
Abstract
Vascular cell survival is compromised under pathological conditions such as abdominal aortic aneurysm (AAA). We have previously shown that the nuclear receptor NOR-1 is involved in the survival response of vascular cells to hypoxia. Here, we identify the anti-apoptotic protein cIAP2 as a downstream effector of NOR-1. NOR-1 and cIAP2 were up-regulated in human AAA samples, colocalizing in vascular smooth muscle cells (VSMC). While NOR-1 silencing reduced cIAP2 expression in vascular cells, lentiviral over-expression of this receptor increased cIAP2 mRNA and protein levels. The transcriptional regulation of the human cIAP2 promoter was analyzed in cells over-expressing NOR-1 by luciferase reporter assays, electrophoretic mobility shift analysis and chromatin immunoprecipitation, identifying a NGFI-B site (NBRE-358/-351) essential for NOR-1 responsiveness. NOR-1 and cIAP2 were up-regulated by hypoxia and by a hypoxia mimetic showing a similar time-dependent pattern. Deletion and site-directed mutagenesis studies show that NOR-1 mediates the hypoxia-induced cIAP2 expression. While NOR-1 over-expression up-regulated cIAP2 and limited VSMC apoptosis induced by hypoxic stress, cIAP2 silencing partially prevented this NOR-1 pro-survival effect. These results indicate that cIAP2 is a target of NOR-1 and suggest that this anti-apoptotic protein is involved in the survival response to hypoxic stress mediated by NOR-1 in vascular cells.
Similar content being viewed by others


Introduction
Vascular remodelling enables the healing and adaptation of blood vessels to mechanical injury or hemodynamic changes and underlies pathogenic processes such as atherosclerosis, restenosis and abdominal aortic aneurysm (AAA)1,2. Apoptosis of vascular smooth muscle cells (VSMC) is critical in vascular remodelling and it is significantly increased in vascular pathologies such as AAA1,3. Indeed, AAA is a complex age-related degenerative disease with high mortality rate, characterized by the degradation of vascular extracellular matrix (ECM) components and by the loss of the vascular cellularity due to increased VSMC apoptosis1.
Apoptosis can be initiated through two main pathways (the extrinsic and the intrinsic) and involves an amplifying proteolytic cascade which leads to the consummation of apoptosis4. The members of the inhibitor of apoptosis (IAP) family are critical proteins regulating apoptosis5. IAPs are structurally related proteins that promote pro-survival signalling pathways and prevent the activation of the effector phase of apoptosis by interfering caspase activity. All IAPs contain the signature baculoviral IAP repeat (BIR), some of them have carboxy-terminal RING domains that function as ubiquitin ligases5,6,7 and cIAP1 and cIAP2 also possess a caspase recruitment domain (CARD) and an ubiquitin-associated domain (UBA)5,8. cIAP2 (also known as HIAP1 or BIRC3) is a potent inhibitor of apoptotic death that, in contrast to other members of the IAP family, is transcriptionally inducible by a number of triggers in different cell types including vascular cells9,10,11 and is up-regulated in human tissues such as atherosclerotic plaques12,13.
We and others have recently involved neuron-derived orphan receptor 1 (NOR-1) in vascular remodelling and coronary artery disease (CAD)14,15,16,17,18. NOR-1 (NR4A3) is a member of the NR4A subfamily of nuclear receptors19,20,21. These nuclear receptors seem to be constitutively active, ligand-independent transcription factors22, expressed at low levels in resting vascular cells but quickly induced by extracellular cues, acting as early-response genes19,20,21. NOR-1, that is up-regulated by a variety of stimuli including growth factors and molecules with mitogen-like activity such as lipoproteins or thrombin14,23,24,25,26,27, regulates vascular cell spreading, migration and proliferation14,17,26,27,28,29. Furthermore, we have shown that NOR-1 is implicated in the survival response of endothelial cells to hypoxia11. Current information on NOR-1 target genes in the vasculature, however, is very limited. By knockdown experiments in endothelial cells we early suggested that cIAP2 could be a target of NOR-111. In the present study we show that cIAP2 is a direct target of NOR-1 and analyze the role of this anti-apoptotic protein in the pro-survival effects of NOR-1 in response to hypoxic stress.
Results
The expression of NOR-1 and cIAP2 is increased in AAA tissues
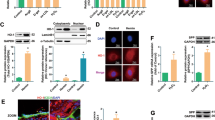
We analyzed the expression of NOR-1 and cIAP2 in human AAA tissues, samples in which vascular cells are exposed to conditions that compromise their survival and in aortas from healthy donors. The expression of both genes was significantly enhanced in AAA samples as we showed by real-time PCR and Western blot (Fig. 1a,b). Interestingly, in these tissues NOR-1 mRNA levels significantly correlated with those of cIAP2 (n = 112; r = 0.454; P < 0.000001; Fig. 1c). Immunohistochemistry analysis of consecutive AAA sections evidenced that NOR-1 colocalizes with cIAP2 in VSMC in the tunica media (Fig. 1d).

NOR-1 and cIAP2 are over-expressed in human AAA tissues.
(a) NOR-1 and cIAP2 mRNA levels determined by real-time PCR in human AAA samples (black bars; n = 96) and human healthy aortic wall (Donors; white bars; n = 16). Data are expressed as mean ± s.e.m. *P < 0.01 vs. Donors; #P < 0.0001 vs. Donors. β-actin was used as endogenous control. (b) Representative western blot showing NOR-1 and cIAP2 protein levels in these tissues. Levels of β-actin were used as a loading control. (c) Positive statistical correlation between mRNA levels of NOR-1 and cIAP2 in patients and donors. The Spearman Rank Order Correlation after logarithmic transformation of data was applied (n = 112; r = 0.454; P < 0.000001). (d) Representative high magnification images showing the immunohistochemical analysis of NOR-1 and cIAP2 in consecutive sections from human healthy aortic wall samples (Donors; upper panels) and human AAA (middle panels). Large lower panels show low magnification images from AAA immunostaining. The position of the corresponding high power views squared. Bar = 50 μm (in high magnification images); Bar = 200 μm (in low magnification images). A: adventitia, m: media and l: lumen.
NOR-1 is necessary for cIAP2 expression in human vascular cells
We have previously shown that NOR-1 silencing decreased cIAP2 mRNA levels in human endothelial cells11. Here we show that, in human VSMC, NOR-1 knockdown significantly decreased cIAP2 mRNA and protein levels (Fig. 2a,b). The inhibitory effect of NOR-1 silencing on cIAP2 expression was confirmed using a pool of siRNAs against NOR-1 (Supplementary Fig. S1). Further, lentiviral over-expression of NOR-1 induced cIAP2 expression (mRNA and protein) in both human VSMC (Fig. 3a,b) and endothelial cells (Fig. 3c,d). The over-expression of NOR-1 in these cells was verified by real-time PCR and western blot (Fig. 3a–d).

NOR-1 knockdown down-regulates cIAP2 expression in VSMC.
Human VSMC were transfected with a siRNA against NOR-1 (siNOR-1, ID s15541; black bars) or a random siRNA (siRandom; white bars). (a) The expression of NOR-1 and that of cIAP2 in these samples was analyzed by real-time PCR (n = 6). *P < 0.0001 vs. siRandom. (b) The down-regulation of cIAP2 by siNOR-1 was also confirmed by western blot. Levels of β-actin were used as a loading control.

NOR-1 over-expression increases cIAP2 expression in vascular cells.
Analysis of NOR-1 and cIAP2 mRNA (a,c) and protein levels (b,d) in human VSMC (a,b) and endothelial cells (c,d) transfected to over-express NOR-1 (pNOR-1) or GFP (pGFP; control cells) (n = 5). *P < 0.0001 vs. control cells. Levels of β-actin are shown as a loading control in western blot analysis.
NOR-1 induces cIAP2 promoter activity through a NBRE
The data described above suggest that NOR-1 modulates cIAP2 transcription. In silico analysis revealed the presence of a putative NBRE binding site in the proximal region of cIAP2 promoter (Fig. 4a). To analyze the functionality of this element, cells were transiently co-transfected with a pGL3 luciferase reporter plasmid harbouring approximately 1.8 Kbp of cIAP2 promoter (pcIAP2-1808) (Fig. 4b) and an expression vector for NOR-1 (pCMV5/NOR-1) or the empty vector (pCMV5). As shown in Fig. 4c, NOR-1 induced cIAP2 transcriptional activity about three-fold. NOR-1 responsiveness of cIAP2 promoter was lost in a deletion that excluded this putative NBRE site (−358/−351). Moreover, the mutation of this element by site-direct mutagenesis completely abrogated the NOR-1-dependent induction of cIAP2 promoter activity.

NOR-1 regulates cIAP2 promoter activity through a NBRE site.
(a) cIAP2 proximal promoter sequence. Cis-acting regulatory elements and the putative NBRE are indicated. (b) Scheme showing the putative NBRE(−358/−351) site present in the cIAP2 promoter region cloned into the pGL3 reporter vector (pcIAP2-1808). The consensus NBRE site (WT-NBRE) is shown and mutated base pairs (m-NBRE) are indicated in bold. (c) Luciferase activity (normalized by Renilla) from cells co-transfected with a NOR-1 expression vector (pCMV5/NOR-1; black bars) or the corresponding empty plasmid (pCMV5; white bars) together with different pGL3/cIAP2 constructs. The activity of a construct mutated in the NBRE site (white triangle) is also shown (n = at least 6). *P < 0.0001 vs. cells co-transfected with pCMV5; #P < 0.0001 vs. cells co-transfected with pCMV5/NOR-1 and pcIAP2-1808. (d) Representative autoradiograms of EMSA performed with a cIAP2 probe containing the NBRE(−358/−351) site (NBRE) and nuclear protein extracts from VSMC transduced with lentiviral vectors to express NOR-1-FLAG (pNOR-1F) or EGFP (pGFP). The position of the complexes up-regulated by NOR-1 (I and II) is indicated. Competition assays with a molar excess of unlabelled probe (100-fold; Competitor) and supershift assays with a specific antibody against the FLAG sequence (anti-FLAG Ab) were performed. EMSA carried out with a mutated NBRE probe (m-NBRE) is also shown. SS: supershifted complex. (e) Representative image of an agarose gel electrophoresis corresponding to a ChIP assay showing the relative in vivo association of NOR-1 with the human cIAP2 promoter in VSMC that over-expressed NOR-1-FLAG (pNOR-1F), GFP (pGFP) or pLVX vector (pLVX). Sheared chromatin was immunoprecipitated with an anti-FLAG antibody (IP:FLAG) or a non-specific IgG (IP:IgG). The enrichment of NOR-1 was assessed by PCR using cIAP2 promoter specific primers. Equal input DNA and control IgG inmunoprecipitations are shown.
To further characterize the functionality of the NBRE(−358/−351) site, we performed electrophoretic mobility shift assays (EMSAs) using nuclear extracts from human VSMC transduced with a FLAG-tagged form of NOR-1. As shown in Fig. 4d, a differential pattern of retarded bands was observed in EMSAs carried out using nuclear extracts from pLVX/NOR-1-transduced cells respect those from control cells (transduced with pLVX/EGFP). NOR-1 over-expression increased the intensity of two of these complexes, that were competed by an excess of unlabelled probe and one of them (complex II) was supershifted by an anti-FLAG antibody, indicating the binding of NOR-1 to this probe. Conversely, mutation of the NBRE probe abrogated the enhanced binding found in NOR-1-transduced cells. To confirm that NOR-1 actually binds to the cIAP2 promoter in vivo, Chromatin immunoprecipitation (ChIP) assays were performed in VSMC expressing FLAG-tagged NOR-1. NOR-1 directly bound to chromatin was immunoprecipitated using an anti-FLAG antibody. Conventional PCR was used to amplify a 129 bp genomic region (−393 to −265) surrounding the NBRE site of cIAP2 promoter. Consistent with data from EMSA, ChIP assays demonstrated that NOR-1 specifically binds to the cIAP2 promoter region encompassing the NBRE site (Fig. 4e). A control IgG did not precipitate detectable DNA and pre-immunoprecipitation samples evidenced equivalent DNA input. Therefore, NOR-1 specifically binds to this NBRE site in cIAP2 promoter.
In contrast to human cIAP2, mouse cIAP2 seems to be unresponsive to NOR-1, as far as no NOR-1 response elements could be detected in mouse cIAP2 promoter and regulation of cIAP2 by NOR-1 was neither observed in mouse aorta from NOR-1 transgenic mice (TgNOR-1) nor in VSMC from these animals (Supplementary Fig. S2a–c).Moreover, lentiviral over-expression of NOR-1 in mouse VSMC did not up-regulate cIAP2 (Supplementary Fig. S2d,e).
NOR-1 mediated the up-regulation of cIAP2 induced by hypoxic stress
We have previously shown that hypoxia and, more potently, the hypoxia mimetic cobalt chloride (CoCl2), up-regulate NOR-1 in human endothelial cells11. In agreement with previous studies30 we detected an up-regulation of the hypoxia-inducible factor-1α (HIF-1α) in human AAA (Supplementary Fig. S3). In human VSMC, hypoxia induced both NOR-1 and cIAP2 (mRNA and protein levels) (Fig. 5a–c) and cobalt chloride was also a stronger inducer of both genes compared to hypoxia (Fig. 5d). Thus, we preferably used this compound to analyze the regulation of cIAP2 by NOR-1 in vascular cells exposed to hypoxic stress. Treatment of VSMC with CoCl2 resulted in similar time- and dose-dependent expression patterns of NOR-1 and cIAP2 (Fig. 6a,b). This parallelism was also observed in HUVEC (Supplementary Fig. S4), although in these cells both genes were less responsive than in VSMC. CoCl2 time-dependently increased NOR-1 and cIAP2 protein levels in VSMC as evidenced by Western blot (Fig. 6c) and confocal immunofluorescence (Fig. 6d).

NOR-1 and cIAP2 are modulated by hypoxia in VSMC.
(a) (upper panel) NOR-1 (white bars) and cIAP2 (black bars) mRNA levels analyzed by real-time PCR in VSMC exposed to hypoxia (0.2% O2) for 4 hours (n = at least 5), *P < 0.001 vs. cells maintained in normoxia. (lower panels) Representative western blot showing protein levels of NOR-1 and cIAP2 in VSMC exposed to hypoxia for 6 hours. Levels of β-actin are shown as a loading control. (b) Immunofluorescence confocal microscopy analysis showing cIAP2 (green) and nuclei (Hoechst stain, blue) in VSMC exposed to hypoxia for 6 hours. Bars: 25 μm. (c) NOR-1 (white bars) and cIAP2 (black bars) mRNA levels analyzed by real-time PCR in VSMC exposed to hypoxia for increasing times. (d) CoCl2 is a stronger inducer of NOR-1 and cIAP2 expression than hypoxia. mRNA levels of NOR-1 and cIAP2 analyzed by real-time PCR in VSMC exposed to hypoxia (Hyp) or CoCl2 (0.5 mM) for 4 hours (n = at least 6). *P < 0.0001 vs. cells maintained in normoxia (Norm).

NOR-1 and cIAP2 are modulated by CoCl2 in VSMC in a dose- and time-dependent manner.
(a,b) NOR-1 (white bars) and cIAP2 (black bars) mRNA levels analyzed by real-time PCR in VSMC exposed to increasing concentrations of CoCl2 (a) or CoCl2 (0.5 mM) for increasing times (b) (n = at least 5). *P < 0.0001 vs. cells maintained in normoxia (Norm). (c) Representative western blot showing protein levels of NOR-1 and cIAP2 in VSMC exposed to CoCl2 (0.5 mM) for increasing times. Levels of β-actin are shown as a loading control in western blot analysis. (d) Immunofluorescence confocal microscopy analysis showing cIAP2 (green) and nuclei (Hoechst stain, blue) in VSMC exposed to CoCl2 for 6 hours. Bar = 25 μm.
NOR-1, which has a functional hypoxia response element in its promoter11, is up-regulated by hypoxia in a HIF-1α-dependent manner. Therefore, as expected, HIF-1α silencing prevented the induction of NOR-1 in response to the hypoxia mimetic CoCl2 and consequently also abrogated the increase of cIAP2 mRNA levels (Supplementary Fig. S5). Finally, we analyzed whether NOR-1 was directly responsible for the up-regulation of cIAP2 by hypoxia and CoCl2. Actinomycin D abrogated the CoCl2-induced cIAP2 up-regulation (Fig. 7a) suggesting the involvement of a transcriptional mechanism. To assess the contribution of NOR-1, VSMC were transiently transfected with the pcIAP2-1808 construct and exposed to hypoxia or CoCl2. The increase in the transcriptional activity of this construct mediated by hypoxia or CoCl2 was significantly reduced when the NBRE (−358/−351) site was deleted or mutated (Fig. 7b). Finally, hypoxia and more evidently CoCl2 increased the binding to the NBRE (−358/−351) site in EMSA (Fig. 7c). These data indicate that NOR-1 is directly involved in the modulation of cIAP2 by hypoxic stress.

NOR-1 mediates the regulation of cIAP2 by hypoxia and CoCl2.
(a) cIAP2 mRNA levels analyzed by real-time PCR in VSMC treated with CoCl2 (black bars) for 4 hours in the presence or absence of actinomycin D (ActD, 4 μM). Results are expressed relative to mRNA levels from VSMC maintained in normoxia and non-treated with ActD (controls) (n = 6). *P < 0.0001 vs. controls; #P < 0.0001 vs. CoCl2 without ActD. 18S rRNA was used as endogenous control. (b) cIAP2 promoter activity in VSMC transfected with the constructs pcIAP2-1808, pcIAP2-349 or pcIAP2-1808-mut and maintained in normoxia (white bars), or exposed to hypoxia (grey bars) or to CoCl2 for 6 hours (black bars). Luciferase activity (normalized by Renilla) was measured and results are expressed relative to transcriptional activity in control VSMC (cells under normoxia conditions; white bars) (n = at least 8). *P < 0.0001 vs. cells maintained in normoxia; †P < 0.001 vs. cells transfected with pcIAP2-1808 and exposed to hypoxia; #P < 0.001 vs. cells transfected with pcIAP2-1808 and exposed to CoCl2. (c) Representative autoradiograms of EMSA performed with a cIAP2 probe containing the NBRE(−358/−351) site (NBRE) and nuclear protein extracts from VSMC maintained under normoxia, or exposed to hypoxia or CoCl2 for 4 hours. The position of the complex up-regulated by hypoxia and CoCl2 (arrowhead) is indicated. Competition assays with a molar excess of unlabelled probe (100-fold; Competitor) were performed. EMSA carried out with a mutated NBRE probe (m-NBRE) is also shown.
The NOR-1 anti-apoptotic effect is mediated by cIAP2
We have previously reported that NOR-1 is a pro-survival transcription factor for endothelial cells exposed to hypoxic stress11. In agreement with this, the rate of apoptosis (estimated either as the level of caspase-3/7 activity or analyzed by fluorescence-activated cell sorting [FACS]) in VSMC exposed to hypoxic stress (0.5 mM CoCl2) was lower in cells over-expressing NOR-1 and this effect was partially abrogated when cIAP2 was silenced (Fig. 8a,b). The efficiency of cIAP2 silencing in these experimental conditions was assessed by real-time PCR (Fig. 8c).

cIAP2 is involved in the pro-survival effect of NOR-1 in VSMC exposed to CoCl2.
VSMC were lentiviral transduced to over-express NOR-1 (pNOR-1) or transduced with the pLVX empty vector (pLVX), transfected with a siRNA against cIAP2 (sicIAP2; black bars) or a random siRNA (siRandom; white bars) and exposed to CoCl2 for 16 hours. (a) Analysis of caspase 3/7 activity in VSMC treated as indicated above. Results are relative to caspase activity in VSMC control (pLVX transduced cells transfected with siRandom) (n = 7). *P < 0.001 vs. pLVX and siRandom; †P < 0.0001 vs. pLVX and sicIAP2; #P < 0.0001 vs. pNOR-1 and siRandom. (b) Apoptosis evaluated by FACS after annexin V-PC5 staining in VSMC treated as indicated above. Data were expressed as percentages of the total cell population (n = 6). *P < 0.0001 vs. pLVX and siRandom; †P = 0.016 vs. pLVX and siRandom; †P < 0.001 vs. pLVX and sicIAP2; #P = 0.004 vs. pNOR-1 and siRandom. (c) mRNA levels of cIAP2 analyzed by real-time PCR in cells as indicated above. Results are expressed relative to cIAP2 mRNA levels in control cells (VSMC transduced with pLVX and transfected with siRandom) (n = 6). *P < 0.005 vs. pLVX and siRandom; †P < 0.05 vs. pLVX and sicIAP2; #P < 0.0001 vs. pNOR-1 and siRandom.
Discussion
NR4A receptors have been implicated in a variety of cellular processes including the transduction of hormonal, inflammatory, mitogenic, apoptotic and differentiative signals. Interestingly, while in some cell types such as myeloid and T cells they seem to play a key pro-apoptotic role31,32,33,34 in other cells and tissues they are pro-survival factors and protect against apoptosis induced by stressful stimuli35,36,37,38. We and others have recently involved NOR-1 in vascular remodelling14,15,16,17,18. NOR-1 regulates vascular cell spreading, migration and proliferation11,14,17,26,28,29 and is implicated in the survival response of vascular cells to hypoxic stress11. In this work, we report that cIAP2, a gene encoding a well-known anti-apoptotic protein, is a downstream target of NOR-1 that mediates NOR-1 pro-survival effects in vascular cells exposed to hypoxic stress. Furthermore, we show that the expression levels of NOR-1 and cIAP2 are increased and positively correlated in AAA, a pathological condition in which vascular cells are forced to adapt to stress conditions that compromise cell survival.
We have previously suggested that cIAP2 could be a target of NOR-111. Interestingly, we observed that both NOR-1 and cIAP2 were up-regulated and colocalized in VSMC in human AAA. To gain more insight into the regulation of cIAP2 by NOR-1 in vascular cells we used both loss- and gain-of-function approaches. NOR-1 knockdown experiments in human VSMC confirmed our previous results from similar approaches in human endothelial cells showing that cIAP2 expression depends on NOR-111. What is more, lentiviral over-expression of NOR-1 significantly up-regulated cIAP2 in both human VSMC and endothelial cells. Finally, in transient co-transfection assays using a luciferase reporter plasmid (harbouring the proximal promoter sequence of cIAP2) and a NOR-1 expression vector we show that this nuclear receptor is able to regulate cIAP2 transcriptional activity. Indeed, promoter deletion and site-directed mutagenesis analysis identified a fully conserved NBRE motif (NBRE −358/−351) critically involved in the NOR-1-mediated induction of cIAP2 promoter activity. The direct binding of NOR-1 to this site was demonstrated in vitro and in vivo by EMSA and ChIP assays respectively. Few transcription factors have been characterized in the control of cIAP2 expression, among them NFκB, CREB or STAT3 39,40. Therefore, our results characterizing a functional NBRE site in cIAP2 promoter and involving NOR-1 in the regulation cIAP2 expand the knowledge on the cIAP2 promoter structure and regulation.
Under pathological conditions such as AAA, hypoxia is a powerful trigger of vascular remodelling30,41. cIAP2 expression is induced by a variety of stimuli including hypoxia; however, no functional hypoxia responsive elements (HREs) in cIAP2 promoter have been characterized and the mechanism by which hypoxia mediates the up-regulation of cIAP2 is unclear11,42,43,44. The transcriptional response of mammalian cells to hypoxia is largely mediated by HIF-145,46, but additional transcription factors cooperate in the complex response of cells to hypoxia in a HIF-dependent or independent manner47. Nowadays, this network of transcription factors that regulate structural genes involved in the adaptation of vascular cells to hypoxic stress is not completely understood. In the present work, we show that hypoxia and more potently a hypoxia mimetic (CoCl2) concomitantly up-regulate NOR-1 and cIAP2 in a time- and dose-dependent manner. CoCl2 was a stronger stimulus stabilizing HIF-1α, the transcription factor responsible for the up-regulation of NOR-1 by hypoxic stress11, than physical hypoxia (Supplementary Fig. S6) resulting in greater induction of both NOR-1 and cIAP2. Moreover, we demonstrate that the NBRE(−358/−351) site is involved in the increase of cIAP2 transcriptional activity triggered by hypoxia. We have previously linked NOR-1 to the survival response of cells to hypoxic stress11. Our present results in VSMC exposed to CoCl2 show that while NOR-1 over-expression limits cell apoptosis associated to high cIAP2 expression levels, cIAP2 silencing partially counteracts this effect. Concerning the potential biological significance of this in AAA, although in cell culture physical hypoxia was a discrete modulator of NOR-1/cIAP2 and a poor inducer of human VSMC apoptosis, it should be taken into account that not only hypoxia but also inflammation, a well known inducer of NOR-1 and a trigger of apoptosis48,49, may work in concert producing additive or synergistic effects on gene expression and cell apoptosis. Therefore, the induction of cIAP2 mediated by NOR-1 seems to be responsible, at least in a part, of the pro-survival effect of NOR-1 in cells exposed to hypoxic stress and probably in more complex pathological settings.
In summary, these results expand to VSMC our previous observations identifying NOR-1 as a pro-survival factor for human endothelial cells, evidence that NOR-1 regulates the expression of cIAP2 through a transcriptional mechanism and show that this anti-apoptotic protein is responsible of the survival response dependent on NOR-1. It should be noted, however, that our in silico analysis identified the NBRE responsive for NOR-1 regulation in the human cIAP2 promoter but not in mouse cIAP2 promoter and our experimental results discard cIAP2 as a NOR-1 target gene in this species. Thus, unfortunately further testing on the functional significance of cIAP2 modulation by NOR-1 using small animal models is not possible. Previous studies from us and others indicate that there are notable differences between human and murine rodents in both the mechanisms that regulate NOR-150 and NOR-1-targeted genes51,52. Consequently, before extrapolating biomedical consequences from NOR-1 biology in mouse models, it is mandatory to corroborate the data in humans. Further studies are needed to determine the relative contribution of the different transcription factors and structural genes responsive to hypoxia to depict the complex network of genes/proteins involved in the adaptive response of mammalian cells to hypoxic stress.
Materials and Methods
Human tissue samples
AAA samples were obtained from patients who underwent open repair surgery for AAA at Hospital de la Santa Creu i Sant Pau (HSCSP). Normal abdominal aortas were obtained from multiorgan donors. Specimens for immunohistochemical studies were fixed overnight in 4% paraformaldehyde/0.1 M PBS (pH 7.4), embedded in paraffin and sectioned into 5 μm sections. Specimens for Western blot and PCR analysis were snap-frozen in liquid nitrogen and stored at −80 °C until processed53. The study was approved by the HSCSP Ethics Committee (13/103/1491) and was conducted according to the Declaration of Helsinki. Written informed consent was obtained from each patient.
Animals
Gene expression was analyzed in aorta from mice that over-expresses NOR-1 in VSMC18 and wild-type control littermates. Mice were bred in the Animal Experimentation Unit (CSIC-ICCC). Animals were euthanized under ketamine (75 mg/kg)/medetomidine (1 mg/kg) anaesthesia and aorta was excised, frozen in liquid nitrogen and stored at −80 °C. All procedures were reviewed and approved by the Ethical Committee at the Centro de Investigación Cardiovascular (Barcelona, Spain; ICCC-055) as stated in Law 5/1995 (Generalitat de Catalunya) and follow the Spanish Policy for Animal Protection RD53/2013, which meets the European Union Directive 2010/63/UE.
Cell cultures
VSMC were obtained from human non-atherosclerotic arteries of hearts removed in transplant surgeries at the HSCSP as described53. All the procedures were approved by the Reviewer Institutional Committee on Human Research of the HSCSP (12/007/1292) and conform to the Declaration of Helsinki and written informed consent was obtained from each patient. VSMC and HUVEC (Lonza) were cultured as described54. Cells were stimulated with CoCl2 or exposed to hypoxia (0.2% O2). When needed, the cells were pre-treated with actinomycin D. VSMCs from mouse aorta were obtained by the explant technique18. Mouse VSMC and HEK293T and HeLa cell lines were cultured in DMEM supplemented with 10% FCS, 2 mM L-glutamine and antibiotics.
Generation of lentiviral particles and transduction
The human NOR-1 cDNA was obtained from a pBlueScript-NOR1 construct, kindly provided by Dr. Ohkura (National Cancer Center Research Institute, Japan)55 and the mouse NOR-1 cDNA linked to a FLAG sequence was obtained from the plasmid pCMV5/NOR-1-FLAG, kindly provided by Dr. Hastie (University of Dundee)56. The pLVX/NOR-1, pLVX/NOR-1-FLAG and pLVX/EGFP were obtained as described48. These constructs and the empty pLVX-Puro vector (pLVX) were transfected in HEK293T cells according to Lenti-XTM Lentiviral Expression System Kit (Clontech). After 48 h, supernatants containing viral particles were harvested and titrated. Lentiviral transductions of VSMC and HUVEC were performed for 48 h at a multiplicity of infection of 15 in presence of polybrene (8 μg/mL). Transduced cell populations were enriched by puromycin selection. pLVX and pLVX/EGFP vectors were used as controls obtaining similar results.
Transfections with small interfering RNA (siRNA)
siRNAs against NOR-1 (Silencer Select Pre-designed siRNA s15541 [Ambion] and ON-TARGET plus SMARTpool L-003428-00-0005 [Dharmacon]), cIAP2 (ON-TARGET plus SMARTpool L-004099-00-0005) and HIF-1α (ON-TARGET plus SMARTpool L-004018-00-0005) were used. The Silencer Select Negative Control #1 was used as a control. LipofectamineTM RNAiMAX (Invitrogen) was used for siRNA delivery57. Cells were transfected with 20–50 nM siRNA, using 7.5 μL of Lipofectamine RNAiMAX Reagent. After transfection (8–24 h), the medium was replaced and cells were incubated for at least 16 h. In some experiments, VSMC were serum-deprived for 24–48 h and exposed to CoCl2.
Gene expression: real-time PCR
Total RNA was extracted using TRIsureTM (Bioline) and reverse-transcribed with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). mRNA levels were assessed by real-time PCR using TaqManTM gene expression assays-on-demand (Applied Biosystems). TATA-binding protein (TBP) was used as endogenous control (unless otherwise stated)18.
Generation of cIAP2 promoter constructs
A 1.8 kb fragment corresponding to nucleotides −1808 to −20 of the human cIAP2 promoter58 was generated by PCR and cloned into the pGL3 vector (pcIAP2-1808). The primers used were: 5′-CGTGCGGTACCACACTTGGCTCATTTTTGT-3′ (forward; KpnI site is underlined) and 5′-GGAGGGCTCGAGTCTCACGCTGTCTTTTAA-3′ (reverse; XhoI site is underlined). The PCR product was digested with KpnI and XhoI and cloned into the pGL3 vector. A promoter deletion construct (−349 to −20; pcIAP2-349) was generated using the reverse primer indicated above and the following forward primer: 5′-TGACCGGTACCAGGCAGGCTAAGCAATGA-3′ (KpnI site is underlined). The putative NBRE(−358/−351) site located in cIAP2 promoter was mutated using the QuikChangeTM Site-Directed Mutagenesis Kit (Stratagene) (pcIAP2-1808-mut) and primers 5′-TGGAGAACAGGGCATATTGttCTTTTCCAGGCAGGCTAAG-3′ and 5′-CTTAGCCTGCCTGGAAAAGaaCAATATGCCCTGTTCTCCA-3′ (NBRE site is underlined and changes are indicated in lower case letters).
Transient transfection and Luciferase assays
VSMC were transfected using Lipofectamine LTXTM and Plus Reagent (Invitrogen). cIAP2 constructs were co-transfected together with pCMV5/NOR-1 expression plasmid or the corresponding empty vector (pCMV5)52. Luciferase activity was determined in cell lysates using the Dual-LuciferaseTM Reporter Assay System (Promega). Results were expressed as the ratio of firefly to renilla activity.
EMSA
EMSA was performed using 5 μg of nuclear extracts obtained from human VSMC59. Double-stranded DNA probes containing the putative wild-type NBRE(−358/−351) site in cIAP2 promoter (5′-GGGCATATTGACCTTTTCCAGGCA-3′) and its mutated form (5′-GGGCATATTGttCTTTTCCAGGCA-3′) were used. DNA probes were labelled with [γ-32P]-ATP using T4 polynucleotide kinase and purified on a Sephadex G-50 column. In competition assays, the unlabelled probe was added before the labelled one and was incubated for 10 min. For supershift assays, nuclear extracts were pre-incubated for 25 min with 2 μg of an anti-FLAG antibody (F1804, Sigma-Aldrich). Protein-DNA complexes were resolved by electrophoresis on 5% polyacrylamide gels in 0.3X TBE. Gels were dried and subjected to autoradiography using a Storage Phosphor Screen (GE Healthcare). Shifted bands were detected using a Typhoon 9400 Scanner (GE Healthcare).
ChIP assay
VSMC were cross-linked with 1% formaldehyde for 10 min. The cross-link reaction was stopped by adding glycine (100 mM). Nuclei were isolated as described52. Chromatin was sheared by sonication and an aliquot was saved and stored as input DNA. Supernatants were then immunoprecipitated with 5 μg of anti-FLAG antibody (F1804) or an IgG as a control. Immune complexes were recovered by addition of A/G-Agarose beads. After washing, complexes were extracted, cross-link was reversed and the DNA was purified and concentrated. Purified DNA was analyzed by PCR amplifying a 129 bp DNA fragment (forward primer: 5′- TGTATGGCGGATGGAGGGTGGA-3′; reverse primer: 5′-AGACATTTGCTTCATTGCTCG-3′). The amplified PCR products were run by electrophoresis on ethidium bromide stained agarose gels60.
Caspase activity assay
Caspase activity was determined in VSMC transduced with the pLVX/NOR-1 construct or the pLVX vector and transfected with a pool of siRNAs against cIAP2 or the corresponding siRandom. After transfection, cells were serum deprived for 48 h and stimulated with CoCl2 16 h. Then, cells were lysed and caspase activity was measured in cell lysates using the Caspase-GloTM 3/7 assay Kit (Promega) and a luminometer. Data were normalized by protein content.
Analysis of apoptosis by FACS
VSMC were processed as indicated in the previous section. Cells were trypsinized, pooled with cells present in the cell supernatants, resuspended in binding buffer and incubated with annexin V conjugated with fluorescein isothiocyanate (FITC) and propidium iodide (FITC Annexin V Apoptosis Detection Kit I, BD Pharmigen) according to the manufacturer11. Annexin V-PC5 and PI binding was analyzed by FACS (Epics XL flow cytometer; Beckman Coulter). Data were gated for viable cells (annexin V− and PI−), damaged cells (annexin V− and PI+), apoptotic cells (annexin V+ and PI−) and late apoptotic cells (annexin V+ and PI+). Data were expressed as percentages of the total cell population.
Western blot analysis
Cellular and tissue extracts were obtained as described53. Lysates were resolved by SDS-PAGE under reducing conditions and electrotransfered onto Immobilon polyvinylidene difluoride membranes. Membranes were probed using antibodies against NOR-1 (H00008013-M06, Abnova), cIAP2 (ab32059, Abcam), HIF-1α (NB-100-449, Novus Biologicals) and β-actin (A5441, Sigma-Aldrich), followed by appropriate horseradish peroxidase-conjugated secondary antibodies and a chemiluminescent detection system. Equal loading was verified by Ponceau staining and by β-actin levels.
Immunocytochemistry
Cells were fixed in ice-cold 4% paraformaldehyde, blocked and incubated with an anti-cIAP2 antibody (ab32059) in PBS containing 5% BSA, overnight at 4 °C. An Alexa fluor 488 goat anti-rabbit immunoglobulin (Molecular Probes) was used as secondary antibody. For nuclei, Hoechst 33342 trihydrochloride trihydrate (H3570, Invitrogen) was used. Controls without the primary antibody were included in all procedures. Cells were mounted with ProLongTM Mounting Medium (Molecular Probes) and analyzed by confocal microscopy.
Immunostaining of human arteries
Consecutive deparaffinized sections were rehydrated, subjected to antigen retrieval in 10 mM citrate buffer pH 6.0 (95 °C for 20 min) and blocked for 30 min with 10% goat or horse serum/PBS. Antibodies against cIAP2 (ab32059), NOR-1 (H00008013-M06) and HIF-1α (NB-100-449) were used. Sections were incubated with the corresponding biotinylated secondary antibodies. Immunocomplexes were detected after incubation with Vectastain Elite ABC reagent (PK6100, Vector) and DAB substrate.
Statistical analysis
Data are expressed as mean ± s.d. (unless otherwise state). Significant differences were established by Student’s t-test or one-way ANOVA, according to the number of groups compared, using the GraphPad Instat program (GraphPad Software V2.03) (GraphPad Software Inc.). When normality failed we used the Mann-Whitney rank sum test to compare two groups. To determine association between variables a Spearman Rank Order Correlation analysis was performed with SigmaPlot software V11.0 (Systat Software Inc.). Differences were considered significant at P < 0.05.
Additional Information
How to cite this article: Alonso, J. et al. NOR-1/NR4A3 regulates the cellular inhibitor of apoptosis 2 (cIAP2) in vascular cells: role in the survival response to hypoxic stress. Sci. Rep. 6, 34056; doi: 10.1038/srep34056 (2016).
References
Nordon, I. M., Hinchliffe, R. J., Loftus, I. M. & Thompson, M. M. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nat. Rev. Cardiol. 8, 92–102 (2011).
Pant, R., Marok, R. & Klein, L. W. Pathophysiology of coronary vascular remodeling: relationship with traditional risk factors for coronary artery disease. Cardiol. Rev. 22, 13–16 (2014).
Galán, M. et al. Induction of histone deacetylases (HDACs) in human abdominal aortic aneurysm: therapeutic potential of HDAC inhibitors. Dis. Model. Mech. 9, 541–552 (2016).
Movassagh, M. & Foo, R. S. Simplified apoptotic cascades. Heart Fail. Rev. 13, 111–119 (2008).
Silke, J. & Vucic, D. IAP family of cell death and signaling regulators. Methods Enzymol . 545, 35–65 (2014).
Vaux, D. L. & Silke, J. IAPs, RINGs and ubiquitylation. Nat. Rev. Mol. Cell. Biol. 6, 287–297 (2005).
Varfolomeev, E. & Vucic, D. (Un)expected roles of c-IAPs in apoptotic and NFkappaB signaling pathways. Cell Cycle Georget. Tex . 7, 1511–1521 (2008).
Gyrd-Hansen, M. et al. IAPs contain an evolutionarily conserved ubiquitin-binding domain that regulates NF-kappaB as well as cell survival and oncogenesis. Nat. Cell. Biol. 10, 1309–1317 (2008).
Conte, D. et al. Inhibitor of apoptosis protein cIAP2 is essential for lipopolysaccharide-induced macrophage survival. Mol. Cell. Biol. 26, 699–708 (2006).
Von Wnuck Lipinski, K. et al. Integrin-mediated transcriptional activation of inhibitor of apoptosis proteins protects smooth muscle cells against apoptosis induced by degraded collagen. Circ. Res. 98, 1490–1497 (2006).
Martorell, L. et al. The hypoxia-inducible factor 1/NOR-1 axis regulates the survival response of endothelial cells to hypoxia. Mol. Cell. Biol. 29, 5828–5842 (2009).
Blanc-Brude, O. P. et al. IAP survivin regulates atherosclerotic macrophage survival. Arterioscler. Thromb. Vasc. Biol. 27, 901–907 (2007).
Moran, E. P. & Agrawal, D. K. Increased expression of inhibitor of apoptosis proteins in atherosclerotic plaques of symptomatic patients with carotid stenosis. Exp. Mol. Pathol. 83, 11–16 (2007).
Martínez-González, J., Rius, J., Castelló, A., Cases-Langhoff, C. & Badimon, L. Neuron-derived orphan receptor-1 (NOR-1) modulates vascular smooth muscle cell proliferation. Circ. Res. 92, 96–103 (2003).
Bonta, P. I. et al. Nuclear receptors Nur77, Nurr1 and NOR-1 expressed in atherosclerotic lesion macrophages reduce lipid loading and inflammatory responses. Arterioscler. Thromb. Vasc. Biol. 26, 2288–2294 (2006).
Nomiyama, T. et al. Deficiency of the NR4A neuron-derived orphan receptor-1 attenuates neointima formation after vascular injury. Circulation 119, 577–586 (2009).
Zhao, Y. et al. Deficiency of the NR4A orphan nuclear receptor NOR1 decreases monocyte adhesion and atherosclerosis. Circ. Res. 107, 501–511 (2010).
Rodríguez-Calvo, R. et al. Over-expression of neuron-derived orphan receptor-1 (NOR-1) exacerbates neointimal hyperplasia after vascular injury. Hum. Mol. Genet. 22, 1949–1959 (2013).
Martinez-Gonzalez, J. & Badimon, L. The NR4A subfamily of nuclear receptors: new early genes regulated by growth factors in vascular cells. Cardiovasc. Res. 65, 609–618 (2005).
Bonta, P. I., Pols, T. W. & de Vries, C. J. NR4A nuclear receptors in atherosclerosis and vein-graft disease. Trends Cardiovasc. Med . 17, 105–111 (2007).
Zhao, Y. & Bruemmer, D. NR4A Orphan Nuclear Receptors in Cardiovascular Biology. Drug Discov. Today Dis. Mech . 6, e43–e48 (2009).
Wang, Z. et al. Structure and function of Nurr1 identifies a class of ligand-independent nuclear receptors. Nature 423, 555–560 (2003).
Liu, D., Jia, H., Holmes, D. I., Stannard, A. & Zachary, I. Vascular endothelial growth factor-regulated gene expression in endothelial cells: KDR-mediated induction of Egr3 and the related nuclear receptors Nur77, Nurr1 and Nor1. Arterioscler. Thromb. Vasc. Biol. 23, 2002–2007 (2003).
Rius, J., Martínez-González, J., Crespo, J. & Badimon, L. Involvement of neuron-derived orphan receptor-1 (NOR-1) in LDL-induced mitogenic stimulus in vascular smooth muscle cells: role of CREB. Arterioscler. Thromb. Vasc. Biol. 24, 697–702 (2004).
Nomiyama, T. et al. The NR4A orphan nuclear receptor NOR1 is induced by platelet-derived growth factor and mediates vascular smooth muscle cell proliferation. J. Biol. Chem. 281, 33467–33476 (2006).
Rius, J., Martínez-González, J., Crespo, J. & Badimon, L. NOR-1 is involved in VEGF-induced endothelial cell growth. Atherosclerosis 184, 276–282 (2006).
Martorell, L., Martínez-González, J., Crespo, J., Calvayrac, O. & Badimon, L. Neuron-derived orphan receptor-1 (NOR-1) is induced by thrombin and mediates vascular endothelial cell growth. J. Thromb. Haemost. 5, 1766–1773 (2007).
Zeng, H. et al. Orphan nuclear receptor TR3/Nur77 regulates VEGF-A-induced angiogenesis through its transcriptional activity. J. Exp. Med. 203, 719–729 (2006).
Thakar, R. G. et al. Cell-shape regulation of smooth muscle cell proliferation. Biophys. J. 96, 3423–3432 (2009).
Erdozain, O. J., Pegrum, S., Winrow, V. R., Horrochs, M. & Stevens, C. R. Hypoxia in abdominal aortic aneurysm supports a role for HIF-1α and Ets-1 as drivers of matrix metalloproteinase upregulation in human aortic smooth muscle cells. J Vasc Res . 48, 163–170 (2011).
Cheng, L. E., Chan, F. K., Cado, D. & Winoto, A. Functional redundancy of the Nur77 and Nor-1 orphan steroid receptors in T-cell apoptosis. EMBO J. 16, 1865–1875 (1997).
Mullican, S. E. et al. Abrogation of nuclear receptors Nr4a3 and Nr4a1 leads to development of acute myeloid leukemia. Nat. Med . 13, 730–735 (2007).
Thompson, J. & Winoto, A. During negative selection, Nur77 family proteins translocate to mitochondria where they associate with Bcl-2 and expose its proapoptotic BH3 domain. J. Exp. Med. 205, 1029–1036 (2008).
Wang, T. et al., Inhibition of activation-induced death of dendritic cells and enhancement of vaccine efficacy via blockade of MINOR. Blood 113, 2906–2913 (2009).
Pönniö, T. & Conneely, O. M. Nor-1 regulates hippocampal axon guidance, pyramidal cell survival and seizure susceptibility. Mol. Cell. Biol. 24, 9070–9078 (2004).
De Léséleuc, L. & Denis, F. Inhibition of apoptosis by Nur77 through NF-kappaB activity modulation. Cell Death Differ . 13, 293–300 (2006).
Saijo, K. et al. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell 137, 47–59 (2009).
Xiao, G., Sun, T., Songming, C. & Cao, Y. NR4A1 enhances neural survival following oxygen and glucose deprivation: an in vitro study. J. Neurol. Sci. 330, 78–84 (2013).
Lanuti, P. et al. Enhancement of TRAIL cytotoxicity by AG-490 in human ALL cells is characterized by downregulation of cIAP-1 and cIAP-2 through inhibition of Jak2/Stat3. Cell. Res. 19, 1079–1089 (2009).
Wu, H. H. et al. cIAP2 upregulated by E6 oncoprotein via epidermal growth factor receptor/phosphatidylinositol 3-kinase/AKT pathway confers resistance to cisplatin in human papillomavirus 16/18-infected lung cancer. Clin. Cancer Res. 16, 5200–5210 (2010).
Vorp, D. A. et al. Association of intraluminal thrombus in abdominal aortic aneurysm with local hypoxia and wall weakening. J Vasc Surg . 34, 291–299 (2001).
Dong, Z. et al. Up-regulation of apoptosis inhibitory protein IAP-2 by hypoxia. Hif-1-independent mechanisms. J. Biol. Chem. 276, 18702–18709 (2001).
Zhang, Q. et al. Treatment with siRNA and antisense oligonucleotides targeted to HIF-1alpha induced apoptosis in human tongue squamous cell carcinomas. Int. J. Cancer 111, 849–857 (2004).
Kilic, M., Kasperczyk, H., Fulda, S. & Debatin, K. M. Role of hypoxia inducible factor-1 alpha in modulation of apoptosis resistance. Oncogene 26, 2027–2038 (2007).
Semenza, G. L. Surviving ischemia: adaptive responses mediated by hypoxia-inducible factor 1. J. Clin. Invest. 106, 809–812 (2000).
Pugh, C. W. & Ratcliffe, P. J. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat. Med. 9, 677–684 (2003).
Cummins, E. P. & Taylor, C. T. Hypoxia-responsive transcription factors. Pflugers Arch . 450, 363–371 (2005).
Calvayrac, O. et al. NOR-1 modulates the inflammatory response of vascular smooth muscle cells by preventing NFκB activation. J. Mol. Cell. Cardiol. 80, 34–44 (2015).
Boyle, J. J., Weissberg, P. L. & Bennett, M. R. Tumor necrosis factor-alpha promotes macrophage induced vascular smooth muscle cell apoptosis by direct and autocrine mechanisms. Arterioscler. Thromb. Vasc. Biol. 23, 1553–1558 (2003).
Li, P. et al. MicroRNA-638 is highly expressed in human vascular smooth muscle cells and inhibits PDGF-BB-induced cell proliferation and migration through targeting orphan nuclear receptor NOR1. Cardiovasc. Res. 99, 185–193 (2013).
Rodríguez-Calvo, R. et al. NR4A receptors up-regulate the antiproteinase alpha-2 macroglobulin (A2M) and modulate MMP-2 and MMP-9 in vascular smooth muscle cells. Thromb. Haemost. 113, 1323–1334 (2015).
Ferrán, B. et al. The nuclear receptor NOR-1 regulates the small muscle protein, X-linked (SMPX) and myotube differentiation. Sci. Rep. 6, 25944 (2016).
Orriols, M. et al. Down-regulation of Fibulin-5 is associated with aortic dilation: role of inflammation and epigenetics. Cardiovasc. Res. 110, 431–442 (2016).
García-Ramírez, M., Martínez-González, J., Juan-Babot, J. O., Rodríguez, C. & Badimon, L. Transcription factor SOX18 is expressed in human coronary atherosclerotic lesions and regulates DNA synthesis and vascular cell growth. Arterioscler. Thromb. Vasc. Biol. 25, 2398–2403 (2005).
Ohkura, N. et al. Structure, mapping and expression of a human NOR-1 gene, the third member of the Nur77/NGFI-B family. Biochim. Biophys. Acta 1308, 205–214 (1996).
Wingate, A. D., Campbell, D. G., Peggie, M. & Arthur, J. S. Nur77 is phosphorylated in cells by RSK in response to mitogenic stimulation. Biochem. J. 393, 715–724 (2006).
Sambri, I. et al. miR-17 and -20a Target the neuron-derived orphan receptor-1 (NOR-1) in vascular endothelial cells. PLoS One . 10, :e0141932 (2015).
Hong, S. Y. et al. Involvement of two NF-kappa B binding elements in tumor necrosis factor alpha -, CD40- and epstein-barr virus latent membrane protein 1-mediated induction of the cellular inhibitor of apoptosis protein 2 gene. J. Biol. Chem. 275, 18022–18028 (2000).
Rodríguez, C., Martínez-González, J., Sánchez-Gómez, S. & Badimon, L. LDL downregulates CYP51 in porcine vascular endothelial cells and in the arterial wall through a sterol regulatory element binding protein-2-dependent mechanism. Circ. Res. 88, 268–274 (2001).
Guadall, A. et al. Fibulin-5 is up-regulated by hypoxia in endothelial cells through a hypoxia-inducible factor-1 (HIF-1α)-dependent mechanism. J. Biol. Chem. 286, 7093–7103 (2011).
Acknowledgements
This work was supported by the Sociedad Española de Cardiología (Proyecto Fundación Española del Corazón para Investigación Básica en Cardiología 2015) and the Spanish Ministerio de Economía y Competitividad (MINECO)-Instituto de Salud Carlos III (ISCIII) [grants SAF2012-40127, SAF2015-64767-R, SAF2013-46707-R, PI15/01016, RD12/0042/0051 and RD12/0042/0053] and Fundació MARATÓ TV3 (2015 23 30). JA was supported by a PhD fellowship (FPU) from the Spanish Ministerio de Educación (MEC). MG is an investigator from the Miguel Servet Fund of the Spanish ISCIII (CP15/00126). The study was co-founded by Fondo Europeo de Desarrollo Regional (FEDER), a way to build Europe. We are indebted to Prof. Naganari Ohkura and Prof. James Hastie for kindly providing us with the human NOR-1 cDNA and the plasmid containing the NOR-1-FLAG, respectively.
Author information
Authors and Affiliations
Contributions
J.M.G. designed and supervised the study, interpreted data and wrote the manuscript. J.A., M.G. and I.M.P. performed experiments and analyzed and interpreted data. J.M.R. and M.C. analyzed data and contributed to manuscript draft writing. C.R. conceived specific experiments, analyzed data and contributed to manuscript draft writing. All authors were involved in writing the paper and gave their final approval of the submitted version.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Alonso, J., Galán, M., Martí-Pàmies, I. et al. NOR-1/NR4A3 regulates the cellular inhibitor of apoptosis 2 (cIAP2) in vascular cells: role in the survival response to hypoxic stress. Sci Rep 6, 34056 (2016). https://doi.org/10.1038/srep34056
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep34056
This article is cited by
-
Potential Roles of Nr4a3-Mediated Inflammation in Immunological and Neurological Diseases
Molecular Neurobiology (2024)
-
Deletion of the NR4A nuclear receptor NOR1 in hematopoietic stem cells reduces inflammation but not abdominal aortic aneurysm formation
BMC Cardiovascular Disorders (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
