of starch-converting enzymes of the α-amylase family. J. Biotechnol. 94, 137–155 (2002).
Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M. & Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495 (2014).
Stam, M. R., Danchin, E. G. J., Rancurel, C., Coutinho, P. M. & Henrissat, B. Dividing the large glycoside hydrolase family 13 into subfamilies: towards improved functional annotations of α-amylase-related proteins. Protein Eng. Des. Sel. 19, 555–562 (2006).
Janeček, Š., Svensson, B. & MacGregor, E. A. α-Amylase: an enzyme specificity found in various families of glycoside hydrolases. Cell. Mol. Life Sci. 71, 1149–1170 (2014).
Liu, Y. et al. Identification and phylogenetic characterization of a new subfamily of α-amylase enzymes from marine microorganisms. Mar. Biotechnol. 14, 253–260 (2012).
Machius, M., Wiegand, G. & Huber, R. Crystal structure of calcium-depleted Bacillus licheniformis α-amylase at 2.2 Å resolution. J. Mol. Biol. 246, 545–559 (1995).
Marchler-Bauer, A. et al. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39, D225–D229 (2011).
Matsuura, Y., Kusunoki, M., Harada, W. & Kakudo, M. Structure and possible catalytic residues of Taka-amylase A. J. Biochem. 95, 697–702 (1984).
Doyon, Y., Home, W., Daull, P. & LeBel, D. Effect of C-domain N-glycosylation and deletion on rat pancreatic alpha-amylase secretion and activity. Biochem. J. 362, 259–264 (2002).
Ben Abdelmalek, I. et al. Structural investigation and homology modeling studies of native and truncated forms of α-amylases from Sclerotinia sclerotiorum. J. Microbiol. Biotechnol. 19, 1306–1318 (2009).
Bozonnet, S. et al. The ‘pair of sugar tongs’ site on the non-catalytic domain C of barley α-amylase participates in substrate binding and activity. FEBS J. 274, 5055–5067 (2007).
Mehta, D. & Satyanarayana, T. Domain C of thermostable α-amylase of Geobacillus thermoleovorans mediates raw starch adsorption. Appl. Microbiol. Biotechnol. 98, 4503–4519 (2014).
Boraston, A. B. et al. A structural and functional analysis of α-glucan recognition by family 25 and 26 carbohydrate-binding modules reveals a conserved mode of starch recognition. J. Biol. Chem. 281, 587–598 (2006).
Machovič, M. & Janeček, Š. Starch-binding domains in the post-genome era. Cell. Mol. Life Sci. 63, 2710–2724 (2006).
Valk, V., Lammerts van Bueren, A., van der Kaaij, R. M. & Dijkhuizen, L. Carbohydrate Binding Module 74 is a novel starch binding domain associated with large and multi-domain α-amylase enzymes. FEBS J. 283, 2354–2368 (2016).
Valk, V., Eeuwema, W., Sarian, F. D., van der Kaaij, R. M. & Dijkhuizen, L. Degradation of granular starch by the bacterium Microbacterium aurum strain B8.A involves a modular α-amylase enzyme system with FNIII and CBM25 domains. Appl. Environ. Microbiol. 81, 6610–6620 (2015).
Sarian, F. et al. Enzymatic degradation of granular potato starch by Microbacterium aurum strain B8.A. Appl. Microbiol. Biotechnol. 93, 645–654 (2012).
Matsubara, T. et al. Degradation of raw starch granules by alpha-amylase purified from culture of Aspergillus awamori KT-11. J. Biochem. Mol. Biol. 37, 422–428 (2004).
Sun, H., Ge, X. & Zhang, W. Production of a novel raw-starch-digesting glucoamylase by Penicillium sp. X-1 under solid state fermentation and its use in direct hydrolysis of raw starch. World J. Microbiol. Biotechnol. 23, 603–613 (2007).
Wankhede, D. B. & Ramteke, R. S. Synergistic digestibility of several native starches by amylolytic enzymes. Starch - Stärke 34, 309–312 (1982).
Wong, D. W. S., Robertson, G. H., Lee, C. C. & Wagschal, K. Synergistic action of recombinant α-amylase and glucoamylase on the hydrolysis of starch granules. Protein J. 26, 159–164 (2007).
MacGregor, E. A., Janeček, Š. & Svensson, B. Relationship of sequence and structure to specificity in the α-amylase family of enzymes. Biochim. Biophys. Acta - Protein Struct. Mol. Enzymol. 1546, 1–20 (2001).
Janeček, Š. How many conserved sequence regions are there in the α -amylase family? Biologia (Bratisl). 57, 29–41 (2002).
Finn, R. D. et al. Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230 (2014).
Chuang, H.-H., Lin, H.-Y. & Lin, F.-P. Biochemical characteristics of C-terminal region of recombinant chitinase from Bacillus licheniformis implication of necessity for enzyme properties. FEBS J. 275, 2240–2254 (2008).
Kahar, U. M., Chan, K. G., Salleh, M. M., Mee Hii, S. & Goh, K. M. A high molecular-mass anoxybacillus sp. SK3-4 amylopullulanase: characterization and its relationship in carbohydrate utilization. Int. J. Mol. Sci. 14, 11302–11318 (2013).
Kim, D. Y. et al. Novel modular endo-β-1,4-xylanase with transglycosylation activity from Cellulosimicrobium sp. strain HY-13 that is homologous to inverting GH family 6 enzymes. Bioresour. Technol. 107, 25–32 (2012).
Kim, D. Y. et al. Novel GH10 xylanase, with a fibronectin type 3 domain, from Cellulosimicrobium sp. strain HY-13, a bacterium in the gut of Eisenia fetida. Appl. Environ. Microbiol. 75, 7275–7279 (2009).
Lin, F. P., Ho, Y. H., Lin, H. Y. & Lin, H. J. Effect of C-terminal truncation on enzyme properties of recombinant amylopullulanase from Thermoanaerobacter pseudoethanolicus. Extremophiles 16, 395–403 (2012).
Lin, F.-P. & Leu, K.-L. Cloning, expression, and characterization of thermostable region of amylopullulanase gene from Thermoanaerobacter ethanolicus 39E. Appl. Biochem. Biotechnol. 97, 33–44 (2002).
Lin, F.-P., Ma, H.-Y., Lin, H.-J., Liu, S.-M. & Tzou, W.-S. Biochemical characterization of two truncated forms of amylopullulanase from Thermoanaerobacterium saccharolyticum NTOU1 to identify its enzymatically active region. Appl. Biochem. Biotechnol. 165, 1047–1056 (2011).
Lin, H.-Y., Chuang, H.-H. & Lin, F.-P. Biochemical characterization of engineered amylopullulanase from Thermoanaerobacter ethanolicus 39E-implicating the non-necessity of its 100 C-terminal amino acid residues. Extremophiles 12, 641–650 (2008).
Mathupala, S. P., Lowe, S. E., Podkovyrov, S. M. & Zeikus, J. G. Sequencing of the amylopullulanase (apu) gene of Thermoanaerobacter ethanolicus 39E, and identification of the active site by site-directed mutagenesis. J. Biol. Chem. 268, 16332–16344 (1993).
Suzuki, K. et al. The third chitinase gene (chiC) of Serratia marcescens 2170 and the relationship of its product to other bacterial chitinases. Biochem. J. 343 Pt 3, 587–596 (1999).
Takasuka, T. E. et al. Biochemical properties and atomic resolution structure of a proteolytically processed β-mannanase from cellulolytic Streptomyces sp. SirexAA-E. PLoS One 9 (2014).
Watanabe, T. et al. The roles of the C-terminal domain and type III domains of chitinase A1 from Bacillus circulans WL-12 in chitin degradation. J. Bacteriol. 176, 4465–4472 (1994).
Watanabe, T., Suzuki, K., Oyanagi, W., Ohnishi, K. & Tanaka, H. Gene cloning of chitinase A1 from Bacillus circulans WL-12 revealed its evolutionary relationship to Serratia chitinase and to the type III homology units of fibronectin. J. Biol. Chem. 265, 15659–15665 (1990).
Benson, D. A., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J. & Sayers, E. W. GenBank. Nucleic Acids Res. 37, D26–D31 (2009).
Yin, X. H., Gerbaud, C., Francou, F. X., Guérineau, M. & Virolle, M. J. amlC, Another amylolytic gene maps close to the amlB locus in Streptomyces lividans TK24. Gene 215, 171–180 (1998).
Sheehan, H. & McCleary, B. V. A new procedure for the measurement of fungal and bacterial α-amylase. Biotechnol. Tech. 2, 289–292 (1988).
Fischer, E. H. & Stein, E. A. Alpha amylases. The enzymes (Academic Press, 1960).
Hashim, S. O. et al. Alkaline active maltohexaose-forming α-amylase from Bacillus halodurans LBK 34. Enzyme Microb. Technol. 36, 139–146 (2005).
Hayashi, T., Akiba, T. & Horikoshi, K. Production and purification from alkalophilic of new maltohexaose-forming amylases from Alkalophilic Bacillus Sp. H-167. Agric. Biol. Chem. 52, 443–448 (1988).
Hayashi, T., Akiba, T. & Horikoshi, K. Properties of new alkaline maltohexaose-forming amylases. Appl. Microbiol. Biotechnol. 28, 281–285 (1988).
Li, Z. et al. AmyM, a novel maltohexaose-forming α-amylase from corallococcus sp. strain EGB. Appl. Environ. Microbiol. 81, 1977–1987 (2015).
Momma, M. Cloning and sequencing of the maltohexaose-producing amylase gene of Klebsiella pneumoniae. Biosci. Biotechnol. Biochem. 64, 428–431 (2000).
Pages, C. Nucleotide sequence of the maltohexaose-producing amylase gene from an alkalophilic Bacillus sp. Biochem. Biophys. Res. Commun. 151, 25–31 (1988).
Zhou, J. H., Baba, T., Takano, T., Kobayashi, S. & Arai, Y. Nucleotide sequence of the maltotetraohydrolase gene from Pseudomonas saccharophila. FEBS Lett. 255, 37–41 (1989).
Zhou, J., Baba, T., Takano, T., Kobayashi, S. & Arai, Y. Properties of the enzyme expressed by the Pseudomonas saccharophila maltotetraohydrolase gene (mta) in Escherichia coli. Carbohydr. Res. 223, 255–261 (1992).
Caddow, A. J. GRAS Notice 000277: G4-amylase enzyme preparation from Bacillus licheniformis expressing the gene encoding a modified maltotetraohydrolase enzyme from Pseudomonas saccharophila. Available at: http://www.fda.gov/ucm/groups/fdagov-public/@fdagov-foods-gen/documents/document/ucm269243.pdf (2009).
Laemmli, U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 (1970).
Yamaguchi, R., Tokunaga, H., Ishibashi, M., Arakawa, T. & Tokunaga, M. Salt-dependent thermo-reversible α-amylase: cloning and characterization of halophilic α-amylase from moderately halophilic bacterium, Kocuria varians. Appl. Microbiol. Biotechnol. 89, 673–684 (2011).
Brückner, J. Estimation of monosaccharides by the orcinol–sulphuric acid reaction. Biochem. J. 60, 200–205 (1955).
Lammerts van Bueren, A., Saraf, A., Martens, E. C. & Dijkhuizen, L. Differential metabolism of exopolysaccharides from probiotic Lactobacilli by the human gut symbiont Bacteroides thetaiotaomicron. Appl. Environ. Microbiol. 81, 3973–3983 (2015).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Yin, Y. et al. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 40, W445–W451 (2012).
Petersen, T. N., Brunak, S., von Heijne, G. & Nielsen, H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786 (2011).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Letunic, I. & Bork, P. Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 39, W475–W478 (2011).
Kelley, L. A. & Sternberg, M. J. E. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4, 363–371 (2009).
Acknowledgements
This study was partly funded by the Top Institute of Food & Nutrition (project B1003) and by the University of Groningen.
Author information
Authors and Affiliations
Contributions
V.V., R.M.v.d.K. and L.D. planned experiments. V.V. performed experiments. V.V., R.M.v.d.K. and L.D. analyzed data. V.V. prepared all figures. V.V., R.M.v.d.K. and L.D. wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Valk, V., van der Kaaij, R. & Dijkhuizen, L. Characterization of the starch-acting MaAmyB enzyme from Microbacterium aurum B8.A representing the novel subfamily GH13_42 with an unusual, multi-domain organization. Sci Rep 6, 36100 (2016). https://doi.org/10.1038/srep36100
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep36100
This article is cited by
-
Aspects and Recent Trends in Microbial α-Amylase: a Review
Applied Biochemistry and Biotechnology (2021)
-
Mangrove soil as a source for novel xylanase and amylase as determined by cultivation-dependent and cultivation-independent methods
Brazilian Journal of Microbiology (2020)
-
Purification of an alpha amylase from Aspergillus flavus NSH9 and molecular characterization of its nucleotide gene sequence
3 Biotech (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
