
Abstract
Obesity is associated with low-grade inflammation that leads to insulin resistance and type 2 diabetes via Toll-like Receptor (TLR) and TNF-family cytokine receptor (TNFR) signaling pathways. Ubc13 is an ubiquitin-conjugating enzyme responsible for non-canonical K63-linked polyubiquitination of TNF receptor-associated factor (TRAF)-family adapter proteins involved in TLR and TNFR pathways. However, the relationship between Ubc13 and metabolic disease remains unclear. In this study, we investigated the role of Ubc13 in insulin resistance and high-fat diet (HFD)-induced obesity. We compared wild-type (WT) and Ubc13 haploinsufficient (ubc13+/−) mice under normal diet (ND) and HFD, since homozygous knockout mice (ubc13−/−) are embryonic lethal. Male and female ubc13+/− mice were protected against age-related insulin resistance under ND and HFD compared to WT mice. Interestingly, only female ubc13+/− mice were protected against HFD-induced obesity and hepatic steatosis. Moreover, only female HFD-fed ubc13+/− mice showed lower expression of inflammatory cytokines that was secondary to reduction in weight gain not present in the other groups. In summary, our results indicate that suppression of Ubc13 activity may play a metabolic role independent of its inflammatory function. Thus, Ubc13 could represent a therapeutic target for insulin resistance, diet-induced obesity, and associated metabolic dysfunctions.
Similar content being viewed by others


Introduction
Obesity is now reaching pandemic proportions worldwide1 and is closely associated with chronic low-grade inflammation, which causes insulin resistance and other metabolic diseases such as hypertension and atherosclerosis2,3. Recent studies suggest that obesity induces immune cell infiltration and activation in liver and adipose tissue accompanied by secretion of inflammatory cytokines and chemokines, adversely affecting insulin sensitivity4. The link between obesity and insulin resistance is disrupted by gene knockout of mediators of inflammatory pathways such as Toll-like Receptor (TLR)5,6,7,8, TNF-family cytokine receptor (TNFR)9, and nucleotide-binding domain, leucine-rich repeats containing family, pyrin domain-containing-3 (Nlrp3) inflammasome10. In addition, aging is associated with chronic low-grade inflammation. Recently, mice lacking Nlrp3 were reported to show improved age-related disorders including type 2 diabetes11, suggesting that suppression of excessive inflammatory action induced by obesity and aging might improve these associated metabolic diseases.
Ubc13 is required for signaling by a variety of receptors important in immune regulation. Ubc13 is a non-canonical ubiquitin-conjugating enzyme (E2) that catalyzes formation of lysine 63 (K63)-linked polyubiquitin chains on various target proteins12,13. Unlike the canonical K48-linked polyubiquitin chains, K63-linked polyubiquitin chains are not substrates for the proteasome14,15. Instead, K63-linked polyubiquitination plays essential roles in protein activation and interaction16,17,18. Ubc13 binds the RING domains of TRAF-family adapter proteins such as TRAF2 and TRAF6, and promotes activation of protein kinases involved in signaling by TNFRs19,20 and by TLRs21. With regard to the role of Ubc13 in TRAF-mediated signaling, lipopolysaccharide (LPS) and cytokines such as TNFα and IL-1β that activate TRAF2 or TRAF6 result in its non-canonical ubiquitination and subsequent downstream activation of kinases, including JNK. Ubc13 is also associated with the activation of NF-κB, via its effect on TRAF6, resulting in activation of the kinase TAK117.
Previously, we have reported in vivo functions of Ubc13 in the context of innate immune responses, using the method of targeted gene ablation in mice22. We reported that Ubc13 homozygous knockout mice were embryonic lethal and that heterozygous ubc13+/− mice have normal phenotypes, despite reduced levels of Ubc13 protein. However, ubc13+/− mice exhibit marked reductions in responsiveness to challenge with LPS22. Moreover, ubc13+/− splenocytes and bone marrow-derived macrophages (BMDMs) display blunted responses to LPS and TNF with respect to cytokine secretion and various TNFR/TLR-mediated signal transduction events22. Although Ubc13 was reported to play a critical role in vivo in inflammatory responses, its role in Ubc13 regulation of chronic low-grade inflammation and insulin resistance is not known.
In the present study, we investigated the role of Ubc13 in insulin resistance and HFD-induced obesity using ubc13+/− mice. We found that insulin resistance was improved in male HFD-fed and male and female ND-fed aged mice without any change in inflammatory status or weight gain. Interestingly, female ubc13+/− mice are protected against HFD-induced obesity, hepatic steatosis, and insulin resistance. Ubc13 haploinsufficiency only had an effect on inflammation in female mice on HFD that was secondary to reduction in weight gain not present in the other groups. Therefore, this study suggests that Ubc13 haploinsufficiency has a metabolic role that is independent of its function in inflammatory signal transduction.
Results
Effect of Ubc13 haploinsufficiency on body composition, food intake, and energy expenditure
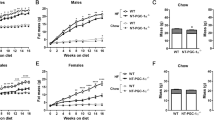
To investigate the functional role of Ubc13 in HFD-induced obesity and insulin resistance, we fed 4-week-old ubc13+/+ (WT) and ubc13+/− mice either ND or HFD for 14 weeks. On ND, no statistical differences were observed in body weight, food intake, and energy expenditure between WT and ubc13+/− mice (Fig. 1A,B, Figs S1 and S2A,B). In contrast, on HFD, male ubc13+/− mice tended to exhibit lower body weight than WT mice, whereas female ubc13+/− mice exhibited significantly lower body weight than WT mice (Fig. 1A and Fig. S1). CT scan imaging revealed decreases in both visceral and subcutaneous adipose tissues of female ubc13+/− mice on HFD (Fig. 1B). Food intake and locomotor activity were similar in female WT and ubc13+/− mice (Fig. 1C,G), whereas energy expenditure, fat oxidation, and rectal temperature were significantly increased in female ubc13+/− mice compared to WT mice on HFD (Fig. 1D,E,H). In addition, uncoupling protein 1(UCP1) expression in visceral adipose tissue (VAT) tended to be increased and respiratory quotient (RQ) tended to be decreased in HFD-fed female ubc13+/− mice compared to WT mice (Fig. 1F,I).

Female ubc13+/− mice display decreased weight gain and increased energy expenditure on HFD.
(A) Body weights of female WT versus ubc13+/− mice on ND or HFD (n = 5 to 6). HFD was started at 4 weeks of age. *p < 0.05 versus HFD-fed WT mice. (B) Representative CT scanning images of abdominal cross-sections of WT and ubc13+/− mice on ND or HFD. Pink, yellow, and blue areas represent visceral fat, subcutaneous fat, and lean mass, respectively. (C–E) Food intake (C), energy expenditure (D), and rectal temperature (E) were measured for female WT and ubc13+/− mice on a HFD (n = 4 to 6). *p < 0.05, **p < 0.01 versus HFD-fed WT mice. (F) qRT-PCR analysis of the expression of the gene encoding UCP1 in VAT of female WT and ubc13+/− mice on HFD (n = 5 to 6). (G–I) activity count (G), fat oxidation (H), and RQ (I) were measured for female WT and ubc13+/− mice on a HFD (n = 4). *p < 0.05, versus HFD-fed WT mice.
Effect of Ubc13 haploinsufficiency on insulin resistance under ND and HFD
We also evaluated the impact of Ubc13 haploinsufficiency on insulin sensitivity using oral glucose tolerance test (OGTT) and insulin tolerance test (ITT). No statistical differences in OGTT and ITT data were observed between 8-week-old WT and ubc13+/− mice after 4 weeks of ND (Fig. 2A–C). Interestingly, plasma insulin levels during OGTT were lower in 18-week-old female ubc13+/− mice versus WT mice despite a lack of difference in plasma glucose levels after 14 weeks of ND (Fig. 2D,E). Consistent with these results, insulin sensitivity was significantly enhanced in 18-week-old female ubc13+/− mice during ITT (Fig. 2F). On HFD, the mice became hyperinsulinemic; however, plasma insulin levels during OGTT and plasma glucose levels during OGTT and ITT were lower in 18-week-old female ubc13+/− mice compared to WT mice after 14 weeks of HFD (Fig. 2G–I). In male mice, plasma glucose and insulin levels during OGTT were also significantly lower in 18-week-old ubc13+/− mice versus WT mice after 14 weeks of ND (Fig. S3A,B). However, plasma glucose levels during ITT tended to be decreased in 18-week-old ubc13+/− mice (Fig. S3C). Moreover, male HFD-fed ubc13+/− mice tended to show improved insulin resistance compared to WT mice after 14 weeks of HFD (Fig. S3D–F), indicating that the effects of Ubc13 haploinsufficiency were mild in male mice compared to female mice. Although the reason for these sex differences is currently unclear, our results demonstrate that Ubc13 haploinsufficiency can ameliorate age-related and HFD-induced insulin resistance.

Female ubc13+/− mice are protected against age-related and HFD-induced insulin resistance.
(A–C) Blood glucose (A) and serum insulin (B) levels during OGTT and blood glucose (C) levels during ITT were measured for female WT and ubc13+/− mice at 8 weeks of age on ND (n = 4). (D–F) Blood glucose (D) and serum insulin (E) levels during OGTT and blood glucose (F) levels during ITT were measured for female WT and ubc13+/− mice at 18 weeks of age on ND (n = 6). *p < 0.05, **p < 0.01 versus WT mice. (G–I) Blood glucose (G) and serum insulin (H) levels during OGTT and blood glucose (I) levels during ITT were measured for female WT and ubc13+/− mice after 14 weeks of HFD (n = 5 to 6). HFD was started at 4 weeks of age. *p < 0.05, **p < 0.01 versus HFD-fed WT mice.
Female ubc13+/− mice are protected against HFD-induced hepatic steatosis and insulin resistance in liver and skeletal muscle
As chronic exposure of mice to HFD causes increased body weight and hepatic lipid accumulation related to insulin resistance4, we examined the status of the liver in female WT and ubc13+/− mice. On ND, no significant differences were observed in liver weight and fat accumulation between 18-week-old female WT and ubc13+/− mice (Fig. S4A,B). On HFD, liver weight and lipid accumulation were reduced in 18-week-old female ubc13+/− mice compared to WT mice (Fig. 3A,B). Measuring liver density by CT scan imaging confirmed clear decreases in lipid accumulation in female ubc13+/− mice on HFD (Fig. 3C).

Female ubc13+/− mice are protected against HFD-induced hepatic steatosis and insulin resistance.
(A) Liver weights of female WT and ubc13+/− mice were measured after 14 weeks of HFD (n = 5 to 6). *p < 0.05 versus HFD-fed WT mice. (B) Representative photographs of H&E staining of liver from female WT and ubc13+/− mice on ND or HFD. (C) Liver densities of female WT and ubc13+/− mice were calculated from abdominal CT scan on ND or HFD (n = 5 to 6). **p < 0.01 versus HFD-fed WT mice. (D) Liver lysates from female WT and ubc13+/− mice on HFD were analyzed by immunoblotting using antibodies for p-Akt (Ser473), total Akt, and Ubc13. The membrane was reprobed with anti-HSP90 antibody as a control. The intensities of phosphorylated-Akt after insulin injection were normalized to total Akt protein levels.
We then examined tissue-specific insulin sensitivity by immunoblotting. Akt phosphorylation was enhanced in livers of female HFD-fed ubc13+/− mice after insulin stimulation (Fig. 3D). Although there was no histological difference in muscle between 18-week-old female WT and ubc13+/− mice on a HFD, Akt phosphorylation was enhanced in muscles of HFD-fed ubc13+/− mice after insulin stimulation (Fig. S5A,B). These results support the interpretation that Ubc13 haploinsufficiency increases insulin sensitivity in female ubc13+/− mice.
Female ubc13+/− mice are protected against HFD-induced adipose tissue inflammation
Recent studies have demonstrated that HFD and aging induce chronic low-grade inflammation in adipose tissue, thereby contributing to insulin resistance4,11. We examined the status of the adipose tissues in female WT and ubc13+/− mice. No significant difference was observed in VAT weight on ND, whereas VAT weight was significantly reduced in 18-week-old HFD-fed female ubc13+/− mice compared to WT mice (Fig. 4A and S4B). Histological analysis of VAT revealed that adipocyte cell size was smaller in female HFD-fed ubc13+/− mice compared to WT mice (Fig. 4B). Because adipose tissue macrophage (ATM) infiltration is closely related to increased VAT weight4, we measured gene expression of F4/80, a macrophage specific marker, in VAT from WT and ubc13+/− mice by qRT-PCR. On ND, F4/80 expression in VAT tended to be reduced in 18-week-old female ubc13+/− mice compared to WT mice. On HFD, F4/80 expression in VAT was significantly reduced in 18-week-old female ubc13+/− mice compared to WT mice (Fig. 4C). ATM is an important source of inflammatory cytokines leading to insulin resistance. We therefore compared insulin sensitivity and cytokine gene expression in 18-week-old HFD-fed female WT and ubc13+/− VAT. Akt phosphorylation was enhanced in VAT of female HFD-fed ubc13+/− mice after insulin stimulation (Fig. 4D). Moreover, TNFα and IKKε expression were significantly reduced while IL-6 and IL-1β expression tended to be reduced in VAT of female HFD-fed ubc13+/− mice (Fig. 4E). In contrast on ND, there was no difference in TNFα, IL-6, IL-1β, and IKKε expression in VAT of ubc13+/− mice (Fig. 4F). Furthermore, no significant differences in F4/80 and inflammatory cytokine expression were observed between male HFD-fed WT and ubc13+/− mice despite improved insulin sensitivity (Fig. S6). These results indicate that Ubc13 haploinsufficiency does not directly regulate adipose tissue inflammation but rather the reduction in HFD-induced adipose tissue inflammation in female ubc13+/− mice is likely secondary to the reduction in weight gain and adipose mass.

Female ubc13+/− mice are protected against HFD-induced adipose tissue inflammation.
(A) VAT weights of female WT and ubc13+/− mice were measured after 14 weeks of HFD (n = 5 to 6). *p < 0.05 versus HFD-fed WT mice. (B) Immunohistochemical analysis of perilipin-2 in VAT from female WT and ubc13+/− mice on HFD. Adipocyte size of female WT and ubc13+/− mice on the HFD (n = 5). **p < 0.01 versus HFD-fed WT mice. (C) qRT-PCR analysis on the expression of the gene encoding F4/80 in VAT of female WT and ubc13+/− mice on HFD (n = 5 to 6). *p < 0.05 versus HFD-fed WT mice. (D) VAT lysates from female WT and ubc13+/− mice on HFD were analyzed by immunoblotting using antibodies for p-Akt (Ser473), total Akt, and Ubc13. The membrane was reprobed with anti-HSP90 antibody as a control. The intensities of phosphorylated-Akt after insulin injection were normalized to total Akt protein levels. (E,F) qRT-PCR analysis of the expression of the gene encoding TNFα, IL-6, IL-1β, and IKKε in VAT of female WT and ubc13+/− mice on HFD (n = 5 to 6) (E) and ND (n = 6) (F). *p < 0.05, **p < 0.01 versus HFD-fed WT mice.
Discussion
Recent studies have reported that obesity and aging can generate a state of chronic low-grade inflammation, leading to insulin resistance predominantly through NF-κB activation mediated by inflammatory signaling pathways such as TNFR, TLR, and Nlrp3 inflammasome5,9,11. Furthermore, Ubc13 is known to play a critical role in TNFR, TLR, and Nlrp3 signaling pathways. In this study, we found that Ubc13 haploinsufficiency improved insulin sensitivity in matured mice under ND. Additionally, female ubc13+/− mice showed increased energy expenditure and improved HFD-induced obesity, hepatic steatosis, and insulin resistance. Recently, it was reported that HFD-fed mice lacking IKKε, a direct transcriptional target of NF-κB, showed similar phenotypes to ubc13+/− mice due to increased energy expenditure23,24. In these mice, increased energy expenditure with increased UCP1 expression but unaccompanied by changes in RQ were observed. On the other hand, our data showed that fat oxidation was significantly increased in HFD-fed female ubc13+/− mice compared to WT mice. The mechanism underlying this discrepancy is unknown, but in our study, expression of NF-κB-inducible genes including IKKε was found to be reduced in HFD-fed female ubc13+/− mice. In addition, UCP1 expression in VAT tended to be increased in HFD fed female ubc13+/− mice compared to WT mice, suggesting that decreased IKKε expression may be one of the molecular mechanisms underlying these phenotypes.
Interestingly, even in ND, 18-week-old, but not 8-week-old, ubc13+/− mice showed improved insulin sensitivity compared to that in WT mice. The precise mechanism of these age-related effects remains unknown. We previously reported that macrophages and splenocytes derived from ND-fed ubc13+/− mice display blunted responses with respect to LPS and TNF-induced cytokine secretion and activation of JNK and NF-κB22. These findings suggest that mild suppression of age-related chronic low-grade JNK and NF-κB activation by Ubc13 haploinsufficiency could be effective to improve insulin sensitivity. However, no significant differences were observed in inflammatory cytokine expression between ND fed WT and ubc13+/− mice. These results suggest that other unknown functions might also exist between Ubc13 and insulin sensitivity. Recently, it was reported that Endoplasmic Reticulum (ER) stress is closely associated with insulin resistance via TRAF2/JNK activation, which cause metabolic diseases25,26,27,28. Since TRAF2 is one of the substrates of Ubc13, ER stress-mediated insulin resistance might be reduced in ubc13+/− mice. Further investigation will be required to identify the detailed mechanisms.
The sex difference that we observed in our study is also interesting. Although both male and female ubc13+/− macrophages and splenocytes had reduced LPS and TNF-induced signal activation22, male ubc13+/− mice showed milder phenotypes than female ubc13+/− mice compared to those in WT mice. Previously, Shi et al. reported that female, not male, mice lacking TLR4 were protected against HFD-induced insulin resistance through an unknown mechanism5. The explanation for the sex difference in our mice is unknown, but the TLR4/Ubc13 signaling pathway could play an important role in the molecular mechanisms in vivo. Recently, Zhang et al. reported that STAT3 restrains RANK- and TLR4-mediated signaling by suppressing expression of Ubc1329. Because estrogen is known to increase STAT3 activation30, the effect of estrogen-mediated STAT3 in Ubc13 may play a partial role in the sex difference. Further investigations are required to determine the relationship between STAT3 and Ubc13.
Since Ubc13 is a unique E2 that catalyzes formation of K63-linked polyubiquitin chains on various substrates, Ubc13 inhibitor is expected to be a novel candidate for the treatment of cancer and inflammatory diseases31. However, homozygous disruption of ubc13 resulted in embryonic lethality at a very early stage of development22. Various tissue-specific Ubc13-deficient mice have been reported and some of these mice show severe phenotypes32,33,34,35. For instance, keratinocyte-specific ubc13−/− mice die by postnatal day 234 and Treg cell-specific ubc13−/− mice develop autoimmune diseases35, indicating that even tissue-specific complete disruption of ubc13 can lead to severe toxicity with systemic ramifications. However, the haploinsufficient ubc13+/− mice used here grow normally and are protected against acute inflammation, suggesting that moderate Ubc13 inhibition may be effective in suppression of excessive inflammation or unknown dysfunction induced by aging and obesity. It remains to be determined whether hypothetical pharmacological inhibitors of Ubc13 could be dosed in a manner that generates an adequate therapeutic index.
In conclusion, we have shown that Ubc13 haploinsufficiency protects against age-related insulin resistance and HFD-induced obesity. Although other functions of Ubc13 still remain unclear and further investigations are required, Ubc13 may have novel potential as a future target for discovering treatments for insulin resistance, obesity, and type 2 diabetes.
Methods
Animals
Ubc13 heterozygous mice were generated as previously described22. The mice were backcrossed at least 6 generations onto a C57BL/6 background, and housed in a specific pathogen-free environment at ambient temperature of 25 °C with a dark-light cycle of 10 and 14 hrs, respectively. WT littermates were used as controls. Animal care and procedures were approved by the Animal Care Committee of Kyoto University. The methods were carried out in accordance with the Animal Care Committee of Kyoto University. The mice were weaned at 4 weeks of age, and fed control fat chow (ND; 10% fat, 20% protein, and 70% carbohydrate by energy) or high fat chow (HFD; 60% fat, 20% protein, and 20% carbohydrate by energy) (Research Diets Inc., New Brunswick, NJ) for 14 weeks.
Oral glucose tolerance test (OGTT)
After a 16 hrs fasting period, OGTTs (0.75 g/kg body weight) were performed. Blood samples were taken at the indicated times (0, 15, 30, 60, and 120 min after glucose loading), and blood glucose levels and plasma insulin levels were measured. Blood glucose levels were determined by the enzyme-electrode method. Plasma insulin levels were determined using enzyme immunoassay (Shibayagi, Gumma, Japan).
Insulin tolerance test (ITT)
At a dose of 0.4 U/kg body weight (for ND fed mice) or 1 U/kg body weight (for HFD fed mice) human insulin (Novonordisk, Copenhagen, Denmark) was injected subcutaneously after a 2-h fasting period. Blood samples were collected at the indicated times (0, 15, 30, 60, 90, and 120 min after the loading). Blood-glucose levels were measured as described above.
Energy expenditure
Energy expenditure was evaluated by measuring respiratory quotient and oxygen consumption by indirect calorimetry every 5 min for 24 hrs under the fed condition36. Air from the room was pumped through the chamber, and expired gas was dried in a cotton thin column and subjected to gas analysis (Alco System model 2000, Chiba, Japan). Oxygen consumption (VO2) and carbon dioxide production (VCO2) were measured, and respiratory quotient (RQ), energy expenditure, and fat oxidation were calculated as follows:

Energy expenditure = 3.816 × VO2 + 1.231 × VCO2 [cal/min] (by using the Lusk equation37).
Fat oxidation = 1.67 × (VO2-VCO2) [mg/min] (by using Frayn equation38).
The locomotor activity of the mice was measured using an automated activity counter (NSAS01; Neuroscience, Tokyo, Japan).
Computed tomography
Computed tomography (CT) scanning was performed as previously described39. Briefly, mice were anesthetized with Nembutal and fixed in a chamber, and transaxially scanned using Latheta (LCT-100M) experimental animal CT system (Aloka, Tokyo, Japan). The whole body was scanned, and contiguous 1-mm slice images of the trunk were used for quantitative assessment (Latheta software, version 1.00). Fat content in liver was quantitatively evaluated.
Real-time PCR analysis
Total RNA from adipose tissue was extracted using TRIzol solution (Invitrogen, Grand Island, NY, USA) according to the manufacturer’s instructions40. For complementary DNA synthesis, 1 μg of total RNA was reverse-transcribed using the reverse transcription system (PrimeScript RT reagent kit, Takara Bio, Shiga, Japan). SYBER Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) was prepared for real-time quantitative PCR using ABI StepOnePlus Real-Time PCR Systems (Applied Biosystems, Foster City, CA, USA). The PCR was performed for 10 min at 90 °C, followed by 50 cycles at 95 °C for 15 sec and at 60 °C for 1 min. The signal of the products was standardized against the GAPDH signal for each sample. Primer pairs for F4/80, TNFα, IL-6, IL-1β, IKKε, UCP1, and GAPDH are shown as follows.: F4/80 (165 bp): 5′-ctt tgg cta tgg gct tcc agt c-3′ and 5′-gca agg agg aca gag ttt atc gtg-3′; TNFα (146 bp): 5′-aaa tgg gct ttc cga att ca-3′ and 5′-cag gga aga atc tgg aaa ggt-3′; IL-6 (78 bp): 5′-gct acc aaa ctg gat ata atc agg a-3′ and 5′-cca ggt agc tat ggt act cca gaa-3′; IL-1β (89 bp): 5′-gca act gtt cct gaa ctc aac t-3′ and 5′-atc ttt tgg ggt ccg tca act-3′; IKKε (148 bp): 5′-aca agg ccc gaa aca aga aat-3′ and 5′-act gcg aat agc ttc acg atg-3′; UCP1 (107 bp): 5′-gga ttg gcc tct acg act ca-3′ and 5′-tgc cac acc tcc agt cat ta-3′; GAPDH (100 bp): 5′-aaa tgg tga agg tcg gtg tg-3′ and 5′-tcg ttg atg gca aca atc tc-3′.
Immunohistochemistry
Liver and visceral fat samples were fixed in 10% formalin buffer, embedded in paraffin, and sectioned at 3 μm. The paraffin sections of the liver were stained with hematoxylin and eosin (HE). Images were taken using a microscope with the BZ-8100 system (KEYENCE Corp., Osaka, Japan). The sections of visceral fat were blocked with 3% bovine serum albumin and then incubated overnight at 4 °C with a monoclonal rabbit anti-perilipin 2 antibody (LSBio, Seattle, WA) and afterward with a secondary antibody at room temperature at 1 h. After immunostaining, the mean adipocyte size (surface areas of 15 representative adipocytes per mouse) was analyzed by BZ Analyzer software (KEYENCE Corp.).
Immunoblotting and antibodies
To compare Akt-phosphorylation levels, insulin (2 U per kg body weight) was injected via inferior vena cava under regular fed condition. 3 min after the injection, adipose tissue, liver tissue, and muscles were collected. Frozen tissue extracts were minced and homogenized in cold RIPA buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% NP40, 0.1% SDS, 1 mM EDTA, phosphatase inhibitor cocktail (Sigma-Aldrich), and protease inhibitor cocktail (Roche), and then were centrifuged at 12,000 × g for 5 min. Equal amounts of cell lysates were subjected to immunoblot analysis. Anti-Akt, anti-phospho-Akt (Ser473) antibodies were obtained from Cell Signaling. Quantification of bands on western blot was accomplished by scanning the blots, then determining the densities of the bands using ImageJ software (National Institutes of Health, Bethesda, MD, http://imagej.nih.gov/ij/, 1997–2012). Anti-Ubc13 was obtained from Zymed laboratories. Anti-HSP90 antibody was purchased from Santa Cruz Biotechnology.
Statistical analysis
All data were expressed as mean ± SE. Statistical analysis was performed using Student’s t-test. Significant difference was considered to be present at p < 0.05.
Additional Information
How to cite this article: Joo, E. et al. Ubc13 haploinsufficiency protects against age-related insulin resistance and high-fat diet-induced obesity. Sci. Rep. 6, 35983; doi: 10.1038/srep35983 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Hossain, P., Kawar, B. & El Nahas, M. Obesity and diabetes in the developing world–a growing challenge. The New England journal of medicine 356, 213–215, 10.1056/NEJMp068177 (2007).
Hotamisligil, G. S. Inflammation and metabolic disorders. Nature 444, 860–867, 10.1038/nature05485 (2006).
Schenk, S., Saberi, M. & Olefsky, J. M. Insulin sensitivity: modulation by nutrients and inflammation. The Journal of clinical investigation 118, 2992–3002, 10.1172/JCI34260 (2008).
Johnson, A. M. & Olefsky, J. M. The origins and drivers of insulin resistance. Cell 152, 673–684, 10.1016/j.cell.2013.01.041 (2013).
Shi, H. et al. TLR4 links innate immunity and fatty acid-induced insulin resistance. The Journal of clinical investigation 116, 3015–3025, 10.1172/JCI28898 (2006).
Tsukumo, D. M. et al. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes 56, 1986–1998, 10.2337/db06-1595 (2007).
Saberi, M. et al. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell metabolism 10, 419–429, 10.1016/j.cmet.2009.09.006 (2009).
Himes, R. W. & Smith, C. W. Tlr2 is critical for diet-induced metabolic syndrome in a murine model. Faseb J 24, 731–739, 10.1096/Fj.09-141929 (2010).
Uysal, K. T., Wiesbrock, S. M., Marino, M. W. & Hotamisligil, G. S. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 389, 610–614 (1997).
Wen, H. T. et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol 12, 408–U461, 10.1038/Ni.2022 (2011).
Youm, Y. H. et al. Canonical Nlrp3 Inflammasome Links Systemic Low-Grade Inflammation to Functional Decline in Aging. Cell metabolism 18, 519–532, 10.1016/j.cmet.2013.09.010 (2013).
Spence, J., Sadis, S., Haas, A. L. & Finley, D. A Ubiquitin Mutant with Specific Defects in DNA-Repair and Multiubiquitination. Mol Cell Biol 15, 1265–1273 (1995).
Hofmann, R. M. & Pickart, C. M. Noncanonical MMS2-encoded ubiquitin-conjugating enzyme functions in assembly of novel polyubiquitin chains for DNA repair. Cell 96, 645–653, 10.1016/S0092-8674(00)80575-9 (1999).
Laine, A. & Ronai, Z. Ubiquitin chains in the ladder of MAPK signaling. Science’s STKE: signal transduction knowledge environment 2005, re5, 10.1126/stke.2812005re5 (2005).
Chen, Z. J., Bhoj, V. & Seth, R. B. Ubiquitin, TAK1 and IKK: is there a connection? Cell Death Differ 13, 687–692, 10.1038/sj.cdd.4401869 (2006).
Deng, L. et al. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 103, 351–361 (2000).
Wang, C. et al. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412, 346–351, 10.1038/35085597 (2001).
Habelhah, H. et al. Ubiquitination and translocation of TRAF2 is required for activation of JNK but not of p38 or NF-kappa B. Embo J 23, 322–332, 10.1038/sj.emboj.7600044 (2004).
Baud, V. & Karin, M. Signal transduction by tumor necrosis factor and its relatives. Trends in cell biology 11, 372–377 (2001).
Aggarwal, B. B. Signalling pathways of the TNF superfamily: a double-edged sword. Nature reviews. Immunology 3, 745–756, 10.1038/nri1184 (2003).
Akira, S. & Takeda, K. Toll-like receptor signalling. Nature Reviews Immunology 4, 499–511, 10.1038/Nri1391 (2004).
Fukushima, T. et al. Ubiquitin-conjugating enzyme Ubc13 is a critical component of TNF receptor-associated factor (TRAF)-mediated inflammatory responses. P Natl Acad Sci USA 104, 6371–6376, 10.1073/pnas.0700548104 (2007).
Chiang, S. H. et al. The protein kinase IKKepsilon regulates energy balance in obese mice. Cell 138, 961–975, 10.1016/j.cell.2009.06.046 (2009).
Reilly, S. M. et al. An inhibitor of the protein kinases TBK1 and IKK-varepsilon improves obesity-related metabolic dysfunctions in mice. Nature medicine 19, 313–321, 10.1038/nm.3082 (2013).
Ozcan, U. et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science (New York, N.Y.) 306, 457–461, 10.1126/science.1103160 (2004).
Tcherpakov, M. et al. Regulation of endoplasmic reticulum-associated degradation by RNF5-dependent ubiquitination of JNK-associated membrane protein (JAMP). J Biol Chem 284, 12099–12109, 10.1074/jbc.M808222200 (2009).
Hotamisligil, G. S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140, 900–917, 10.1016/j.cell.2010.02.034 (2010).
Ozcan, U. et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science (New York, N.Y.) 313, 1137–1140, 10.1126/science.1128294 (2006).
Zhang, H. et al. STAT3 restrains RANK- and TLR4-mediated signalling by suppressing expression of the E2 ubiquitin-conjugating enzyme Ubc13. Nature communications 5, 5798, 10.1038/ncomms6798 (2014).
Dziennis, S. et al. Role of signal transducer and activator of transcription-3 in estradiol-mediated neuroprotection. The Journal of neuroscience: the official journal of the Society for Neuroscience 27, 7268–7274, 10.1523/jneurosci.1558-07.2007 (2007).
Madiraju, C. et al. TR-FRET-based high-throughput screening assay for identification of UBC13 inhibitors. Journal of biomolecular screening 17, 163–176, 10.1177/1087057111423417 (2012).
Yamamoto, M. et al. Key function for the Ubc13 E2 ubiquitin-conjugating enzyme in immune receptor signaling. Nat Immunol 7, 962–970, 10.1038/ni1367 (2006).
Yamamoto, M. et al. Cutting Edge: Pivotal function of Ubc13 in thymocyte TCR signaling. Journal of immunology 177, 7520–7524 (2006).
Sayama, K. et al. E2 Polyubiquitin-conjugating Enzyme Ubc13 in Keratinocytes Is Essential for Epidermal Integrity. J Biol Chem 285, 30042–30049, 10.1074/jbc.M110.106484 (2010).
Chang, J. H. et al. Ubc13 maintains the suppressive function of regulatory T cells and prevents their conversion into effector-like T cells. Nat Immunol 13, 481–U484, 10.1038/Ni.2267 (2012).
Naitoh, R. et al. Inhibition of GIP signaling modulates adiponectin levels under high-fat diet in mice. Biochem Bioph Res Co 376, 21–25, 10.1016/j.bbrc.2008.08.052 (2008).
Lusk, G. Animal calorimetry. Twenty-fourth paper. Analysis of the oxidation of mixtures of carbohydrate and fat. A correction. J Biol Chem 59, 41–42 (1924).
Frayn, K. N. Calculation of substrate oxidation rates in vivo from gaseous exchange. Journal of applied physiology: respiratory, environmental and exercise physiology 55, 628–634 (1983).
Nasteska, D. et al. Chronic Reduction of GIP Secretion Alleviates Obesity and Insulin Resistance Under High-Fat Diet Conditions. Diabetes 63, 2332–2343, 10.2337/Db13-1563 (2014).
Joo, E. et al. Enteral supplement enriched with glutamine, fiber, and oligosaccharide attenuates experimental colitis in mice. Nutrition 29, 549–555, 10.1016/j.nut.2012.09.007 (2013).
Acknowledgements
We thank S. Yasui and M. Akazawa for technical assistance and S. Asano for helpful advice. This study was supported by Scientific Research grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan and by the Kyoto University Global COE Program, Center for Frontier Medicine.
Author information
Authors and Affiliations
Contributions
E.J., T.F., N.H., S.-i.M., J.C.R. and N.I. designed research; E.J. and T.F. performed research; E.J., T.F. and N.I. analyzed data; and E.J., T.F., S.-i.M., J.C.R. and N.I. wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Joo, E., Fukushima, T., Harada, N. et al. Ubc13 haploinsufficiency protects against age-related insulin resistance and high-fat diet-induced obesity. Sci Rep 6, 35983 (2016). https://doi.org/10.1038/srep35983
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep35983
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
