
Abstract
Nucleic acid amplification is the core technology of molecular biology and genetic engineering. Various isothermal amplification techniques have been developed as alternatives to polymerase chain reaction (PCR). However, most of these methods can only detect single stranded nucleic acid. Herein, we put forward a simple solution for opening double-stranded DNA for isothermal detection methods. The strategy employs recombination protein from E. coli (RecA) to form nucleoprotein complex with single-stranded DNA, which could scan double-stranded template for homologous sites. Then, the nucleoprotein can invade the double-stranded template to form heteroduplex in the presence of ATP, resulting in the strand exchange. The ATP regeneration system could be eliminated by using high concentration of ATP, and the 3′-OH terminal of the invasion strand can be recognized by other DNA modifying enzymes such as DNA polymerase or DNA ligase. Moreover, dATP was found to be a better cofactor for RecA, which make the system more compatible to DNA polymerase. The method described here is a general solution to open dsDNA, serving as a platform to develop more isothermal nucleic acids detection methods for real DNA samples based on it.
Similar content being viewed by others


Introduction
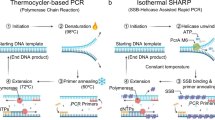
Highly sensitive and selective DNA detection methods are important scientific tools in molecular biology and medical research. Many analytical methods for specific nucleic acid quantification have been developed1,2,3,4,5,6,7. Polymerase chain reaction (PCR) is a widely used technology because of its remarkably high rapidity, precision and reproducibility8,9,10,11,12,13,14. Based on the thermal cycling system (PCR machine), duplex DNA targets and amplicons could be separated at denaturation temperature, yielding two single stranded DNA as templates for next round of amplification. The following primer annealing and elongation on the template will carry out at different temperatures. Therefore, sophisticated equipment for precise temperature control and thermal stable DNA polymerase are indispensable for this method, which has limited its application in unspecialized laboratory and on-site tests. These limitations in PCR-based techniques have spurred the development of a new molecular-biological technique known as isothermal DNA amplification15,16,17,18,19,20,21. As only a single optimal reaction temperature is required for the entire detection, isothermal amplification could satisfy rapid on-site detection of environmental, foodborne, and water-borne pathogens as well as for point-of-care clinical diagnostics, providing simpler and more effective reaction conditions without expensive equipment. So far, a lot of isothermal DNA amplification technologies, including NASBA15, SDA16,RCA18, HDA19, LAMP21 and so on ref. 22, have been developed as promising alternatives of thermal cycling based technique. However, most of isothermal amplification methods can only detect single-stranded nucleic acid. To apply those methods in the detection of double-stranded DNA, extra steps to obtain the single stranded DNA through heat denaturation are required. One solution to open double stranded DNA is to preheat the DNA sample at 95 °C with two pairs of primers before isothermal amplification (Fig. 1A). Then the outer primers and inner primers are annealed to corresponding unwound single stranded template which would be extended by DNA polymerase with strand displacement activity to displace the elongated product of inner primers, yielding the single-stranded DNA to initiate the following isothermal amplification. However, the pre-denaturation step is dependent of heating equipment, making it impossible for these methods to achieve in one step. Recently, a novel isothermal DNA amplification method named recombinase polymerase amplification (RPA) has been reported20. Instead of using high temperature to achieve the denaturation of the double-stranded DNA target, RPA utilize recombinase enzyme to form complex with ssDNA primer that could search for DNA sequence homology on the double stranded DNA target, facilitating the strand exchange between dsDNA and single stranded primer to form D-loop. Then the primer can be extended by DNA polymerase with strand-displacement activity (Fig. 1B). Compared to other isothermal amplification methods likeHDA19 and LAMP21 that need preheating to open dsDNA or constant high temperature for entire processes, RPA is the only isothermal method that could work at 37 °C because of the application of recombinase. To facilitate the DNA replication, two more proteins besides recombinase uvsX are indispensable in RPA: uvsY for stabilizing interactions between uvsX and ssDNA and gp32 for stabilization of the displaced ssDNA (Fig. 1B), moreover, an ATP regeneration system are required to supplement the ATP consumed in the process of DNA substitution. Three proteins applied in RPA are unavailable commercially and all those enzymes are obtained from T4 phage that was a considerable threat for widely used engineered bacterium for large-scale preparation. As the critical component in RPA,uvsX is the recombinase of T4 phage. Recombinase are ubiquitous proteins found in almost all organisms, including RecA from bacteria, Rad51 andDmc1 from eukaryotes, and RadA from archaea. They are important proteins participating in many biological processes such as double-strand DNA break (DSB) repair, rescue of stalled or collapsed replication forks, chromosomal rearrangements and horizontal gene transfer23. Up to now, RecA ofE. coli is the only commercial available recombinase and has been extensively studied. And the substantial function of RecAin vivo is to form nucleoprotein complex with transient single-stranded DNA and facilitate strand exchange with homologous duplex24. Therefore, we ask this question: Can we utilize the recombinase RecA of E. coli as a tool to open double-stranded DNA for isothermal detection methods. To achieve this goal, there are three challenges we need to address: 1. Even it have been reported that RecA can promote the formation of D-loop in the presence of ATP25, whether the complex formed by RecA and short ssDNA can invade into duplex DNA to substitute the identical strand? 2. It is known that RecA, as a single-stranded DNA dependent ATPase, could bind with single-stranded DNA to hydrolyze ATP into ADP immediately and dissociates from single-stranded DNA as a result. However, it is still ambiguous and controversial whether hydrolysis of ATP is needed to realize DNA strand exchange26,27,28,29. 3. Though RecA could open dsDNA target with short ssDNA, it is not clear whether the 3′-OH terminal of the invasion strand can be recognized by other DNA modifying enzymes such as DNA polymerase or DNA ligase. Herein, we report the research to figure out these problems and establish a general solution to open double-stranded DNA for the different isothermal detection methods using RecA protein from E. coli.

(A) The strategy to open dsDNA to produce ssDNA for isothermal detection methods. After the double-stranded DNA is denatured by heating, two pairs of ssDNA primer could anneal to the corresponding single-stranded DNA. Then the primers are extended by DNA polymerase with strand displacement activity. The extension of the outer primers results in the displacement of the elongated products of the inner primers, forming ssDNA. (B) Dynamic processes in recombinase polymerase amplification (RPA). In the presence of ATP, uvsX bind with oligonucleotides cooperatively, upon ATP hydrolysis, the nucleoprotein disassembles and uvsX is displaced by gp32, but uvsY can assist uvsX rebind with oligonucleotides. The stable nucleoprotein search on the double-stranded DNA and promote strand exchange at homologous sites with the displaced strand that will be stabilized bygp32. Then the oligonucleotides are extended by DNA polymerase with strand displacement activity and a single-stranded DNA was released.
Results
Stoichiometry of binding oligonucleotides by RecA
It was reported that RecA and ssDNA could form a compressed nucleoprotein complex in the absence of ATP. The complex is inactive for strand exchange, while in the presence of ATP the nucleoprotein will extend to functional structure to perform strand exchange30. In addition, there was a constant stoichiometry ratio between RecA and nucleotides for forming nucleoprotein. To begin with, we demonstrated the ssDNA binding activity and the stoichiometry of RecA either purchased or expressed by ourselves (Protocol S1) using gel mobility shift assay31. As we can see, RecA and ssDNA (54 nt) indeed formed nucleoprotein and generated a low mobility band in the native agarose gel in the absence of ATP (lane 1, Fig. 2A). However, RecA disassemble from ssDNA when ATP was added (lane 2, Fig. 2A), which could be caused by ADP produced from hydrolysis of ATP as RecA/ssDNA has the strong ATPase activity. This assumption was verified by adding ADP into the reaction, where we found that RecA disassemble from ssDNA when ADP was added (lane 3, Fig. 2A). To prevent disassemble of the nucleoprotein we introduced a modified nucleotide adenosine-5′-o-(3-thio-triphosphate) (ATPγS) which could not be hydrolyzed by RecA. By incubating various concentration ofRecA with ssDNA (12 pmol) and then analyzed on native agarose gel, the corresponding binding ratio of ssDNA by RecA was obtained. Through calculation, we obtained the average stoichiometry of about 3 nucleotides per RecA in the presence of ATPγS (Fig. 2B). In the same way, the stoichiometry of about 5 nucleotides per RecA was obtained in the absence of ATP (Figure S1B), consistent with previous research32,33,34,35. Therefore, in the following experiment RecA was used to completely cover ssDNA based on the 3 nt/RecA in the presence of ATP and 5 nt/RecA in the absence of ATP.

(A) 160 pmol RecA was incubated with 12 pmol 54 nt oligonucleotides in the presence of different nucleotide cofactor. Lane 1, no nucleotide. Lane 2, ATP. Lane 3, ADP. Lane 4, ATPγS. Lane 5, no RecA was added. (B) Incremental concentration of RecA was incubated with 12 pmol 54 nt oligonucleotides in the presence of ATPγS. Lane 1–6 correspond 20, 40, 80, 120, 160, 0 pmol RecA was added respectively. (C) Schematic of the process of strand exchange promoted by RecA. RecA and isotope-labeled ssDNA (ss) can form a nucleoprotein complex, then this complex will recognize its homologous double-stranded DNA (ds) and displace the identical strand to form a new isotope-labeled heteroduplex. RecA will disassemble from the product upon ATP hydrolysis and recycle. (D) Result of RecA promoted strand exchange between ssDNA oligonucleotides and homologous double-stranded DNA, reaction details could be found in Materials and Methods except the nucleotides cofactor variation: Lane 1, no nucleotides; Lane 2, 1 mM ATP; Lane 3, 1 mM ATP with 10 mM phosphocreatine and 1 U creatine kinase; Lane 4, 5 mM ATP; Lane 5, 5 mM dATP; Lane 6, 5 mM ATPγS; Lane 7, no RecA; Lane 8 and 9 are isotope-labeled ds and ss as markers.
RecA promote exchange between oligonucleotides with homologous double-stranded DNA
As nearly all the nucleic acid detection methods depend on hybridization of primer or probe with template, we then check the capability of nucleoprotein complex formed by RecA and ssDNA to substitute the identical strand of the homologous double stranded DNA. Here we designed an experiment to demonstrate the strand exchange activity of RecA, as shown in Fig. 2C: The single stranded DNA (ss) was isotope-labeled, which could form nucleoprotein with RecA. If the complex (RecA/ss) could invade into homologous double stranded DNA (ds), the isotope-labeled ds should be detected based on the native PAGE analysis. So strand exchange of ssDNA oligonucleotides and their homologous double stranded DNA was tested in our experiment. As illustrated inFigure S2C, no isotope-labeled dsDNA products was detected in the absence of ATP (lane 3, Figure S2C), while the exchanged dsDNA containing isotope-labeled ss was observed on the native PAGE in the presence of ATP and the exchange efficiency increased as the ATP concentration increased (lane 4–9, Figure S2C), which verified the DNA strand-substitution catalyzed byRecA requires ATP as cofactor. However, the efficiency of strand exchange was very low in the presence of 1 mM ATP, which could be caused by the hydrolysis of ATP by RecA in the presence of ssDNA. AsRecA can form stable nucleoprotein with ssDNA in the presence of ATPγS as shown in the experiment above, we wonder if ATPγS can replace ATP to increase the efficiency. However, when ATPγS was employed, no exchange products were obtained (lane 6,Fig. 2D). Those results implied that ATP hydrolysis was indispensable in the process of strand exchange promoted by RecA. Therefore, It was supposed that the low efficiency of strand exchange was caused by hydrolysis of ATP by RecA. It is well known that, RecA hydrolyze ATP into ADP continuously when it bind with ssDNA, so in all the strand-exchange reaction in vitro, an ATP regeneration system (composed of phosphocreatine and creatine kinase) that convert ADP into ATP was included, such as in RPA. When ATP regeneration system was introduced into the reaction mixture with 1 mM ATP, the efficiency of strand exchange between ssDNA oligonucleotides and homologous double-stranded DNA increased dramatically (lane 3, Fig. 2D). However, the addition of ATP regeneration system made the reaction more complex and costly. We are very glad to found that this problem could be solved through increasing the concentration of ATP. In the presence of 5 mM ATP, the exchanging efficiency reach to the same level with the reaction containing ATP regeneration system (lane 4 vs. lane 3 in Fig. 2D). It has been reported that dATP is a better cofactor for RecA36. Therefore, we investigate the feasibility to exchange the ATP in our system with dATP. As illustrated in Fig. 2D, the efficiency of strand exchange catalyzed by RecA in the presence of dATP (lane 5) was as same as that in the presence of ATP (lane 4). Besides, the capacity to perform strand exchange of ssDNA of varying length based on this system was investigated, revealing that ssDNA as short as 24 nt was enough for RecA promoted strand exchange (Figure S2B). In short, we realized the opening of dsDNA and substitution with a ssDNA by using RecA of E. coli. And we found that ATP hydrolysis was essential for the whole process of strand exchange. The ATP regeneration system could be eliminated by using high concentration of ATP. Moreover, dATP could be applied as cofactor for RecA to perform strand exchange between short ssDNA and homologous double stranded DNA with same efficiency.
Ligation of two oligonucleotides after strand exchange
As research showed that RecA remain associated with the heteroduplex after strand exchange37, so after strand exchange, what we care about is whether 3′-OH terminal of the invasion strand can be manipulated by DNA modifying enzyme like DNA ligase. Some isothermal detection methods likeRCA adopt the strategy of ligation to initiate the nucleic acid detection. Therefore, we designed the experiment to verify the feasibility of ligation reaction of two ssDNA in the presence of duplex template with the aid of RecA-based dsDNA opening system. As shown in Fig. 3A, we designed two ssDNA oligonucleotides that were complementary to one strand of the duplex template in juxtaposition. The first one was labeled with isotope and the second one with 5′-phosphate group that is necessary for ligation reaction catalyzed by T4 DNA ligase. Then the nucleoprotein complexes formed by two oligonucleotides with RecA could scan double stranded template and invade at the homologous sites to form hetroduplex with a nick in the middle, which would be recognized and ligated by T4 DNA ligase to yield an isotope-labeled product with expected length. However, the ligation efficiency was very low in the presence of high concentration of ATP (lane 1, Figure S4B). We speculated that after nucleoprotein complex invaded into duplex template to form D-loop the displaced strand was likely to re-anneal with the complementary strand and exclude the invasion ssDNA oligonucleotides before they were ligated by T4 DNA ligase. Previous studies reported that single-stranded DNA binding protein (SSB) can aid in the formation of nucleoprotein complexes and stimulate DNA strand exchange by binding to the displaced single-stranded DNA38,39,40. Single-strand DNA-binding protein (SSB) is a 178-amino-acid-long protein found in E. coli that binds to single-stranded regions of DNA. It has important function during all aspects of DNA metabolism: replication, recombination, and repair. As well as stabilizing the single-stranded DNA, SSB proteins bind to and modulate the function of numerous proteins involved in all of these processes. Moreover SSB protein is easily obtained from commercial sources or direct expression fromE. coli (Protocol S1 and Figure S3). When SSB was introduced into the reaction, the efficiency of ligation increased greatly (lane 2, Figure S4B). Out of expectation, when dATP was employed into the same reaction, the ligation efficiency was further improved (Fig. 3B). As it is well known that ATP is a cofactor of T4 DNA ligase and studies showed that although adenylation enzyme could form in the presence of dATP, it is inactive in the ligation reaction41. However, it is surprising to find that dATP could also serve as a cofactor for T4 DNA ligase to work in the condition employed in our experiment (Figure S5). Eventually, we had performed ligation of two oligonucleotides using double-stranded DNA as template at a constant temperature. This would be a great help for the isothermal detection methods which rely on ligation reaction such as rolling circle amplification (RCA) to realize the direct detection of double-stranded DNA isothermally.

(A) Schematic of the process of RecA mediated ligation of two ssDNA oligonucleotides using double-stranded DNA as template. Firstly,RecA bind with two ssDNA oligonucleotides to form nucleoprotein complex respectively to recognize and unwound double-strand template homologously. Then, the two oligonucleotides annealed with complementary strand in juxtaposition and are ligated by T4 DNA ligase. (B) PAGE analysis of RecA mediated ligation reaction: Lane 1, the reaction was conducted in presence of dATP. Lane 2, RecA was not included. Lane 3, RecA was included while the double-stranded DNA template was removed. Lane 4 and 5, 32P-labeled 108 nt and 54 nt oligonucleotide as markers.
Primer extension after strand exchange
Besides ligation, most of the isothermal detection methods, such as SDA and LAMP and so on, rely on generation of single stranded DNA as target, which could be served as template of the primers for DNA polymerase to start the isothermal amplification. So we wonder if the 3′-OH terminal of the invasion strand can be recognized by the DNA polymerase with strand displacement activity, like Bst and Bsm DNA polymerase, to displace one strand of the duplex template after extension. In order to figure out the accessibility of the 3′-OH terminal of the invasion strand by the DNA polymerase, we designed a 5′-ssDNA tailed duplex DNA to serve as template (left,Fig. 4A). In the presence of RecA and ATP, almost all the labeled primers were extended to corresponding length (lane 3,Fig. 4B). However, in the absence of RecA, only a little background products were observed (lane 4, Fig. 4B). Therefore, it can be concluded that the 3′-OH terminal of the primer was accessible for DNA polymerase to elongate after strand exchange. However, the natural DNAs are always intact duplex, unlike the case of 5′-ssDNA tailed duplex DNA where the displaced strand was completely stripped off the heteroduplex. As shown in Fig. 4A (right), after RecA assisted the ssDNA oligonucleotide to invade into intact duplex to form D-loop, the displaced strand was not completely dissociated and the downstream still paired42,43,44,45. This structure was not stable because the primer may be excluded through re-annealing of the displaced strand as referred above. So we designed an blunt-ended duplex as a target to explore if the polymerase could recognize at the 3′-OH terminal and overcome the barrier of downstream base-pair through displacing it (right, Fig. 4A). In the presence of RecA, almost half of the labeled primers were extended to corresponding length (lane 1, Fig. 4B), while in the absence of RecA only a little products were obtained as background (lane 2, Fig. 4B). The results revealed that isotope-labeled primer have invaded into double-stranded template, and it could be extended by the DNA polymerase with strand displacement activity to release a ssDNA. Although the efficiency of extension using blunt-ended duplex as template was lower than that of 5′-ssDNA tailed duplex DNA, it can be concluded that strand exchange promoted by RecA is compatible to DNA polymerase with strand displacement activity and ssDNA will be released in the process of DNA replication.

(A) Schematic of the process of primer extension after strand exchange on 5′-ssDNA tailed duplex and intact blunt-ended duplex template. Left, after complete strand substitution of the isotope-labeled primer with one strand of the 5′-ssDNA tailed duplex, the isotope-labeled primer was elongated by DNA polymerase. Right, after strand exchange of isotope-labeled primer with one strand of blunt-ended duplex, D-loop formed. Then the isotope-labeled primer was extended by DNA polymerase with strand displacement activity and a single-stranded DNA was released (B) Results of primer extension after strand exchange. Lane 1, intact blunt-ended duplex served as template in the presence ofRecA. Lane 2, intact blunt-ended duplex served as template in the absence of RecA. Lane 3, 5′-ssDNA tailed duplex served as template in the presence of RecA. Lane 4, 5′-ssDNA tailed duplex served as template in the absence of RecA. Lane 5, isotope-labeled primer was extended on the complementary 78 nt single-stranded template to serve as a positive control. Lane 6,32P-labeled 78 nt oligonucleotide. Lane 7,32P-labeled 54 nt oligonucleotide as marker respectively. (C) Schematic of RecA based DNA amplification. First, single-stranded primers bind with RecA to form nucleoprotein complex, which could scan the double-stranded template until the homologous sites are located. Then, following strand exchange, the displaced strand is stabilized by SSB and primers are extended by Bsm DNA polymerase. After extension, two ssDNA are generated. (D) Results ofRecA-based DNA amplification using blunt-ended duplex as template. Lane 1, in the presence of 5 mM ATP. Lane 2, in the presence of 5 mM dATP. Lane 3 and 4 are 54 nt and 118 nt 32P-labeled oligonucleotides as marker respectively.
The RecA based DNA amplification
We have obtained ssDNA through RecA promoted strand exchange followed by extension of the invading strand with DNA polymerase. However, the yield was not very high (lane 1, Fig. 4B). So we wonder if the displaced ssDNA can serve as template for another ssDNA primer, the amplification of templates should carry on (Fig. 4C). As a result, more ssDNA for isothermal amplification template could be produced in the process of amplification, which would be a great advantage for isothermal detection methods. However, no amplification products were obtained in the presence of ATP (lane 2, Figure S6B). We deduced that when two primers were used, a lot of ssDNA that were complementary to each other were generated. As mentioned before, SSB can be used to bind with these ssDNA which could serve as template for the primers before they annealed to each other. Consistent with our expectations, amplification products were obtained when SSB was employed (lane 1, Figure S6B). However, the efficiency of amplification was still not very high, we speculated that RecA remain associated with the heteroduplex after strand exchange37 and the homologous sites could be open by nucleoprotein complex only when RecA dissociated from the heteroduplex by hydrolysis of ATP. This may be improved by adding dATP, which was hydrolyzed faster than ATP36. It was very glad to find that in the presence of dATP the amplification efficiency greatly increased (lane 2, Figure 4D). Besides, we demonstrated oligonucleotides with 54 nt or 35 nt in length could all be used as primers for amplification of blunt-ended duplex(Figure S6C). In addition to blunt-ended duplex, plasmid DNA could also serve as template for RecA based DNA amplification, and the amplification products were also visible on stained native agarose gel (Figure S7). Through analysis on multifunctional laser scanning imaging system, we calculated that the templates were amplified 240 times, which implies that more than 240 fold ssDNA were produced in the process of amplification. Although previous research have established an amplification method which can target double-stranded DNA isothermally by introducing recombinase from T4 phage20. However, as mentioned before, the proteins they used were all from T4 phage and unavailable commercially. Besides, recombinase would continuously hydrolyze ATP, so an ATP regeneration system should be included. Here, by using commercially available recombinase of E. coli (RecA), we realized the multiplex ssDNA templates could be produced and have eliminated the need for ATP regeneration system by introducing dATP.
Discussion
In summary, we have put forward a simple solution for opening double-stranded DNA for isothermal detection methods through invasion of ssDNA into double-stranded DNA in assist of RecA protein of E. coli. After the ssDNA annealed with complementary strand of the double-stranded template, it can be manipulated by other DNA modified enzyme like DNA ligase and DNA polymerase. The RecA-based dsDNA opening system described here appears to have several promising features for research and diagnostic applications as follows: All proteins (RecA andSSB) used in our method are commercially available, and they can be expressed easily in ordinary laboratory for large-scale preparation; RecA has been reported to promote strand exchange reaction with a high sequence specificity46, so once combined with other isothermal amplification strategies it could improve the specificity of whole method; The ATP regeneration system is eliminated in our system, which make this method simple and cost-efficient. Moreover, dATP could serve as the cofactor of RecA to perform strand exchange in place of ATP, offering more flexibility to our system; After the strand exchange, the 3′-OH terminal of the invasion strand was proved to be accessible to other DNA modifying enzymes like DNA ligase or polymerase, which is compatible with almost all reported isothermal detection methods. By designing a padlock probe with two binding arms complementary to one strand of double stranded template in juxtaposition, rolling circle amplification (RCA) can detect dsDNA18. Based on our solution, starting structure of loop-mediated isothermal amplification (LAMP) can produce at a constant temperature21 and the double-stranded DNA with Nickase site at both ends are obtained using only one pair of primers at a constant temperature16. By introducing T7 promoter sequence on the 5′ terminal of primers, Nucleic Acid Sequence Based Amplification (NASBA) can conduct using dsDNA as substrate15. Therefore, The RecA-based dsDNA opening system described here is a general solution to open dsDNA, serving as a platform to develop more isothermal nucleic acids detection methods for real DNA samples.
Materials and Methods
Oligonucleotides, enzymes, and other reagents
All oligonucleotides were purchased from Sangon Biotech (Shanghai, China) and are shown in Table S1. Bsm DNA Polymerase (Large Fragment), T4 polynucleotide kinase were purchased from Thermo Scientific. Plasmid pET-28(a) was purchased from Novagen, now part of Merck Biosciences. Trans5α Chemically Competent Cell, BL21(DE3) Chemically Competent Cell, FastPfu DNA Polymerase and dNTP were purchased from TransGen Biotech (Beijing, China). Ni–agarose His tag protein purification kit was bought from Beijing CoWin Biotech. [γ-32P]ATP was purchased from Furui Biological Engineering (Beijing, China).
Labelling Reaction
A reaction mixture containing oligonucleotides X1 and X1-35 with 50 mM Tris-HCl (pH 7.8), 40 mM NaCl, 10 mM MgCl2, 1 mg/mL BSA, 10 μCi [γ-32P]ATP and 10 units of Polynucleotide kinase (PNK) was incubated for 1 h at 37 °C for DNA phosphorylation. The labelled product was purified by 10% denaturing polyacrylamide gel.
DNA binding assays
Reactions were performed by incubating indicated concentration of purified proteins with 12 pmol X1 (Table S1) at 37 °C for 10 min. Standard conditions were 25 mM Tris (pH 7.6), 1 mM Magnesium chloride and 1 mM DTT. The reaction volume of all binding assays was 10 μl. After the reaction, 2.5 ul 6* loading buffer (0.25% bromophenol blue, 36% sucrose and 30 mM EDTA) was added into each tube. Subsequently, all the samples were loaded on 2% native agarose gel. The results were analyzed by Typhoon FLA 7000 IP and the stoichiometry of RecA to oligonucleotides was determined through calculation (GE Healthcare).
DNA exchange assays
Incubate 0.05 μM 32P-labelled oligonucleotides X1 with 18 pmol RecA that completely cover the ssDNA oligonucleotides based on the stoichiometry in the standard condition as follows: 25 mM Tris (pH 7.6), 10 mM Magnesium chloride, 1 mM DTT, nucleotide cofactors used were as shown in the Figure. Then 0.05 μM homologous double-stranded DNA formed by annealing of the same unlabeled oligonucleotides X1 with their complementary strand X1C (Table S1) was added. When ATP regeneration system was included, phosphocreatine was 15 mM, creatine kinase was 1 U. The reaction volume was 10 μl. The incubation last for 10 min at 37 °C. After the reaction, the mixture was extracted by phenol-chloroform and precipitated with ethanol. The sediment was dried by Vacuum Drier and loaded on 12% native polyacrylamide gel. The results were analyzed by Typhoon FLA 7000 IP (GE Healthcare).
Ligation of two oligonucleotides after strand exchange
Incubate 0.05 μM 32P-labelled X1 and 0.05 μM 5′-phosphorylated X1f with 18 pmol RecA in condition as follows: 25 mM Tris (pH 7.6), 10 mM Magnesium chloride, 1 mM DTT, nucleotide cofactors used were as shown in the Figure. Then 0.05 μM double-stranded DNA(formed by annealing of T1 and T2), 15 pmolSSB, 5 U T4 DNA ligase was added followed by incubating for 1 h. After that, 1% SDS and 1 U proteinase K were added and incubated for another 15 min. Afterwards, the reactions were extracted by phenol-chloroform and precipitated with ethanol. The sediment was dried by Vacuum Drier and loaded on 10% denaturing polyacrylamide gel. The results were analyzed by Typhoon FLA 7000 IP(GE Healthcare).
Extension after DNA exchange
We designed a 5′-ssDNA tailed duplex DNA which was formed by unlabeled X1 annealed with X3 and a blunt-ended duplex DNA annealed with X3 and X4 (Table S1). First, we demonstrated 32P-labelled X1 can substitute the unlabeled X1 in the 5′-ssDNA tailed duplex DNA and extended by DNA polymerase. The reaction condition were as follows: 25 mM Tris(PH 7.6), 10 mM Magnesium chloride, 5 mM ATP, 0.25 mM dNTP, 210 pmolRecA, 4 U Bsm, 1 mM DTT. Primer and template used in this reaction were 32P-labelled X1 0.05 μM, 5′-ssDNA tailed duplex DNA 0.05 μM. Then, we demonstrated32P-labelled X1 can exchange with the blunt-ended duplex and extended by DNA polymerase with strand displacement activity. The reaction condition were as follows: 25 mM Tris(PH 7.6), 10 mM Magnesium chloride, 5 mM ATP, 0.25 mM dNTP, 210 pmol RecA, 4 U Bsm, 1 mM DTT. Primer and template used in this reaction was 32P-labelled X1 0.05 μM, blunt-ended duplex 0.05 μM. After incubation at 37 °C for 1 h, the reactions were extracted by phenol-chloroform and precipitated with ethanol. The sediment was dried by Vacuum Drier and loaded on 10% denaturing polyacrylamide gel. The results were analyzed by Typhoon FLA 7000 IP(GE Healthcare).
Amplification using two primers
Pre-incubation of the mixture of 25 mM Tris(pH 7.6), 8% (m/v)PVP(Polyvinyl pyrrolidone), 6 mM Magnesium chloride, 1 mM DTT, 420 pmol RecA, 0.3 μM forward primer X1, 0.3 μM reverse primer X1-re and 5 mM nucleotide cofactors shown in the Figure for 5 min at 37 °C. Subsequently the mixture of double-stranded template formed by annealing of T1 and T2 (blunt-ended duplex), 250 μM dNTP, 360 pmolSSB and 4 U Bsm was added, the final concentration of template was 625 pM. The incubation continued at 37 °C for 1 h and the reaction was extracted by phenol-chloroform and precipitated with ethanol. The sediment was dried by Vacuum Drier and loaded on 10% denaturing polyacrylamide gel or 4% agarose gel. The results were analyzed by Typhoon FLA 7000 IP(GE Healthcare).
Additional Information
How to cite this article: Chen, G. et al. A general solution for opening double-stranded DNA for isothermal amplification. Sci. Rep.6, 34582; doi: 10.1038/srep34582 (2016).
References
Zhang, K., Lin, G. & Li, J. Quantitative nucleic acid amplification by digital PCR for clinical viral diagnostics. Clin Chem Lab Med, doi: 10.1515/cclm-2015-1101 (2016).
Koppelman, M. H., Cuypers, H. T., Emrich, T. & Zaaijer, H. L. Quantitative real-time detection of parvovirus B19 DNA in plasma. Transfusion 44, 97–103 (2004).
Piorkowski, G. et al. Development of generic Taqman PCR and RT-PCR assays for the detection of DNA and mRNA of beta-actin-encoding sequences in a wide range of animal species. J Virol Methods 202, 101–105, doi: 10.1016/j.jviromet.2014.02.026 (2014).
Theves, C. et al. Detection and quantification of the age-related point mutation A189G in the human mitochondrial DNA. J Forensic Sci 51, 865–873, doi: 10.1111/j.1556-4029.2006.00163.x (2006).
He, N. et al. Chemiluminescence analysis for HBV-DNA hybridization detection with magnetic nanoparticles based DNA extraction from positive whole blood samples. J Biomed Nanotechnol 9, 267–273 (2013).
Ceccarelli, M., Galluzzi, L., Migliazzo, A. & Magnani, M. Detection and characterization of Leishmania (Leishmania) and Leishmania (Viannia) by SYBR green-based real-time PCR and high resolution melt analysis targeting kinetoplast minicircle DNA. Plos One 9, e88845, doi: 10.1371/journal.pone.0088845 (2014).
Poole, C. B. & Williams, S. A. A rapid DNA assay for the species-specific detection and quantification of Brugia in blood samples. Mol Biochem Parasitol 40, 129–136 (1990).
Whelen, A. C. & Persing, D. H. The role of nucleic acid amplification and detection in the clinical microbiology laboratory. Annu Rev Microbiol 50, 349–373, doi: 10.1146/annurev.micro.50.1.349 (1996).
Farrell, D. J. et al. Rapid-cycle PCR method to detect Bordetella pertussis that fulfills all consensus recommendations for use of PCR in diagnosis of pertussis. J Clin Microbiol 38, 4499–4502 (2000).
Louie, M., Louie, L. & Simor, A. E. The role of DNA amplification technology in the diagnosis of infectious diseases. Can Med Assoc J 163, 301–309 (2000).
Matsuda, K., Mitsuhashi, M., Ogura, M., Powell, S. & Brooks, T. Rapid RT-PCR quantitation of gene expression in human whole blood. Mol Biol Cell 11, 16a–16a (2000).
Ratge, D., Scheiblhuber, B., Nitsche, M. & Knabbe, C. High-speed detection of blood-borne hepatitis C virus RNA by single-tube real-time fluorescence reverse transcription-PCR with the LightCycler. Clin Chem 46, 1987–1989 (2000).
Klein, D. Quantification using real-time PCR technology: applications and limitations. Trends Mol Med 8, 257–260, doi: Pii S1471-4914(02)02355-9, doi: 10.1016/S1471-4914(02)02355-9 (2002).
Yang, S. & Rothman, R. E. PCR-based diagnostics for infectious diseases: uses, limitations, and future applications in acute-care settings. Lancet Infect Dis 4, 337–348, doi: 10.1016/S1473-3099(04)01044-8 (2004).
Compton, J. Nucleic-Acid Sequence-Based Amplification. Nature 350, 91–92, doi: 10.1038/350091a0 (1991).
Walker, G. T. et al. Strand Displacement Amplification - an Isothermal, In vitro DNA Amplification Technique. Nucleic acids research 20, 1691–1696, doi: 10.1093/nar/20.7.1691 (1992).
Walker, G. T., Little, M. C., Nadeau, J. G. & Shank, D. D. Isothermal Invitro Amplification of DNA by a Restriction Enzyme DNA-Polymerase System. Proceedings of the National Academy of Sciences of the United States of America 89, 392–396, doi: 10.1073/pnas.89.1.392 (1992).
Lizardi, P. M. et al. Mutation detection and single-molecule counting using isothermal rolling-circle amplification. Nat Genet 19, 225–232, doi: 10.1038/898 (1998).
Vincent, M., Xu, Y. & Kong, H. M. Helicase-dependent isothermal DNA amplification. Embo Rep 5, 795–800, doi: 10.1038/sj.embor.7400200 (2004).
Piepenburg, O., Williams, C. H., Stemple, D. L. & Armes, N. A. DNA detection using recombination proteins. Plos Biol 4, 1115–1121, doi: 10.1371/journal.pbio.0040204 (2006).
Tomita, N., Mori, Y., Kanda, H. & Notomi, T. Loop-mediated isothermal amplification (LAMP) of gene sequences and simple visual detection of products. Nat Protoc 3, 877–882, doi: 10.1038/nprot.2008.57 (2008).
Zhao, Y. X., Chen, F., Li, Q., Wang, L. H. & Fan, C. H. Isothermal Amplification of Nucleic Acids. Chem Rev 115, 12491–12545, doi: 10.1021/acs.chemrev.5b00428 (2015).
Lee, J. Y. et al. DNA RECOMBINATION. Base triplet stepping by the Rad51/RecA family of recombinases. Science 349, 977–981, doi: 10.1126/science.aab2666 (2015).
Holthausen, J. T., Wyman, C. & Kanaar, R. Regulation of DNA strand exchange in homologous recombination. DNA repair 9, 1264–1272, doi: 10.1016/j.dnarep.2010.09.014 (2010).
Ohtani, T., Shibata, T., Iwabuchi, M., Nakagawa, K. & Ando, T. Hydrolysis of ATP dependent on homologous double-stranded DNA and single-stranded fragments promoted by RecA protein of Escherichia coli. J Biochem 91, 1767–1775 (1982).
Cunningham, R. P., Dasgupta, C., Shibata, T. & Radding, C. M. Homologous Pairing in Genetic-Recombination - Reca Protein Makes Joint Molecules of Gapped Circular DNA and Closed Circular DNA.5. Cell 20, 223–235, doi: 10.1016/0092-8674(80)90250-0 (1980).
Shibata, T., Dasgupta, C., Cunningham, R. P. & Radding, C. M. Homologous Pairing in Genetic-Recombination - Formation of D-Loops by Combined Action of Reca Protein and a Helix-Destabilizing Protein 4. P Natl Acad Sci-Biol 77, 2606–2610, doi: 10.1073/pnas.77.5.2606 (1980).
Cox, M. M. & Lehman, I. R. Reca Protein of Escherichia-Coli Promotes Branch Migration, a Kinetically Distinct Phase of DNA Strand Exchange. P Natl Acad Sci-Biol 78, 3433–3437, doi: 10.1073/pnas.78.6.3433 (1981).
Hsieh, P., Cameriniotero, C. S. & Cameriniotero, R. D. Pairing of Homologous DNA-Sequences by Proteins - Evidence for 3-Stranded DNA. Genes & Development 4, 1951–1963, doi: 10.1101/gad.4.11.1951 (1990).
Yu, X. & Egelman, E. H. Structural data suggest that the active and inactive forms of the RecA filament are not simply interconvertible. J Mol Biol 227, 334–346 (1992).
Taylor, J. D., Ackroyd, A. J. & Halford, S. E. The gel shift assay for the analysis of DNA-protein interactions. Methods Mol Biol 30, 263–279, doi: 10.1385/0-89603-256-6:263 (1994).
Zlotnick, A., Mitchell, R. S., Steed, R. K. & Brenner, S. L. Analysis of two distinct single-stranded DNA binding sites on the recA nucleoprotein filament. The Journal of biological chemistry 268, 22525–22530 (1993).
Zlotnick, A., Mitchell, R. S. & Brenner, S. L. RecA protein filaments bind two molecules of single-stranded DNA with off rates regulated by nucleotide cofactor. The Journal of biological chemistry 265, 17050–17054 (1990).
Bryant, F. R., Taylor, A. R. & Lehman, I. R. Interaction of the recA protein of Escherichia coli with single-stranded DNA. The Journal of biological chemistry 260, 1196–1202 (1985).
Ruigrok, R. W. et al. The inactive form of recA protein: the ‘compact’ structure. Embo J 12, 9–16 (1993).
Bell, J. C., Plank, J. L., Dombrowski, C. C. & Kowalczykowski, S. C. Direct imaging of RecA nucleation and growth on single molecules of SSB-coated ssDNA. Nature 491, 274–278, doi: 10.1038/nature11598 (2012).
Pugh, B. F. & Cox, M. M. recA protein binding to the heteroduplex product of DNA strand exchange. The Journal of biological chemistry 262, 1337–1343 (1987).
Lavery, P. E. & Kowalczykowski, S. C. A postsynaptic role for single-stranded DNA-binding protein in recA protein-promoted DNA strand exchange. The Journal of biological chemistry 267, 9315–9320 (1992).
Morrical, S. W., Lee, J. & Cox, M. M. Continuous association of Escherichia coli single-stranded DNA binding protein with stable complexes of recA protein and single-stranded DNA. Biochemistry-Us 25, 1482–1494 (1986).
Soltis, D. A. & Lehman, I. R. recA protein promoted DNA strand exchange. The Journal of biological chemistry 258, 6073–6077 (1983).
Rossi, R., Montecucco, A., Ciarrocchi, G. & Biamonti, G. Functional characterization of the T4 DNA ligase: a new insight into the mechanism of action. Nucleic acids research 25, 2106–2113 (1997).
Biet, E., Maurisse, R., Dutreix, M. & Sun, J. S. Stimulation of RecA-mediated D-loop formation by oligonucleotide-directed triple-helix formation: Guided homologous recombination (GOREC). Biochemistry-Us 40, 1779–1786, doi: 10.1021/Bi001605a (2001).
Jayasena, V. K. & Johnston, B. H. Complement-Stabilized D-Loop - Reca-Catalyzed Stable Pairing of Linear DNA-Molecules at Internal Sites. J Mol Biol 230, 1015–1024, doi: 10.1006/jmbi.1993.1216 (1993).
Register, J. C. & Griffith, J. Direct Visualization of Reca Protein-Binding to and Unwinding Duplex DNA Following the D-Loop Cycle. Journal of Biological Chemistry 263, 11029–11032 (1988).
Shibata, T., Ohtani, T., Iwabuchi, M. & Ando, T. D-Loop Cycle - a Circular Reaction Sequence Which Comprises Formation and Dissociation of D-Loops and Inactivation and Reactivation of Superhelical Closed Circular DNA Promoted by Reca Protein of Escherichia-Coli. Journal of Biological Chemistry 257, 3981–3986 (1982).
Shigemori, Y., Haruta, H., Okada, T. & Oishi, M. Marking of specific sequences in double-stranded DNA molecules–SNP detection and direct observation. Genome Res 14, 2478–2485, doi: 10.1101/gr.2789604 (2004).
Acknowledgements
This study was supported by National Sciences Foundation of China (Grant No. 21572222, 21402189 and 21322208).
Author information
Authors and Affiliations
Contributions
G.C. and Z.T. conceived and designed the research. G.C., Y.Y., N.L., X.H. and X.C. performed the experiments. G.C., J.D., X.H., X.C. and Z.T. analyzed the data and wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Chen, G., Dong, J., Yuan, Y. et al. A general solution for opening double-stranded DNA for isothermal amplification. Sci Rep 6, 34582 (2016). https://doi.org/10.1038/srep34582
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep34582
This article is cited by
-
Physical and chemical template-blocking strategies in the exponential amplification reaction of circulating microRNAs
Analytical and Bioanalytical Chemistry (2020)
-
Aluminosilicate Nanocomposite on Genosensor: A Prospective Voltammetry Platform for Epidermal Growth Factor Receptor Mutant Analysis in Non-small Cell Lung Cancer
Scientific Reports (2019)
-
A DNA based visual and colorimetric aggregation assay for the early growth factor receptor (EGFR) mutation by using unmodified gold nanoparticles
Microchimica Acta (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
