
Abstract
The S385Y/D469T/R520Q variant of E. coli transketolase was evolved previously with three successive smart libraries, each guided by different structural, bioinformatical or computational methods. Substrate-walking progressively shifted the target acceptor substrate from phosphorylated aldehydes, towards a non-phosphorylated polar aldehyde, a non-polar aliphatic aldehyde, and finally a non-polar aromatic aldehyde. Kinetic evaluations on three benzaldehyde derivatives, suggested that their active-site binding was differentially sensitive to the S385Y mutation. Docking into mutants generated in silico from the wild-type crystal structure was not wholly satisfactory, as errors accumulated with successive mutations, and hampered further smart-library designs. Here we report the crystal structure of the S385Y/D469T/R520Q variant, and molecular docking of three substrates. This now supports our original hypothesis that directed-evolution had generated an evolutionary intermediate with divergent binding modes for the three aromatic aldehydes tested. The new active site contained two binding pockets supporting π-π stacking interactions, sterically separated by the D469T mutation. While 3-formylbenzoic acid (3-FBA) preferred one pocket, and 4-FBA the other, the less well-accepted substrate 3-hydroxybenzaldehyde (3-HBA) was caught in limbo with equal preference for the two pockets. This work highlights the value of obtaining crystal structures of evolved enzyme variants, for continued and reliable use of smart library strategies.
Similar content being viewed by others
Introduction
Transketolase (TK) (EC 2.2.1.1) is a ubiquitous thiamine diphosphate (ThDP)-dependent enzyme linking the non-oxidative pentose phosphate pathway and the Calvin cycle1. It catalyses the reversible transfer of a two-carbon moiety from a ketol donor to an aldehyde acceptor2,3. Transketolase has been used to form asymmetric carbon-carbon bonds in biocatalytic organic synthesis4,5,6, where the use of β-hydroxypyruvate (HPA) as the ketol donor renders the donor half-reaction irreversible. The stereoselective synthesis of α,α’-dihydroxyketones can be directed to the synthesis of ketosugars6, chiral aminodiols7,8 and other high-value synthons9,10,11,12.
Besides the biocatalytic potential, TK has proven to be a suitable model to evaluate new directed-evolution approaches13,14,15, in which small libraries were found to be effective with several targeting strategies, such that by altering even highly-conserved sites, the substrate specificity of an enzyme could be modified significantly. Using relatively small libraries, 100-fold improvements in efficiency can be achieved compared to error-prone PCR (EP-PCR)16 approaches. Such methods have been successfully applied to lipases, for example, to expand substrate-range via combinatorial active-site saturation test (CASTing) or improve thermostability via iterative saturation mutagenesis (ISM)17,18. Likewise, constrained alphabet approaches have proven successful tools to maximise directed evolution efficiency by creating smaller and/or smarter libraries14,19,20,21.
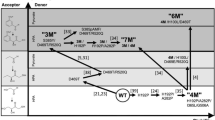
For E. coli TK a series of smart library approaches were combined previously in an overall substrate-walking approach, to shift the substrate specificity from phosphorylated to non-phosphorylated polar acceptors13, then to non-polar aliphatic substrates22,23,24, and on to hetero-aromatic25 and non-polar aromatic substrates15,26. As shown in Fig. 1, a different library design strategy was used at each stage to guide saturation mutagenesis, including the use of structural and phylogenetic information13,22, statistical coupling analysis (SCA) methods to target networks for re-stabilisation14, and substrate docking15. In the latter stage, saturation mutagenesis of two TK active-site residues led to variants, including S385Y/D469T/R520Q, that were active on three benzaldehyde derivatives15 (Fig. 2), in contrast to wild-type TK which was active only on non-aromatic aldehydes25.

Successive library design strategies used to evolve E. coli TK towards aromatic aldehydes.

Structures of aromatic aldehydes used with TK variants.
Interestingly, kinetic analysis of S385Y/D469T/R520Q (Table 1) showed that this variant improved the activities towards the three benzaldehyde analogues, 3-formylbenzoic acid (3-FBA), 4-formylbenzoic acid (4-FBA) and 3-hydroxybenzaldehyde (3-HBA), in three different ways15. For 3-FBA, the variant displayed a 10-fold improved (lower) Km, yet only marginally increased kcat by 16%, relative to double mutant D469T/R520Q, to give an 11.5-fold increase in kcat/Km at 5400 ± 1490 s−1 M−1. By contrast, the same variant increased kcat towards 4-FBA 8.5-fold, and decreased only Km marginally, for a 13.6-fold improved kcat/Km. Finally for 3-HBA, the kcat and Km were both improved from previously unmeasurable values, to give a respectable kcat/Km of 5.4 s−1 M−1, albeit still with a relatively poor Km of 390 mM. These contrasting observations suggested that the positioning and orientation of each of the tested substrates within the active-site was differentially sensitive to the S385Y mutation, and that this was impacting both Km and kcat. Thus evolutionary divergence into distinct binding modes was speculated to have arisen through different orientations of the aldehyde moiety for the three aromatic-aldehyde substrates, relative to the cofactor. Such a mechanism was difficult to confirm by substrate docking into an enzyme structure generated by modelling of three active-site mutations. This also made further rounds of directed evolution more challenging when using structure-docking approaches to guide small library designs, particularly where the different substrates potentially require the design of distinct libraries. To elucidate the impact of the three active-site mutations, we have now determined the crystal structure of S385Y/D469T/R520Q E. coli TK. We have then used molecular docking to identify potential substrate-binding mechanisms that led to the divergent kinetic behaviour observed with the benzaldehyde analogues.
Results and Discussion
Structural comparison of S385Y/D469T/R520Q and wild-type E. coli TK
The crystal structure of S385Y/D469T/R520Q E. coli TK in the presence of its cofactors was determined to 1.50 Å resolution with very good statistics (Rwork: 12,58%, Rfree: 15,69%) (Table 2).
The structural analysis of EcTK S385Y/D469T/R520Q could unambiguously confirm the introduced mutations (Fig. 3).

X-ray crystal structure of EcTK S385Y/D469T/R520Q highlighting the mutation sites at the active site (encircled).
The homodimer is shown in cartoon representation with individual colors for the two chains (A in yellow, B in magenta). Mutated residues Tyr385, Thr469 and Gln520 are shown in ball-and-stick representation. The calculated electron density of the three amino acid residues is shown at a contour level of 1.5σ in a 2Fo-Fc map.
A structural alignment of the variant with wild-type E. coli TK in the resting state (pdb code 1QGD, resolution 1.90 Å)27 and in complex with substrate D-xylulose-5-phosphate (pdb code 2R8O, 1.47 Å) determined previously28 revealed an RMSD (using all 662 residues per chain, and 99% of sequence in PDBeFold29) for the dimer of 0.37 Å and 0.40 Å, respectively. This indicates very little structural change resulting from the three mutations. In fact, there was no evidence of any significant shifts in backbone, sidechain or cofactor structure compared to the wild-type protein. Comparison with our previously reported energy-minimised model showed structural differences at all 3 positions, with the aromatic ring of S385Y rotated nearly 90° in the model. Therefore, the observed changes in substrate affinity and catalysis are very likely all directly attributable to the mutational sites themselves. A superposition of EcTK S385Y/D469T/R520Q with EcTK wild-type in covalent complex with substrate D-xylulose-5-phosphate (pdb code 2R8O) provides structural insights that can explain the observed functional changes (Fig. 4). The aromatic side chain of introduced residue Y385 is accommodated almost coplanar 7–8 Å above the thiazolium ring of ThDP thus generating a snug π-π stacking binding pocket for aromatic substrates (sandwiched between Y385 and ThDP). The binding of aliphatic phospho-sugar substrates is clearly impeded as the Y385 side chain would clash with the substrate phosphate moiety, and the S385Y mutation also removes a hydrogen-bonding site to the S385 hydroxyl moiety, which is known to interact with phosphate groups in the natural substrates of TK28. In the triple variant, the side chain of Y385 is held in place through a network of H-bonding interactions involving 3 water molecules and residue G262. The D469T mutation removed a salt bridge to R91, and replaced it with a hydrogen bond from the T469 hydroxyl moiety. The mutation also created a small pocket in which the R91 guanidinium moiety is more solvent accessible within the enzyme active site, and also available for hydrogen bonding to potential substrates. The phenyl hydroxyl moiety of Y385 instead formed a hydrogen bond to the backbone carbonyl of Gly262. The R520Q mutation created a wider opening to the active-site funnel, which may compensate for the decreased steric access for substrates, introduced by the S385Y mutation.

Superposition of the active site of EcTK S385Y/D469T/R520Q (yellow) with that of wild-type EcTK in complex with substrate D-xylulose-5-phosphate (grey).
The mutation sites are highlighted in magenta. Note that the salt bridge between Asp469 and Arg91 (red dashes) has been removed in the variant. The OH of introduced Tyr385 H-bonds to residue Gly262 via three water molecules with no known equivalent in the wild-type structure (black dashes).
Computational docking of aromatic aldehydes into S385Y/D469T/R520Q
Computational molecular docking of polar aromatic substrates 3-FBA, 4-FBA and 3-HBA into the active site of the TK triple-mutant, each produced clusters with very different binding behaviours. After energetic curation, the poses generated for 3-FBA and 4-FBA each produced a single cluster, distinctly different from each other, and with 100% of poses in catalytically productive orientations. Catalytically productive was broadly defined as having aldehyde moieties placed within 4Å of the enamine-ThDP cofactor intermediate. By contrast, 3-HBA resolved into two distinct clusters (Fig. 5), with 77% of poses clustered into a potentially productive orientation (cluster A). The other 23% of 3-HBA poses clustered into a distinctly different binding location (cluster B), with a calculated affinity only slightly higher, but in a non-productive orientation.

Energy clusters generated by molecular docking of 3-HBA, 3-FBA and 4-FBA into the active site of the triple-mutant transketolase.
Two distinct active-site binding pockets were observed overall (Fig. 6). The contacts they make with docked substrates are shown schematically in Fig. 7, and summarised in Table 3. Pocket 1 was formed on one face of the T469 sidechain, and docked 3-FBA in a productive orientation, and the less populated 3-HBA cluster B in an unproductive orientation. The aldehyde O-atom of 3-FBA, and the hydroxyl moiety of 3-HBA in cluster B, formed hydrogen bonds to the sidechains of H26 and H216. The carboxylate of 3-FBA, and the aldehyde carbonyl of 3-HBA formed hydrogen bonds to the guanidine moiety of R91, and for 3-FBA only, also to the T469 hydroxyl moiety. The substrate aromatic ring bound edge-to-edge to the F434 ring, in a π-π stack with an interplanar angle between the aromatic ring faces of 90°, and with the F434 ring-edge placed 3.3–3.6 Å below the face of the substrate ring. The Y385 aromatic ring-edge interacted with the substrate ring face, with an interplanar angle of 45°, and the substrate ring-edge positioned 3.7–4.4 Å above the Y385 ring face. This form of π-π stacking at a 45° interplanar angle is suboptimal and lies at an energetic saddle point between the more stable π-π sandwich (0°) and edge-on T-shaped (90°) orientations.

Computational docking of 3-HBA (yellow), 3-FBA (orange) and 4-FBA (blue) into the active site of S385Y/D469T/R520Q E. coli transketolase.
Oxygen atoms (red) identify the position of the aldehyde moiety relative to ThDP. 3-FBA and the unproductive pose of 3-HBA (cluster B, cyan surface), each hydrogen bond to the R91 and T469 sidechains, while their aromatic rings are positioned at 90° to the F434 ring, and 45° to the Y385 ring. 4-FBA and the productive pose of 3-HBA (cluster A, magenta surface) hydrogen bond to the T469 sidechain, while their aromatic rings are positioned at 45° to the F434 ring, and 90° to the Y385 ring. The two binding pockets are separated by the T469 sidechain.

Non-covalent interactions involved in the ligand-TK complex of (a) unproductive 3-HBA (b) productive 3-HBA (c) 3-FBA and (d) 4-FBA. Black and green dashed lines respectively refer to hydrogen bond and π-π stacking interactions. Hydrophobic interactions are shown as green solids lines next to the effecting amino acid.
Pocket 2 was formed on the opposite face of T469 to pocket 1, and docked both 4-FBA and the productive pose of 3-HBA, with the aldehyde O-atoms hydrogen bonded to the sidechains of H26 and H216. The hydroxyl moiety of 3-HBA, or the carboxyl moiety of 4-FBA, was oriented towards L466, in close proximity to the T469 β-methyl group, but also with the potential to hydrogen bond to the T469 hydroxyl moiety. The substrate aromatic ring bound edge-to-edge to the Y385 ring, with an interplanar angle of 90°, and with the substrate ring edge placed 3.9 Å below the face of the Y385 ring. The F434 aromatic ring-edge interacted with the substrate ring face, with an interplanar angle of 45°, and the F434 ring edge was positioned 3.3 Å above the substrate ring.
Overall, there was a clear similarity between the binding modes of the two pockets, which docked the substrates with the aldehyde moieties in similar positions, but with an approximately 45° rotation of the ring faces relative to each other, pivoted around the aldehyde moiety position. This 45° rotation resulted from binding of the aromatic substituents into the pockets on either face of T469, where both docking modes provided similar π-π stacking to the F434 and Y385 aromatic rings, with interplanar angles of 90° and 45° respectively for pocket 1, but 45° and 90° respectively for pocket 2. The additional hydrogen bonding to R91 in pocket 1, provided an additional interaction for 3-FBA (and the unproductive 3-HBA mode) that may explain the lower Km observed for 3-FBA (Table 1).
The unproductive 3-HBA orientation in pocket 1 described, should not be disregarded entirely because a simple 180° coplanar rotation of the aromatic ring would produce a catalytically productive pose, and also retain a hydrogen bond to R91. This may be a docking artefact based on assigned energy contributions for hydrogen bonding to the carbonyl and hydroxyl moieties of 3-HBA. It could be that either or both orientations of 3-HBA in pocket 1 can populate. Either way, the potential to bind a population of 3-HBA into pocket 1 in an unproductive orientation, in addition to the productive orientations in at least pocket 2, would contribute to the relatively high Km observed for 3-HBA. The two clusters may yet resolve into a single binding orientation for 3-HBA if further directed evolution was targeted specifically towards improving activity with 3-HBA.
Inspection of the docked substrates allows us to examine why 3-FBA was only found in pocket 1, and why 4-FBA was only found in pocket 2. 3-FBA cannot bind into pocket 2 with the productive aldehyde-moiety conformation, and the π-π stacking arrangement to F434 and Y385 predicted for 4-FBA as the carboxylate moiety in 3-FBA would be sterically constrained either by H473, or by Y385 for the 180° rotation of the 3-FBA aromatic ring around the bond to the aldehyde moiety. While 3-HBA was predicted to dock in a productive conformation into pocket 2, this was at the expense of a displacement of the aromatic ring edge from the optimal position for π-π stacking to Y385 found for 4-FBA. This observation may also contribute towards the higher Km of 3-HBA when compared to 4-FBA.
Conversely, 4-FBA would not favour binding into pocket 1 while retaining the productive aldehyde-moiety conformation, and the π-π stacking arrangement predicted for 3-FBA, as it would not then be able to form the hydrogen bond to R91. Instead, the carboxylate moiety of 4-FBA would be oriented out towards the active-site entrance. Alternatively, to form the hydrogen bond with R91, 4-FBA would not be able to form the same π-π stacking interactions with Y385 and F434 as predicted for 3-FBA.
Role of mutations in evolution of catalytic activity and specificity
The structural formation of two binding-pockets in the TK triple-mutant, and prediction of differential substrate docking between the two pockets, can be used to rationalise the impact of the combined mutations upon the observed kinetics for the three substrates. We also suggest plausible explanations for the contributions of each mutation as they occurred along the evolutionary trajectory from wild type, to D469T, D469T/R520Q, and finally S385Y/D469T/R520Q.
In previous work, the wild-type enzyme was found to be inactive on the three aromatic aldehyde substrates15,26. The D469T mutation introduced activity towards 3-FBA and to a 10-fold lesser degree towards 4-FBA, which had a 5-fold higher Km. The acceptance of aromatic substrates induced by the D469T mutation can be explained sterically, as above, by the formation of two binding pockets on either side of T469 in which the γ-hydroxyl and γ-methyl groups of T469 formed a steric barrier between the pockets (Fig. 8a). D469T also created the space for the benzene rings of all three aldehyde substrates, that was otherwise hindered by D469 in the wild type. The lower Km for 3-FBA compared to 4-FBA would result from the increased access to R91, which then forms a hydrogen bond to the carboxylate moiety of 3-FBA in pocket 1. The carboxyl moieties of 3-FBA and 4-FBA can each form a hydrogen bond with the γ-hydroxyl group of T469 as this moiety can orient towards either pocket 1 or pocket 2 as necessary.

Alignment of the crystal structures of wild-type E. coli transketolase, the S385Y/D469T/R520Q triple-mutant, and the pose for 3-FBA (orange) docked into the triple-mutant using Autodock.
(a) D469 (green) points towards the inside of the active site while D469T (magenta) provides an alternative binding pocket for a phenyl ring. (b) The R520 guanidinium group (blue) obstructs substrate access through the binding funnel compared to R520Q (buried in the green surface). (c) I189 (purple), L382 (pink), S385Y (magenta) and F434 (red) form a planar hydrophobic pocket which S385Y encloses.
The R520Q mutation improved the Km for both substrates 5–10 fold, yet also decreased kcat 2-fold and 25-fold for 3-FBA and 4-FBA respectively. Improvements in Km can be explained through improved steric access to the active site (Fig. 8b). However, the mechanism of influence of this mutation on kcat is not explained by steric access. Also R520 is 13 Å away from the ThDP cofactor, and hence too far to electronically influence the catalytic residues. However, interaction of R520 with the carboxylate moiety of 3-FBA or 4-FBA would increase the reactivity of the aldehydes by withdrawing electrons, particularly for 4-FBA. Hence the R520Q mutation would be expected to decrease the activity of 4-FBA in particular, as was observed by the decreases in kcat. However, this effect would also be convoluted with any shift that may have occurred to the binding position and orientation of the aldehydes relative to the cofactor and catalytic residues. If R520 did interact directly with the carboxylate moieties of 3-FBA or 4-FBA in the D469T variant, then this would necessarily pull these substrates further from ThDP and also orient them differently to the docked poses predicted in the triple-mutant. Hence removing this interaction with the R520Q mutation, could also allow more favourable binding close to the ThDP, and in this way contribute to an improved kcat.
Introduction of the S385Y mutation into D469T/R520Q to produce the triple-mutant creates an enclosed hydrophobic binding-pocket for all three benzaldehyde substrates. All three substrates docked with their aldehyde moieties hydrogen bonded to H26 and H261. As seen in Fig. 8c, this also brings part of their benzene rings into the hydrophobic pocket formed by three coplanar residues I189, L382 and F434. These are members of a previously identified coevolved network of six residues thought to be important for transketolase activity and stability14. The Y385 phenyl ring then forms a cap above these residues to enclose the hydrophobic pocket, and the substrate. In Fig. 8c, 3-FBA is shown with the aromatic ring perpendicular to F434. The S385Y mutation, led previously to a 10-fold decrease in Km for 3-FBA, without influencing kcat. This suggests that the binding position and orientation predicted in the docking of 3-FBA to S385Y/D469T/R520Q, was essentially already in place with D469T/R520Q. The improved Km thus likely resulted from the new 45° π-π stacking interaction with Y385. By contrast, the Km for 4-FBA decreased less than two-fold, but the kcat increased almost 10-fold. As above, a strong influence on kcat for 4-FBA suggests that the S385Y mutation repositioned this substrate relative to the ThDP-enamine intermediate for more efficient catalysis. 4-FBA formed a favourable 90° π-π stacking arrangement with Y385, which would be expected to improve the Km by more than that observed for 3-FBA which only formed the less optimal 45° π-π stacking arrangement. However, the opposite was observed and this is also consistent with an additional movement of the position of the 4-FBA substrate due to the S385Y mutation.
Regardless of the evolutionary route to the kinetics observed in the final triple-mutant S385Y/D469T/R520Q, the end result was a divergence into two binding modes that separate 3-FBA from 4-FBA, but where 3-HBA was still able to bind into both pockets. The structural insights obtained for S385Y/D469T/R520Q and the divergent binding modes, now present different strategies for further engineering of the specificities of each substrate. For example, engineering the specificity towards 4-FBA and 3-HBA, over that for 3-FBA, might best be explored by targeting residues only in binding pocket 2, such as L466. By contrast, re-targeting T469 may allow further fine-tuning of the specificity to all three substrates.
In summary, docking into the new crystal structure for the S385Y/D469T/R520Q variant of TK supports the original hypothesis that directed evolution towards 3-FBA and 4-FBA had led to a divergence in their binding modes. While D469T had already provided a steric boundary between the two binding pockets observed, S385Y introduced π-π stacking interactions differentially for 3-FBA and 4-FBA, and improved the newly evolved activities towards both substrates. The less well accepted substrate 3-HBA, not specifically evolved for, was found to be caught in limbo with the potential for binding to both pockets. Furthermore, this work provides a novel enzyme engineering paradigm whereby a series of semi-rational directed evolution strategies are sequentially built upon and then resolved through crystallography. This will then allow protein engineers to avoid the accumulation of computational errors inherent to models, and provide an informed basis for further semi-rational directed evolution, again guided by molecular docking.
Methods
All chemical reagents were purchased from Sigma-Aldrich (Aldrich Chemistry, UK) unless otherwise stated.
Triple-mutant construction
The triple-mutant S385Y/D469T/R520Q was constructed from plasmid pQR791 containing the tktA gene as described previously13. Mutations were introduced with mutagenic primers according to the manufacturer’s instructions using the Quickchange kit (Agilent Technologies, USA). Mutations were confirmed by DNA sequencing on both strands.
Crystallisation and Structure Determination
TK triple variant S385Y/D469T/R520Q was expressed and purified to homogeneity as detailed for the wild-type enzyme28. The freshly purified protein was crystallized by the hanging-drop vapor-diffusion method relying on established crystallization conditions28 with some minor changes. The triple variant as apo-enzyme was concentrated to 16–20 mg/ml in 50 mM glycyl-glycine buffer, pH 7.9. Afterwards 5 mM ThDP and 5 mM CaCl2 were added to the protein solution. 3 μl of protein solution were mixed at a 1 + 1 ratio with reservoir solution containing 17–22% (w/v) PEG 6000, 2% (v/v) glycerol, 50 mM glycyl-glycine buffer, pH 7.9. Typically, crystal growth occurred within 4–6 weeks at 8 °C. Before flash-cooling a crystal in liquid nitrogen, it was incubated in a cryoprotectant solution consisting of 20% (w/v) PEG 6000, 30% (v/v) ethylene glycol, 10 mM ThDP and 5 mM CaCl2 in 50 mM glycyl-glycine, pH 7.9.
A diffraction data set of a single crystal of EcTK S385Y/D469T/R520Q was collected at a wavelength of 1.00 Å at MAX II BEAMLINE I911–3 at Lund (Sweden). Software package Xds was used for data reduction and scaling30. The atomic structure of the variant was determined by molecular replacement using the previously determined structure of wild-type EcTK in complex with substrate D-xylulose-5-phosphate (pdb code 2R8O) as search model. The crystal structure was built and refined to a resolution of 1.5 Å (Rwork: 12,58%, Rfree: 15,69%) using programs Coot31 and Phenix32 respectively. The final refined model and the corresponding structure factor amplitudes have been deposited in the Research Collaboratory for Structural Biology (http://www.rcsb.org) under accession number 5HHT. Figures were prepared using Pymol software.
Molecular docking
Computational protocol
The enamine-ThDP intermediate present in previously docked crystal structures was aligned into the triple-mutant in Wincoot31. The transketolase structure was stripped of all crystallographic waters and docked in Autodock 4.233. Ligands were obtained as SMILEs in PubChem and assigned three-dimensional coordinates in PRODRG34. The explorable space for docking was defined as a cube 15 Å in length centred at 23.489, 15.398, 5.953 including the entire active-site and omitting surface hydrophobic pockets. For each search, a Lamarckian genetic algorithm was run 200 times with a maximum number of 25 million energy evaluations. Resulting poses were analysed and checked for hydrogen bonding in PyMOL Molecular Graphics System (Schrödinger, USA) and Poseview35.
Pose curation
Only the most energetically favourable pose-clusters were retained where the energy difference was significant (>0.5 kcal/mol). When a structurally different cluster with similar energy was produced, both were selected. This methodology retained poses that were observed to be potentially “catalytically unproductive”. Catalytically unproductive was taken to mean orientations of the benzene ring that point the reactive aldehyde group away from the enamine-ThDP intermediate.
Additional Information
Accession codes: The final refined model and the corresponding structure factor amplitudes have been deposited in the Research Collaboratory for Structural Biology (www.rcsb.org) under accession number 5HHT.
How to cite this article: Affaticati, P. E. et al. Structural Analysis of an Evolved Transketolase Reveals Divergent Binding Modes. Sci. Rep. 6, 35716; doi: 10.1038/srep35716 (2016).
References
Sprenger, G. A., Schörken, U., Sprenger, G. & Sahm, H. Transketolase A of Escherichia coli K12. Purification and properties of the enzyme from recombinant strains. Eur. J. Biochem. 230, 525–532 (1995).
Schenk, G., Duggleby, R. G. & Nixon, P. F. Properties and functions of the thiamin diphosphate dependent enzyme transketolase. Int. J. Biochem. Cell Biol. 30, 1297–1318 (1998).
Schneider, G. & Lindqvist, Y. Crystallography and mutagenesis of transketolase: Mechanistic implications for enzymatic thiamin catalysis. Biochim. Biophys. Acta - Protein Struct. Mol. Enzymol. 1385, 387–398 (1998).
Morris, K. G. et al. Transketolase from Escherichia coli: A practical procedure for using the biocatalyst for asymmetric carbon-carbon bond synthesis. Tetrahedron Asymmetry 7, 2185–2188 (1996).
Breuer, M. & Hauer, B. Carbon-carbon coupling in biotransformation. Curr. Opin. Biotechnol. 14, 570–576 (2003).
Wohlgemuth, R. C2-Ketol elongation by transketolase-catalyzed asymmetric synthesis. J. Mol. Catal. B Enzym. 61, 23–29 (2009).
Ingram, C. U. et al. One-pot synthesis of amino-alcohols using a de-novo transketolase and beta-alanine: pyruvate transaminase pathway in Escherichia coli. Biotechnol. Bioeng. 96, 559–569 (2007).
Smith, M. et al. A multi-disciplinary approach toward the rapid and preparative scale biocatalytic synthesis of chiral amino alcohols: a concise transketolase/ω-transaminase-mediated synthesis of (2S,3S)-2-aminopentane-1,3-diol. Org. Process Res. Dev. 14, 99–107 (2010).
Myles, D. C., Andrulis, P. J. & Whitesides, G. M. A transketolase-based synthesis of (+)-exo-brevicomin. Tetrahedron Lett. 32, 4835–4838 (1991).
Humphrey, A. J., Parsons, S. F., Smith, M. E. & Turner, N. J. Synthesis of a novel N-hydroxypyrrolidine using enzyme catalysed asymmetric carbon–carbon bond synthesis. Tetrahedron Lett. 41, 4481–4485 (2000).
Hecquet, L., Bolte, J. & Demuynck, C. Enzymatic synthesis of ‘natural-labeled’ 6-deoxy-L-sorbose precursor of an important food flavor. Tetrahedron 52, 8223–8232 (1996).
Ziegler, T., Straub, A. & Effenberger, F. Enzyme-Catalyzed Synthesis of 1-Deoxymannojirimycin, 1-Deoxynojirimycin, and 1,4-Dideoxy-1,4-imino-D-arabinitol. Angew. Chem. Int. Ed. 27, 716–717 (1988).
Hibbert, E. G. et al. Directed evolution of transketolase activity on non-phosphorylated substrates. J. Biotechnol. 131, 425–432 (2007).
Strafford, J. et al. Directed evolution to re-adapt a co-evolved network within an enzyme. J. Biotechnol. 157, 237–245 (2012).
Payongsri, P., Steadman, D., Hailes, H. C. & Dalby, P. A. Second generation engineering of transketolase for polar aromatic aldehyde substrates. Enzyme Microb. Technol. 71, 45–52 (2015).
Paramesvaran, J., Hibbert, E. G., Russell, A. J. & Dalby, P. A. Distributions of enzyme residues yielding mutants with improved substrate specificities from two different directed evolution strategies. Protein Eng. Des. Sel. 22, 401–411 (2009).
Reetz, M. T., Bocola, M., Carballeira, J. D., Zha, D. & Vogel, A. Expanding the range of substrate acceptance of enzymes: Combinatorial active-site saturation test. Angew. Chemie - Int. Ed. 44, 4192–4196 (2005).
Reetz, M. T., Carballeira, J. D., Carballeira, D. & Reetz, M. T. Iterative saturation mutagenesis (ISM) for rapid directed evolution of functional enzymes. Nat. Protoc. 2, 891–903 (2007).
Jochens, H. & Bornscheuer, U. T. Natural diversity to guide focused directed evolution. ChemBioChem 11, 1861–1866 (2010).
Sun, Z. et al. Reshaping an Enzyme Binding Pocket for Enhanced and Inverted Stereoselectivity: Use of Smallest Amino Acid Alphabets in Directed Evolution. Angew. Chem. Int. Ed. 1–7 10.1002/anie.201501809 (2015).
Reetz, M. T. & Wu, S. Greatly reduced amino acid alphabets in directed evolution: making the right choice for saturation mutagenesis at homologous enzyme positions. Chem. Commun. 5499–5501, 10.1039/b813388c (2008).
Hibbert, E. G. et al. Directed evolution of transketolase substrate specificity towards an aliphatic aldehyde. J. Biotechnol. 134, 240–245 (2008).
Smith, M. E. B., Hibbert, E. G., Jones, A. B., Dalby, P. a. & Hailesa, H. C. Enhancing and reversing the stereoselectivity of Escherichia coli transketolase via single-point mutations. Adv. Synth. Catal. 350, 2631–2638 (2008).
Cázares, A. et al. Non-alpha-hydroxylated aldehydes with evolved transketolase enzymes. Org. Biomol. Chem. 8, 1301–1309 (2010).
Galman, J. L. et al. α,α’-Dihydroxyketone formation using aromatic and heteroaromatic aldehydes with evolved transketolase enzymes. Chem. Commun. 46, 7608–7610 (2010).
Payongsri, P. et al. Rational substrate and enzyme engineering of transketolase for aromatics. Org. Biomol. Chem. 10, 9021–9029 (2012).
Littlechild, J. et al. Crystallization and preliminary X-ray crystallographic data with Escherichia coli transketolase. Acta Crystallogr. Sect. D Biol. Crystallogr. 51, 1074–1076 (1995).
Asztalos, P. et al. Strain and near attack conformers in enzymic thiamin catalysis: X-ray crystallographic snapshots of bacterial transketolase in covalent complex with donor ketoses xylulose 5-phosphate and fructose 6-phosphate, and in noncovalent complex with acceptor aldo. Biochemistry 46, 12037–12052 (2007).
Krissinel, E. & Henrick, K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. Sect. D Biol. Crystallogr. 60, 2256–2268 (2004).
Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 125–132 (2010).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 213–221 (2010).
Morris, G. M. et al. Software news and updates AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 (2009).
Schüttelkopf, A. W. & Van Aalten, D. M. F. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 60, 1355–1363 (2004).
Stierand, K., Maaß, P. C. & Rarey, M. Molecular complexes at a glance: Automated generation of two-dimensional complex diagrams. Bioinformatics 22, 1710–1716 (2006).
Acknowledgements
We thank our colleague Oliver Kupski for X-ray crystallographic data collection as well as the Deutsche Forschungsgemeinschaft (DFG-FOR 1296/TP3 to K.T.) and the Chinese Scholarship Council (stipend to S.D.) for financial support. P.E.A. was supported by an EPSRC studentship. P.P. was supported by the Royal Thai Government.
Author information
Authors and Affiliations
Contributions
P.A.D., K.T., H.C.H., P.P. and P.E.A. wrote the main manuscript text. S.-B.D. obtained the crystal structure and P.E.A. carried out the molecular docking. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Affaticati, P., Dai, SB., Payongsri, P. et al. Structural Analysis of an Evolved Transketolase Reveals Divergent Binding Modes. Sci Rep 6, 35716 (2016). https://doi.org/10.1038/srep35716
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep35716
This article is cited by
-
Two-substrate enzyme engineering using small libraries that combine the substrate preferences from two different variant lineages
Scientific Reports (2024)
-
Biophysical characterization of the inactivation of E. coli transketolase by aqueous co-solvents
Scientific Reports (2021)
-
Modelling of substrate access and substrate binding to cephalosporin acylases
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
