
Abstract
Changes in the size of cellular organelles are often linked to modifications in their function. Endothelial cells store von Willebrand Factor (vWF), a glycoprotein essential to haemostasis in Weibel-Palade bodies (WPBs), cigar-shaped secretory granules that are generated in a wide range of sizes. We recently showed that forcing changes in the size of WPBs modifies the activity of this cargo. We now find that endothelial cells treated with statins produce shorter WPBs and that the vWF they release at exocytosis displays a reduced capability to recruit platelets to the endothelial cell surface. Investigating other functional consequences of size changes of WPBs, we also report that the endothelial surface-associated vWF formed at exocytosis recruits soluble plasma vWF and that this process is reduced by treatments that shorten WPBs, statins included. These results indicate that the post-exocytic adhesive activity of vWF towards platelets and plasma vWF at the endothelial surface reflects the size of their storage organelle. Our findings therefore show that changes in WPB size, by influencing the adhesive activity of its vWF cargo, may represent a novel mode of regulation of platelet aggregation at the vascular wall.
Similar content being viewed by others


Introduction
The relationship between an organelle’s size and its function has become recognized as an important element in the modulation of cellular behaviour, both in physiology and pathology1,2,3. We have been investigating this phenomenon in endothelial cells, using their pro-haemostatic granules as a model system of clinical relevance. The major cargo of these granules, the Weibel-Palade bodies (WPBs), is the haemostatic protein von Willebrand Factor (vWF)4. Formed by concatamers (often referred to as multimers) of its ~270 kDa monomer, plasma vWF is the largest soluble protein in vertebrates and plays a fundamental role in primary haemostasis5. This function depends on the multimeric structure of vWF, which in the circulation is stretched by shear to expose binding sites that anchor it at the vascular wall and recruit platelets, thus localizing formation of primary haemostatic plugs6,7. The fundamental role of vWF in haemostasis is highlighted by genetic or acquired deficiencies, qualitative or quantitative, that cause von Willebrand disease (vWD), the most common inherited bleeding disorder in humans5. vWF’s contribution to pro-thrombotic conditions is dramatically demonstrated by thrombotic thrombocytopenic purpura (TTP), a condition in which the ultra-large vWF multimers released by endothelial cells are not reduced in size by the circulating metalloprotease ADAMTS13. Genetic deficiency or autoimmune inhibition of this protease results in the formation of disseminated thrombi in the microcirculation, a life-threatening condition.
Within WPBs, highly multimerized vWF is coiled into tubules8; a structural arrangement that imparts the elongated, cigar-like morphology of these secretory granules and is necessary to the orderly release of vWF9. This arrangement requires that the structures of vWF and of the WPB are closely related, a major reason why the relationship of organelle size and function are especially transparent in this system. In addition, in cultured endothelial cells, WPBs are present with lengths between 0.5 and 5 μm, providing a large range for study.
vWF plays a multifaceted role in platelet adhesion and thrombus formation. vWF is synthesized not only by endothelial cells but also megakaryocytes and stored in platelets; however, experimental evidence shows that the endothelial vWF contribution to haemostasis outweighs that of platelets10,11 Soluble plasma vWF, derived from ADAMTS13 processing of the highly multimerized vWF secreted from endothelial cells can bind to substratum-adhered vWF, a homotypic association regulated by shear stress12,13. At pathologically high shear rates, soluble vWF can self-associate on collagen coated substrata (a model of the sub-endothelial matrix) and generates, in a concentration-dependent manner, string-like structures capable of recruiting platelets14. Within physiological shear stress values observed in the venous vasculature (2.5–20 dynes/cm2 or 0.25–2 MPa)15, vWF released by exocytosis of WPBs forms transient strings that are tens to hundreds of microns long16. These vWF strings result from the self-association of ultra-large (UL) vWF multimers into cables that are anchored at the endothelial surface and have been visualized at the ultra-structural level17. In order to form strings, UL-vWF multimers likely interact side by side and in an out-of-register fashion18, the only possible arrangement that explains both the cable structure and the vast length these structures can achieve. vWF strings, by adhering to the cell surface, provide a transient scaffold for platelet recruitment before being degraded by ADAMTS1316,19. Platelet-decorated vWF strings can further increase in number and complexity in the presence of soluble plasma vWF18,20. vWF function is thus closely linked to its quaternary, supra-multimeric structure.
We recently investigated how WPBs acquire their size21. During their biogenesis, the size of WPBs is dictated by the general architecture of the Golgi apparatus. A continuous Golgi ribbon allows for co-packaging of vWF cargo units, called “quanta”, into nascent WPBs. The final size of a WPB, therefore, depends on the number of quanta that are encased into each organelle at biogenesis. Unlinking (a.k.a. fragmentation) of the Golgi ribbon prevents such co-packaging, resulting in formation almost exclusively of short WPBs, termed mini-WPBs (Fig. 1a)21. In the same study, the functional significance of organelle size was investigated. Long WPBs (which we define as >2 μm) represent a minority of the organelle population but contain a significant fraction of the vWF stored by endothelial cells (Fig. 1b). This observation raised the possibility that long WPBs might be disproportionally important in defining the activity of the vWF released by endothelial cells. Indeed, we found that at exocytosis mini-WPBs generate shorter platelet-decorated vWF strings on the endothelial surface. These findings raised the possibility that endothelial cells may respond to physiological and pathological cues by modulating their haemostatic phenotype through changes in the size of these secretory organelles21. On one side, endothelial dysfunction might result in the production of a higher fraction of long WPBs and thus lead to increased pro-thrombotic propensity; on the other, a higher proportion of short organelles could result in an antithrombotic phenotype. At present, little is known about whether and how WPB size is regulated and our conclusions were based on experimental manipulations to generate mini-WPBs that are not physiological. Here, to validate our hypothesis, we addressed whether WPB size can be pharmacologically regulated. We find that statin treatment of cultured endothelial cells reduces the size of their WPBs, and like other treatments with the same effect, this impacts on both platelet recruitment and the recruitment of circulating plasma vWF to the endothelial surface. These new results lend further support to the idea that WPB size can play a role in the regulation of platelet aggregation.

(a) The functional unit of the Golgi apparatus is the mini-stack, a pile of flat membrane cisternae. At its entry side (the cis-side) the mini-stack receives secretory cargo from the endoplasmic reticulum and at its exit side (trans-side) this cargo is delivered to the trans-Golgi network. At the trans-Golgi network, cargos are packaged into transport carriers and sorted to their final destination within the cell. We have found that WPB size is determined by the structural status of the Golgi apparatus21. Mini-stacks, whose diameter ranges between 500 and 1000 nm, impart a limit to the size of the transiting vWF cargo, generating what we have defined vWF “quanta”. When the mini-stack are linked to form the structure known as Golgi ribbon (route 1), both short and long WPBs are formed, since vWF quanta can be co-packaged together at the trans-Golgi network. When the mini-stacks are unlinked (route 2), co-packaging of quanta is prevented and short WPBs are produced. The micrographs exemplify these conditions. HUVECs were transfected with vWF-GFP to label the WPBs being made during the treatment. In untreated cells (control cells) mini-stacks are linked into a ribbon (GM130) and WPBs of various sizes are generated. In cells with unlinked mini-stacks (route 2, obtained by treatment with nocodazole), only short (or mini-) WPBs are produced. Scale: 5 um. (b) WPBs have a typical cylindrical morphology and in untreated cultured HUVECs (as the Control in panel a) their length ranges between 0.5 and 5 μm21. The total area covered by WPBs indirectly measures the vWF they store. Morphological analysis indicates that long WPBs (defined as those longer than 2 μm; red-coded in the cartoon) represent a minor fraction of total number of organelles but account for a large fraction of the area they cover (bar graph, black vs red bar). Therefore, while a minority, long WPBs store a significant fraction of vWF. The quantifications displayed were obtained by High-throughput morphometric analysis (see text) of 4 sets (means ± SD) of untreated cells. Number of WPBs measured ~105.
Results
Statin treatment of endothelial cells reduces the size of WPBs and the length of platelet-decorated vWF strings
Statins reduce cholesterol plasma levels and decrease morbidity and mortality in patients with or at risk of cardiovascular disease22,23,24. More recently, in vitro, animal and clinical studies have provided evidence that statins have also antithrombotic effects25,26,27,28,29,30,31,32,33,34. Given our previous findings about the size of WPBs affecting platelet recruitment at the endothelial surface, and the antithrombotic phenotypes of statins, we investigated the effect on WPB size of this class of drugs. We treated human umbilical vein endothelial cells (HUVECs) for 24 h with two statins, simvastatin and fluvastatin, at concentrations usually used in in vitro studies31,35,36,37,38. Microscopic inspection showed that, at all concentrations tested, both statins reduced the size of WPBs. (Figs 2a and S1a). These effects were measured with an approach that we recently developed and named high-throughput morphometry (HTM), which allows the unbiased morphological quantification of tens to hundreds of thousands of organelles21. Induction of mini-WPB production was concentration-dependent: at 2.5 and 12.5 μmol/L both statins shortened the size of WPB to lengths similar to those generated by nocodazole, our positive control for mini-WPB generation, with an intermediate effect at 0.5 μmol/L (Fig. 2b–e). These effects on WPB size are explained by the unlinking of the Golgi ribbon in statin treated cells (Fig. 2a the Golgi marker GM130, S1a and 2f,g; also refer to Fig. 1a). Statin treatment generated Golgi fragments that clustered in proximity to the nucleus, similar to the treatment with acidic pH/acetate (Figs 2a, S1a and 5g), another treatment that we have previously shown to reduce WPB size through Golgi ribbon unlinking21. The Golgi unlinking induced by statins was morphologically different from that caused by nocodazole; statins do not induce microtubule depolymerisation, the basis for Golgi mini-stack scattering seen with nocodazole (Figs 2a and S1a,b). We conclude that statins cause WPB size reduction through unlinking of the Golgi ribbon, in agreement with the current model for the biogenesis of these organelles (Fig. 1a)21.

(a) HUVECs were grown for 24 h in medium containing vehicle (DMSO), statins or nocodazole at the indicated concentrations before being fixed and processed for immunofluorescence of the indicated markers (vWF for WPBs, GM130 for Golgi complexes). Nocodazole was used as a positive control treatment to induce mini-WPBs. Scale bar: 20 μm; magnified insets: 5 μm. (b,c) High-throughput morphometric (HTM) analysis of the whole population of WPBs in HUVECs treated for 24 h as indicated. Cumulative frequency (C.F.) plots of the total number of WPBs as a function of the organelles’ length. Relative to the control plot (DMSO), left-shifted curves indicate WPB size shortening, as shown by the nocodazole traces (the positive control for mini-WPB production). The number of WPBs analysed was ~105 per condition. £, P < 10−15 for the indicated pairwise comparisons (Mann-Whitney test). (d) The fraction of long WPBs (>2 μm) relative to DMSO controls is shown. Bars: mean ± range for two replicate HTM experiments with simvastatin and fluvastatin. (e) HTM analysis of the area covered by long organelles (>2 μm) as a fraction of the total area covered by all WPBs (see Fig. 1b) in HUVECs treated as indicated for 24 h. N = 16 for each treatment; median and interquartile ranges are shown; ****P < 0.0001. Comparisons are to the DMSO control (Mann-Whitney test). (f,g) HTM analysis of the whole population of the Golgi objects in cells treated with the indicated concentrations of simvastatin and fluvastatin. Cumulative frequency (C.F.) plots of the number of Golgi objects as a function of the their area. The number of Golgi objects analysed was ~104−105 in all conditions. £, P < 10−15 for the indicated pairwise comparisons (Mann-Whitney test).
If our hypothesis that WPB size correlates with the length of platelet-recruiting adhesive strings that vWF forms under flow on the endothelial surface is correct, then statin incubation should result in a reduction in the size of these structures. Statins have antiplatelet effects in vitro, ex vivo, in animal models and in patients29,32,39. Therefore, in order to distinguish their effects on endothelial cells from those on platelets, we used washed platelets from healthy non-medicated volunteers for our experiments. HUVECs treated with simvastatin at 0.5 and 2.5 μmol/L (concentrations that, respectively, induce submaximal and maximal WPB shortening) and challenged with histamine generated shorter platelet-decorated vWF strings (Fig. 3a,b), with the longest strings (>100 μm) being especially affected (Fig. 3c).

(a) HUVEC monolayers treated with vehicle (DMSO), 0.5 or 2.5 μmol/L simvastatin for 24 h were perfused with washed platelets from healthy non-medicated volunteers in the presence of histamine (100 μmol/L) to stimulate vWF secretion. Left, video-microscopy frames taken during perfusion show platelet-decorated strings (highlighted by dotted boxes). Right, samples were fixed and immuno-stained to visualize vWF strings and platelets (CD41). Scale bar: 50 μm. (b) Analysis of the length of platelet-decorated vWF strings, pooled from two independent experiments, in duplicate for each treatment. Median and interquartile ranges are shown. Number of strings measured: DMSO, 271; simvastatin 0.5 μmol/L, 217; simvastatin 2.5 μmol/L, 278. ****P < 0.0001 (Mann-Whitney test). (c) Statin treatment primarily affected the longest (>100 μm) strings. Error bars represent the range of variation for the two experiments. (d) Integrin β3, which in endothelial cell is present in complex with integrin αν and functions as an anchor for vWF strings, was quantified from western blots by densitometry following incubation for 24 h with 0.5, 2.5 and 12.5 μmol/L statins (indicated by the triangle symbols). n = 3; mean ± SEM. *P < 0.05 or lower (not shown for simplicity) for all treatments compared to DMSO control (Student’s t-test). γ-Adaptin (Ad-γ) was used as a loading control. (e) Cell-free WPBs prepared from cells treated for 24 h as indicated were detergent-permeabilised and the resulting vWF filaments immuno-stained and imaged (insets show magnifications of the dashed boxes). Scale bar: 25 μm. (f) vWF multimerization in cell-free WPBs was analysed by agarose gel electrophoresis. Highly multimerized vWF is indicated (high-molecular weight, HMW). The multimeric patterns of plasma vWF (p-vWF) untreated or reduced (dithiothreitol, DTT) and alkylated (N-ethylmaleimide, NEM) to generate monomers are shown for comparison.
This reduced length of the strings might be due to a reduced capability of secreted vWF to adhere to the endothelial surface. A known anchor of vWF strings to the endothelial surface is αvβ3 integrin17. Statin treatment might down-regulate of αvβ3 integrin, thus reducing the ability of the endothelial surface to bind secreted vWF. In fact, αvβ3 levels were slightly up-regulated following treatment with both simvastatin and fluvastatin (Fig. 3d), ruling out that the reduced platelet recruitment was due to a defect in string anchoring to the endothelial surface.
Shorter vWF strings could also result from a reduction in vWF multimerization40. The possibility thus exists that the production of mini-WPBs might influence the multimeric state of the vWF they store and therefore the length of the resulting strings. To address this, we prepared cell-free WPBs, from control-, statin- and nocodazole-treated cells. Permeabilization of cell-free WPBs results in the formation of vWF filaments, the likely precursors of the strings formed upon exocytosis21. During WPB biogenesis, vWF undergoes pH- and Ca2+-dependent conformational rearrangements necessary for inter-dimer disulphide formation and therefore required for multimerization41,42. Cell-free organelles from control and mini-WPB producing cells were equally capable of releasing vWF filaments in vitro (Fig. 3e), indicating that vWF folding and inter-dimer disulphide bond formation were unaffected by organelle size. In addition, WPB size did not have any obvious effect on the multimeric pattern of vWF, with highly multimerized protein present in comparable abundance in all conditions (Fig. 3f). These data rule out that the mini-WPBs generated following statin or nocodazole treatment impair vWF multimerization. Therefore, changes in vWF multimer size within mini-WPBs cannot explain the reduced length of the platelet-decorated strings. In summary, pharmacological treatment can prompt changes in WPB size and thus influence the adhesive properties of its haemostatic cargo, vWF, towards platelets.
The role of KLF2 in statin-mediated effects on WPB size
One of the mechanistic explanation for the antithrombotic effects of statins is that they up-regulate the expression of Krüppel-like Factor 2 (KLF2)31,35, a transcription factor known to coordinate a compounded anti-inflammatory and anticoagulant response43,44. Intriguingly, virus-mediated overexpression of KLF2 in endothelial cells results in the production of mini-WPBs45. Due to this link between statins and KLF2, we investigated whether this transcription factor may be responsible for the effects of statins on WPB size. As reported by others31,35, at all the concentrations we tested, both simvastatin and fluvastatin resulted in KLF2 mRNA up-regulation in HUVECs (Figure S2a) and induction of two of its target genes, vWF (Figure S2b,c) and Thrombomodulin (TM)46,47 (Figure S2d). KLF2 overexpression was also reported to increase the number of WPBs in endothelial cells45. Statin treatment for 24 h had the same effect (Figure S2e,f). Finally, we confirmed that up-regulation of KLF2 results in WPB size shortening45, by overexpressing an EGFP-KLF2 fusion protein. Although EGFP-KLF2 expressing cells were fewer than EGFP-expressing controls (Figure S2g,h), HTM analysis at a single-cell level allowed us to identify and separately analyse the WPB population in transiently transfected cells and clearly showed that the size of WPBs was reduced in EGFP-KLF2 positive cells (Fig. 4a,b). Together, these data suggest that KLF2 up-regulation by statins might indeed play a key role in mediating their effects on WPB size. If this is the case, then KLF2 knockdown should prevent the effects of statins on WPB size. Simvastatin and fluvastatin treatment for 24 h increased both KLF2 mRNA and protein levels in Luciferase-siRNA treated cells; this up-regulation was however blocked in KLF2-siRNA cells (Figs 4c and S2i,j). Of note, the increase in WPB numbers due to statin incubation was almost completely blocked by KLF2 knockdown, suggesting that this phenomenon is mediated for the most part by KLF2 up-regulation (Figure S2k). As for WPB size, KLF2 knockdown cells treated with DMSO showed a modest increase in WPB size (Fig. 4d,e and compare insets 3 and 4 to 1 and 2 in 4f). However, upon statin treatment, KLF2 knockdown only partially rescued the WPB size reduction (Fig. 4d,e and compare insets 7 and 8 to 5 and 6 in 4f). This indicates that KLF2 plays only a partial and not a major role in the statin-mediated WPB size reduction. Statins cause a reduction in WPB size through Golgi unlinking (Fig. 2a,f,g). We therefore analysed the effects of KLF2 knockdown on this organelle, finding that it did not rescue the fragmentation caused by statin treatment (Fig. 4g,h). The effects of statin on the Golgi complex thus explain why KLF2 knockdown fails to fully rescue the size of WPBs to control (i.e., Luciferase/DMSO treatment) levels. We conclude that other mechanisms besides KLF2 are responsible for the statin-induced Golgi unlinking and WPB shortening.

(a) EGFP and EGFP-KLF2 overexpressing HUVECs. Scale bar, 10 μm; inset magnifications: 2 μm. (b) HTM analysis showing the fraction of the WPB area covered by long organelles in the subpopulations of EGFP or EGFP-KLF2 overexpressing HUVECs; ****P < 0.0001 (Mann-Whitney test). (c) HUVECs were treated with Luciferase or KLF2 targeting siRNAs and after 24 were incubated with DMSO (D), 2.5 μmol/L simvastatin (S) or 2.5 μmol/L fluvastatin (F) for 24 h before cell lysis. Equal protein amounts were analysed as described for Fig. 3d. Bars represent means ± SEM from 3 experiments. *P < 0.05; ***P < 0.001; ****P < 0.0001 (Student’s test). (d) HTM analysis of WPB size in Luciferase- and KLF2-siRNA treated HUVECs incubated with vehicle (DMSO) or 2.5 μmol/L simvastatin for 24 h. Cumulative frequency of WPB number as a function of size (note the right-shift of KLF2 curves in both conditions). The number of WPBs analysed was ~105 per condition. £, P < 10−15 for the indicated pairwise comparisons (Mann-Whitney test). (e) Left, the fraction of long WPBs (>2 μm) was expressed as % of the Luciferase-siRNA/DMSO controls. Bars, mean ± range for two replicate experiments. Right, the fraction of the WPB area covered by long organelles; ****P < 0.0001 (Mann-Whitney test); comparisons are with the Luciferase-siRNA/DMSO controls. (f) WPBs in Luciferase- and KLF2-siRNA treated cells after 24 h treatment with DMSO or 2.5 μmol/L simvastatin. Scale bar: 10 μm; inset magnifications: 2 μm. (g) HTM analysis of Golgi objects. The numbers of Golgi object analysed were similar to those in Fig. 2e,f. £, P < 10−15 (Mann-Whitney test). (h) Cells treated for 24 h with Luciferase- or KLF2-targeting siRNAs were then incubated for 24 h with 2.5 μmol/L simvastatin and processed for immunofluorescence. Scale bar: 20 μm.
Plasma vWF recruitment to the endothelial surface is sensitive to WPB size
We investigated further the functional consequences of WPB size. In vitro, under high wall shear stress conditions, surface immobilized vWF interacts with soluble flowing vWF, promoting its deposition20,48, a process that in vivo may act to amplify the recruitment of platelets and thus growth of platelet aggregates. We therefore tested whether, at physiological venous wall shear stress (2.5 dynes/cm2), the secreted endothelial vWF is able to recruit circulating plasma vWF to the cell surface.
We initially tested the effects of human plasma perfusion, to mimic the blood circulation, on endothelial cell monolayers in the absence or presence of histamine, which stimulates WPB exocytosis and vWF secretion (Fig. 5a). Plasma perfusion alone resulted in low amounts of vWF associated with the endothelial surface (Fig. 5b, plasma). Upon histamine stimulation and in the absence of plasma, the vWF secreted by endothelial monolayers adhered to the endothelial surface, most notably as strings (Fig. 5b, histamine). When plasma was perfused concurrently with histamine stimulation, higher levels of surface-adhered vWF were observed, indicating that soluble plasma is recruited to the endothelial surface (Fig. 5b, histamine and plasma). In this condition, fewer and shorter strings were visible, likely because of the proteolytic activity of ADAMTS13 - and possibly of other plasma proteases - which reduce both length and numbers of vWF strings16,19. The methodology and effects observed in these assays are summarized in the schematic depicted in Fig. 5c. In order to quantify vWF association with endothelial cells and compare treatments, we measured the area covered by its immuno-fluorescent signal. As shown in Fig. 5d, soluble plasma vWF can bind the endothelial surface in the absence of platelets. This recruitment seems to require WPB exocytosis (histamine stimulation), since in un-stimulated cells, even in the presence of plasma, little vWF was associated with endothelial cells (Fig. 5b,d, plasma). Therefore, secretagogue-activated endothelial cells become adhesive to soluble plasma vWF.

(a) HUVECs were subjected to flow assays as indicated. When WPB exocytosis was stimulated, histamine (100 μmol/L) was supplemented to buffer and human normal pooled plasma. (b) At the end of the assays, cells were fixed and the extracellular vWF immuno-stained and imaged. Scale bar: 25 μm. (c) Diagram of the procedure in panels (a) and (b) illustrates the assay and what it measures. (d) vWF associated with the endothelial surface following the assays described in panel (a). The area of the vWF signal on the endothelial surface was measured from 36 fields of view (the data points displayed) per condition. Bars: medians and interquartile ranges; histamine stimulation in buffer and histamine stimulation coupled to plasma were compared to the unstimulated plus plasma condition; ****P < 0.0001 (Mann-Whitney test). (e) Luciferase- or vWF-siRNA treated HUVECs were subjected to the indicated flow assay (left) and vWF deposition on endothelial cells measured (right); ****P < 0.0001 (Mann-Whitney test). (f) vWF plasma recruitment after a brief treatment with recombinant ADAMTS13 (rADAMTS13) at the indicated concentrations; *P < 0.05; ***P < 0.001 (Mann-Whitney test). (g) HUVECs treated for 24 h with vehicle (DMSO, control), medium at pH 6.4 and supplemented with acetate or nocodazole (1 μg/mL) were processed for immunofluorescence. Scale bar: 5 μm. (h) HTM analysis of HUVECS treated as described in (g). Left, Cumulative frequency (C.F.) plots of the total number of WPBs as a function of the organelles’ length. N ~ 105 per condition. £, P < 10−15 for the indicated pairwise comparisons (Mann-Whitney test). Right; long WPBs (>2 μm). Bars: mean ± range for two replicate HTM experiments. (i) HUVECs treated as described and subjected to the indicated flow assay (left) to measure vWF deposition on endothelial cells (right). Quantifications were as described above. ****P < 0.0001 (Mann-Whitney test). (j) vWF deposition on HUVECs treated with vehicle (DMSO), 0.5 or 2.5 μmol/L simvastatin for 24 h and perfused as described (left). ****P < 0.0001 (Mann-Whitney test).
To establish whether this recruitment of soluble plasma vWF depends on the secretion of endothelial vWF, we tested the effects of vWF knockdown in this assay. vWF-targeting siRNA treatment of HUVECs resulted in the reduction of both protein levels and WPB numbers21 (see also Figure S3a). Surface deposition of secreted endothelial and soluble plasma vWF was abolished when vWF was knocked down (Figs 5e and S3b), indicating that endothelial vWF secretion is required for recruitment of circulating plasma vWF to the cell surface.
To test whether plasma vWF recruitment to the endothelial surface is direct, we carried out experiments with purified plasma vWF (Figure S3c) at the same concentration it has in whole plasma (~10 μg/mL). Perfusion of purified vWF was sufficient to induce an increase in endothelial surface associated vWF signal, but this was not as striking as that seen in the presence of whole plasma (Figure S3d). These data suggest that while plasma vWF can bind directly to the secreted endothelial vWF, other plasma components are likely to co-operate to enhance this interaction.
ADAMTS13 promptly degrades highly multimerized vWF, which, by being more sensitive to shear, exposes its cleavage sites to the protease under flow6,49. vWF strings are particularly sensitive to ADAMTS13 activity; in its presence their size and number are progressively reduced16,19. We therefore measured the effects of short treatments with recombinant ADAMTS13 (rADAMTS13) in order to reduce the levels of secreted vWF on the endothelial surface before perfusion with plasma. At concentrations found in plasma of patients with inherited and acquired TTP (0.1 μg/mL)50, rADAMTS13 had no effect on vWF associated to the endothelial surface after buffer perfusion (Fig. S3e) and only slightly reduced it when plasma was added (Fig. 5f). Pre-treatment with normal concentrations of rADAMTS13 (1.0 μg/mL), however, resulted in a clear decrease of endothelial-associated vWF in both conditions (Figs 5f and S3e, 1.0 μg/mL rADAMTS13). Together, these results are consistent with the conclusion that secreted endothelial vWF is the relevant factor for the recruitment of plasma vWF to the endothelial surface. Secreted endothelial vWF thus provides adhesive scaffolds that can locally recruit plasma vWF to the endothelial surface. Under flow, the immobilized plasma vWF might transiently (i.e. before ADAMTS13 counteracts this phenomenon) synergize with the vWF strings generated by WPB exocytosis to increase the recruitment of circulating platelets to the endothelial surface.
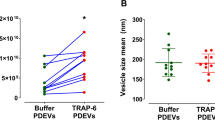
Our findings imply that mechanisms capable of regulating vWF string size or number can modulate the adhesive properties of a vascular bed towards soluble vWF and platelets. We have shown that one mode of regulation of vWF string length is through control of the size of WPBs. We therefore assessed whether the size of these organelles, in addition to the length of platelet-decorated vWF strings, also influences the capability of its vWF cargo to recruit, when secreted, soluble plasma vWF. To this aim, we prompted formation of mini-WPBs in HUVECs by two experimental treatments that we previously used21 to unlink the Golgi ribbon, whose integrity is necessary for the formation of long organelles (Fig. 1a): depolymerisation of microtubules with nocodazole51 and incubation with acetate in acidic conditions21,52 (Figure S3f,g). In both cases, we obtained cells with WPBs that were shorter than controls (Fig. 5g,h). Cells with mini-WPBs showed either no change or a reduced association of secreted vWF with the cell surface, compared to control conditions, when stimulated with buffer containing histamine under flow (Fig. S3h). However, both mini-WPB-producing endothelial monolayers recruited considerably less soluble plasma vWF than controls (Fig. 5i). Of note, upon histamine challenge, one of the mini-WPB-generating treatments (pH 6.4/acetate) released more vWF while the other (nocodazole) released less than control cells (Fig. S3i,j). Thus, the extent of plasma vWF recruitment appears to correlate with the size of WPBs, rather than the total amount of their secreted vWF cargo.
Since statins also reduce WPB size, we tested their effects on the recruitment of soluble plasma vWF to the endothelial surface. We compared controls to cells treated for 24 h with simvastatin at 0.5 and 2.5 μmol/L, concentrations that generate intermediate and maximal effects on mini-WPB production (Fig. 2b–e). At both concentrations, soluble plasma vWF recruitment was dramatically inhibited by statin treatment (Fig. 5j). Thus, consistent with other mini-WPB inducing treatments, statins also reduce plasma vWF recruitment to the endothelium.
Discussion
Endothelial cells produce Weibel-Palade bodies in a wide range of sizes (0.5–5 μm). Long WPBs contain a substantial fraction of the vWF stored by endothelial cells (Fig. 1b) and may therefore be disproportionately important in defining the activity of the vWF secreted by endothelial cells. In a recent study, we tested this by measuring the size of the platelet-decorated vWF strings generated by control cells or cells enriched in mini-WPBs, observing a positive correlation between WPB size and string length21. These findings suggested that endothelial beds may use regulation of WPB size to spatially and temporally modulate their haemostatic propensity as a consequence of physiological cues21. This hypothesis predicts that: (a) size control dysregulation, with production of a higher proportion of long WPBs, may contribute to pathological endothelial thrombotic behaviour; (b) such a condition might be ameliorated or reverted by promoting biogenesis of shorter organelles.
The experimental manipulations we used to induce Golgi unlinking and WPB size changes (microtubule depolymerisation, knockdown of Golgi structural proteins, etc.)21 do not occur physiologically in cells. Therefore, to validate our hypothesis, we investigated whether pharmacological treatment of endothelial cells was capable of inducing changes in WPB size. We focused on a widely used class of drugs: statins. The rationale for this choice was that, besides lowering cholesterol plasma levels, statins display a wide range of “pleiotropic” effects that are independent from lipid regulation. In particular, statins elicit rapid antithrombotic and anticoagulant responses. Statins decrease pro-thrombotic/pro-coagulant mediators, such as NOX2 (a subunit of NADPH oxidase), Tissue Factor, Plasminogen Activator Inhibitor-1 and PAR-1, while increasing antithrombotic molecules, such as Thrombomodulin, tissue Plasminogen Activator, Tissue Factor Pathway Inhibitor, eNOS and its product nitric oxide30,33,34,53. Moreover, statins up-regulate KLF231,35, a “master switch” transcription factor that promotes a quiescent, antithrombotic and anti-inflammatory phenotype in endothelial cells43,44. Interestingly, when overexpressed in endothelial cells, KLF2 induces formation of smaller WPBs45, which to the best of our knowledge, is the only experimental evidence indicating the existence of pathways that regulate WPB size.
We found that statin treatment induces a reduction of WPB size that is dependent on Golgi ribbon unlinking, consistent with the current model of WPB biogenesis (Fig. 1a). We also investigated whether KLF2 mediates these statin effects. KLF2 overexpression does, indeed reduce WPB size, and we also show that KLF2 siRNA-mediated depletion slightly increases organelle size. However, upon statin treatment KLF2 depletion, although efficient, is not sufficient for a full rescue of the WPB size to control levels (i.e. as in DMSO-treated Luciferase-siRNA cells). It is the Golgi unlinking induced by statins, and which is not rescued by KLF2 knockdown, that prevents the biogenesis of long WPBs (Fig. 1a) and therefore explains the reason for the failure to rescue size. We must thus conclude that statin-responsive effectors other than KLF2 are involved in the Golgi ribbon unlinking that mediates the reduction of WPB size. What these effectors might be remains to be investigated. The most obvious is the lack of cholesterol, since its depletion in some cell types does result in fragmentation of the Golgi apparatus54. However, we cultured cells in medium containing 20% serum in all conditions; it is therefore unlikely that cellular cholesterol is severely depleted even in the presence of statins. Importantly, since statins inhibit HMG-CoA reductase activity and thus block the synthesis of mevalonate and its downstream products, the changes to the Golgi structure might also be the results of signalling pathways sensing the depletion of metabolites other than cholesterol.
It is known that under high wall shear stress, vWF immobilized on a substratum can recruit soluble vWF48 and that compared to buffer, at venous shear wall stress, the combined presence of plasma and platelets temporarily increases the size and number of platelet-decorated vWF strings before ADAMTS13 initiates their degradation20. We now directly show that, in the absence of platelets, plasma vWF is recruited to the endothelial surface following WPB secretion. This recruitment is dependent on endothelial, highly-multimerized vWF being secreted and assembling into strings, since it is inhibited by both vWF knockdown and recombinant ADAMTS13 pre-treatment. Crucially, WPB size has similar effects to ADAMTS13 treatment, leading to the conclusion that the dimensions of WPBs do affect the homotypic (i.e. towards plasma vWF) adhesive activity of their vWF cargo.
As in the case of other mini-WPB inducing treatments, statin incubation reduced both plasma vWF recruitment and the length of the platelet-decorated vWF strings. Of note, even a small reduction of the fraction of long WPBs, such as that seen at the lowest concentration of statins used in our experiments, was sufficient to elicit an effect on both the homotypic and the heterotypic (towards platelets) adhesive activity of the secreted vWF. This seems to indicate that functional effects become apparent when the fraction of long WPBs falls below a threshold.
How WPB size regulates the length of the strings generated by vWF secretion is at present unclear. It is known that vWF multimerization can affect string length40. However, we show here that in short WPBs, induced by either statin or nocodazole treatment, multimerization is not impaired and therefore cannot account for the difference in size of the platelet-decorated vWF strings that are generated. We conclude that the size of the organelle, imparting an as yet unknown spatial arrangement to the packaged vWF, results in the formation of strings of differing length.
Statins reduce vWF secretion from endothelial cells55,56. Could this reduction in vWF release explain the effects on plasma vWF recruitment to the endothelial surface and on the length of platelet-decorated vWF strings observed when cells are treated with statins? While this possibility cannot be ruled out, our results suggest otherwise. First, we found that rather than the total amount of vWF released it is the size of WPBs that correlates best with the recruitment of plasma vWF to the endothelial surface (Figs 5i and S3i). Second, independently of the number of the platelet-decorated vWF strings generated at the cell surface, statin treatment does indeed affect their lengths (Fig. 3b).
Statins’ effect on WPB size is rapid. Interestingly, as shown in animal and clinical studies, the antithrombotic response elicited by statins also have a fast onset that is unexplained by their effects on plasma cholesterol29,30,32,33,34,57,58. It is therefore tantalizing to suggest, based on the results we present, that statins’ effect on WPB size could contribute to the overall antithrombotic phenotype induced by these drugs. However, whether our in vitro findings regarding the statin effects on WPB size can be translated to in vivo models and clinical settings remains to be tested experimentally.
Nonetheless, this study represents a proof of concept. By confirming and significantly expanding our previous observations, the present findings provide further support to the notion that WPB size plays a role in modulating the adhesive activity of its vWF cargo. Since plasma vWF is required for platelet aggregation48, control of WPB size, by affecting the recruitment of both these factors, may represent a regulatory node for platelet aggregation to the endothelial cell surface (Fig. 6). Considering that signalling pathways that affect the integrity of the Golgi ribbon do exist59, the physiological modulation of WPB size (Fig. 1a) is not only possible but indeed likely. Compounds capable of intercepting these pathways, whether they will turn out to be statins or others, may thus represent new tools in the treatment of thrombotic pathologies.

(A) Cultured endothelial cells produce WPBs with lengths ranging between 0.5 and 5 μm. Under flow, which mimics blood circulation, WPB exocytosis releases highly multimerized vWF generating vWF strings, which function as adhesive platforms for the recruitment of platelets on the endothelial surface. In mini-WPB producing cells, vWF exocytosis generates shorter platelet-decorated vWF strings. (B) Exocytosis of mini-WPBs, such as those produced by treatment with statins, deploys vWF that is less active in recruiting the soluble vWF present in circulating plasma. This diminished self-adhesion may synergize with the reduced platelet recruitment.
Materials and Methods
Cells
Human umbilical vein endothelial cells (HUVECs) pooled from donors of both sexes were commercially obtained from Lonza or PromoCell. Cells were used within passage 5 (~15 population doublings since isolation from umbilical cord).
Antibodies and reagents
Four antibodies to vWF were used: a rabbit polyclonal antibody to vWF pro-peptide region, a kind gift by Dr. Carter (St. George’s University, London), was previously described60; a rabbit polyclonal antibody from DAKO (cat. no. A0082); a sheep polyclonal antibody from AbD Serotec (cat. no. AHP062); and a HRP-conjugated rabbit polyclonal antibody from DAKO (cat. no. P0226). The antibody to pan-Tubulin (sheep polyclonal, cat. no. ATN02) was from Cytoskeleton. Anti-Thrombomodulin (mouse monoclonal, D-3) antibody was from Santa Cruz biotechnology. Mouse monoclonal antibodies to GM130, VE-Cadherin, and γ-Adaptin were from BD Bioscience. Anti-β3 integrin antibody (rabbit polyclonal, cat. no. 4702) was from Cell Signalling Technology. Anti-GPIIb/CD41 (clone 5B12) mouse monoclonal antibody was from Millipore. Anti-KLF2 raabbit polyclonal antibody was from Novus Biologicals (cat. no. NBP2-31619). Histamine was either from Sigma-Aldrich (cat. no. H7250) or Enzo Life Sciences (cat. no. ALX-550-132). Tyrode salts (T2145), nocodazole (M1404), simvastatin (S6196) and fluvastatin sodium hydrate (SML0038) were all from Sigma-Aldrich. Human recombinant full-length ADAMTS13 was from R&D Systems (cat. no. 6156-AD-020). Purified plasma vWF was kindly provided by Dr. Mckinnon and Prof. Laffan (Imperial College London). The plasmids used in this study are described in the Supplementary Information.
WPB secretion
WPB secretion was stimulated by challenging HUVECs with histamine at a final concentration of 100 μmol/L.
High-throughput morphometry (HTM)
Image processing, organelle segmentation and measurement of morphological parameters (high-throughput morphometry, HTM) have been described in detail elsewhere21. Briefly, images of the organelle channel (i.e., WPBs or Golgi) were processed with ImageJ using the “rolling ball” background subtraction and then subjected to segmentation with an auto local threshold, the “Bernsen’s method” (https://fiji.sc). This high-throughput imaging approach provides an unbiased, deep probing of samples, detecting quantitative morphological differences with a high level of confidence. Details of the HTM analysis of EGFP and EGFP-KLF2 expressing cells are provided in the Supplementary Information.
Flow assays
HUVECs seeded on Ibidi μ-slides were maintained at 37 °C and stimulated to secrete vWF while perfused with buffer, normal human pooled plasma or washed platelets at a constant wall shear stress of 0.25 MPa (2.5 dynes/cm2). Details are available in the Supplementary Information.
Statistical analysis
Non-parametric, two-tailed, two independent sample Mann-Whitney test was used, except when explicitly stated otherwise. R (http://www.r-project.org/) was used to analyse HTM datasets. In all other cases, we used Prism 6 (GraphPad) statistical package.
Additional Information
How to cite this article: Ferraro, F. et al. Weibel-Palade body size modulates the adhesive activity of its von Willebrand Factor cargo in cultured endothelial cells. Sci. Rep. 6, 32473; doi: 10.1038/srep32473 (2016).
Change history
26 September 2016
A correction has been published and is appended to both the HTML and PDF versions of this paper. The error has been fixed in the paper.
References
Chan, Y. H. & Marshall, W. F. How cells know the size of their organelles. Science (New York, N.Y) 337, 1186–1189, 10.1126/science.1223539 (2012).
Marshall, W. F. Organelle size control systems: from cell geometry to organelle-directed medicine. Bioessays 34, 721–724, 10.1002/bies.201200043 (2012).
Marshall, W. F. Subcellular size. Cold Spring Harb Perspect Biol 7, 10.1101/cshperspect.a019059 (2015).
Ewenstein, B. M., Warhol, M. J., Handin, R. I. & Pober, J. S. Composition of the von Willebrand factor storage organelle (Weibel-Palade body) isolated from cultured human umbilical vein endothelial cells. The Journal of cell biology 104, 1423–1433 (1987).
Sadler, J. E. Biochemistry and genetics of von Willebrand factor. Annu Rev Biochem 67, 395–424, 10.1146/annurev.biochem.67.1.395 (1998).
Springer, T. A. von Willebrand factor, Jedi knight of the bloodstream. Blood 124, 1412–1425, 10.1182/blood-2014-05-378638 (2014).
Springer, T. A. Biology and physics of von Willebrand factor concatamers. J Thromb Haemost 9 Suppl 1, 130–143, 10.1111/j.1538-7836.2011.04320.x (2011).
Zenner, H. L., Collinson, L. M., Michaux, G. & Cutler, D. F. High-pressure freezing provides insights into Weibel-Palade body biogenesis. Journal of cell science 120, 2117–2125 (2007).
Michaux, G. et al. The physiological function of von Willebrand’s factor depends on its tubular storage in endothelial Weibel-Palade bodies. Developmental cell 10, 223–232 (2006).
Nichols, T. C. et al. Function of von Willebrand factor after crossed bone marrow transplantation between normal and von Willebrand disease pigs: effect on arterial thrombosis in chimeras. Proceedings of the National Academy of Sciences of the United States of America 92, 2455–2459 (1995).
Kanaji, S., Fahs, S. A., Shi, Q., Haberichter, S. L. & Montgomery, R. R. Contribution of platelet vs. endothelial VWF to platelet adhesion and hemostasis. J Thromb Haemost 10, 1646–1652, 10.1111/j.1538-7836.2012.04797.x (2012).
Ruggeri, Z. M. Platelet adhesion under flow. Microcirculation 16, 58–83, 10.1080/10739680802651477 (2009).
Ruggeri, Z. M. & Mendolicchio, G. L. Interaction of von Willebrand factor with platelets and the vessel wall. Hamostaseologie 35, 10.5482/HAMO-14-12-0081 (2015).
Barg, A. et al. Soluble plasma-derived von Willebrand factor assembles to a haemostatically active filamentous network. Thromb Haemost 97, 514–526 (2007).
Kroll, M. H., Hellums, J. D., McIntire, L. V., Schafer, A. I. & Moake, J. L. Platelets and shear stress. Blood 88, 1525–1541 (1996).
Dong, J. F. et al. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood 100, 4033–4039, 10.1182/blood-2002-05-1401 (2002).
Huang, J., Roth, R., Heuser, J. E. & Sadler, J. E. Integrin alpha(v)beta(3) on human endothelial cells binds von Willebrand factor strings under fluid shear stress. Blood 113, 1589–1597, 10.1182/blood-2008-05-158584 (2009).
Choi, H., Aboulfatova, K., Pownall, H. J., Cook, R. & Dong, J. F. Shear-induced disulfide bond formation regulates adhesion activity of von Willebrand factor. The Journal of biological chemistry 282, 35604–35611, 10.1074/jbc.M704047200 (2007).
De Ceunynck, K. et al. Local elongation of endothelial cell-anchored von Willebrand factor strings precedes ADAMTS13 protein-mediated proteolysis. The Journal of biological chemistry 286, 36361–36367, 10.1074/jbc.M111.271890 (2011).
Li, Y. et al. Covalent regulation of ULVWF string formation and elongation on endothelial cells under flow conditions. J Thromb Haemost 6, 1135–1143, 10.1111/j.1538-7836.2008.02991.x (2008).
Ferraro, F. et al. A two-tier Golgi-based control of organelle size underpins the functional plasticity of endothelial cells. Developmental cell 29, 292–304, 10.1016/j.devcel.2014.03.021 (2014).
Lipid study group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med 339, 1349–1357, 10.1056/NEJM199811053391902 (1998).
Brugts, J. J. et al. The benefits of statins in people without established cardiovascular disease but with cardiovascular risk factors: meta-analysis of randomised controlled trials. Bmj 338, b2376, 10.1136/bmj.b2376 (2009).
Ross, S. D. et al. Clinical outcomes in statin treatment trials: a meta-analysis. Archives of internal medicine 159, 1793–1802 (1999).
Agarwal, V., Phung, O. J., Tongbram, V., Bhardwaj, A. & Coleman, C. I. Statin use and the prevention of venous thromboembolism: a meta-analysis. Int J Clin Pract 64, 1375–1383, 10.1111/j.1742-1241.2010.02439.x (2010).
Banfi, C. et al. Statins prevent tissue factor induction by protease-activated receptors 1 and 2 in human umbilical vein endothelial cells in vitro . J Thromb Haemost 9, 1608–1619, 10.1111/j.1538-7836.2011.04366.x (2011).
Krysiak, R., Okopien, B. & Herman, Z. Effects of HMG-CoA reductase inhibitors on coagulation and fibrinolysis processes. Drugs 63, 1821–1854 (2003).
Ludman, A., Venugopal, V., Yellon, D. M. & Hausenloy, D. J. Statins and cardioprotection–more than just lipid lowering? Pharmacology & therapeutics 122, 30–43, 10.1016/j.pharmthera.2009.01.002 (2009).
Moraes, L. A. et al. Antithrombotic actions of statins involve PECAM-1 signaling. Blood 122, 3188–3196, 10.1182/blood-2013-04-491845 (2013).
Owens, A. P. & Mackman, N. The antithrombotic effects of statins. Annual review of medicine 65, 433–445, 10.1146/annurev-med-051812-145304 (2014).
Parmar, K. M. et al. Statins exert endothelial atheroprotective effects via the KLF2 transcription factor. The Journal of biological chemistry 280, 26714–26719, 10.1074/jbc.C500144200 (2005).
Pignatelli, P. et al. Immediate antioxidant and antiplatelet effect of atorvastatin via inhibition of Nox2. Circulation 126, 92–103, 10.1161/CIRCULATIONAHA.112.095554 (2012).
Undas, A., Brummel-Ziedins, K. E. & Mann, K. G. Statins and blood coagulation. Arteriosclerosis, thrombosis, and vascular biology 25, 287–294, 10.1161/01.ATV.0000151647.14923.ec (2005).
Violi, F., Calvieri, C., Ferro, D. & Pignatelli, P. Statins as antithrombotic drugs. Circulation 127, 251–257, 10.1161/CIRCULATIONAHA.112.145334 (2013).
Sen-Banerjee, S. et al. Kruppel-like factor 2 as a novel mediator of statin effects in endothelial cells. Circulation 112, 720–726, 10.1161/CIRCULATIONAHA.104.525774 (2005).
Kou, R., Sartoretto, J. & Michel, T. Regulation of Rac1 by simvastatin in endothelial cells: differential roles of AMP-activated protein kinase and calmodulin-dependent kinase kinase-beta. The Journal of biological chemistry 284, 14734–14743, 10.1074/jbc.M808664200 (2009).
Sun, W. et al. Statins activate AMP-activated protein kinase in vitro and in vivo . Circulation 114, 2655–2662, 10.1161/CIRCULATIONAHA.106.630194 (2006).
Kosmidou, I., Moore, J. P., Weber, M. & Searles, C. D. Statin treatment and 3′ polyadenylation of eNOS mRNA. Arteriosclerosis, thrombosis, and vascular biology 27, 2642–2649, 10.1161/ATVBAHA.107.154492 (2007).
Hamada, M. et al. Antithrombotic properties of pravastatin reducing intra-thrombus fibrin deposition under high shear blood flow conditions. Thromb Haemost 105, 313–320, 10.1160/TH10-09-0587 (2011).
Nightingale, T. D., Pattni, K., Hume, A. N., Seabra, M. C. & Cutler, D. F. Rab27a and MyRIP regulate the amount and multimeric state of VWF released from endothelial cells. Blood 113, 5010–5018, 10.1182/blood-2008-09-181206 (2009).
Huang, R. H. et al. Assembly of Weibel-Palade body-like tubules from N-terminal domains of von Willebrand factor. Proceedings of the National Academy of Sciences of the United States of America 105, 482–487 (2008).
Zhou, Y. F. et al. A pH-regulated dimeric bouquet in the structure of von Willebrand factor. The EMBO journal 30, 4098–4111 (2011).
Atkins, G. B. & Jain, M. K. Role of Kruppel-like transcription factors in endothelial biology. Circ Res 100, 1686–1695, 10.1161/01.RES.0000267856.00713.0a (2007).
Nayak, L., Lin, Z. & Jain, M. K. “Go with the flow”: how Kruppel-like factor 2 regulates the vasoprotective effects of shear stress. Antioxidants & redox signaling 15, 1449–1461, 10.1089/ars.2010.3647 (2011).
van Agtmaal, E. L. et al. The shear stress-induced transcription factor KLF2 affects dynamics and angiopoietin-2 content of Weibel-Palade bodies. PLoS One 7, e38399, 10.1371/journal.pone.0038399 (2012).
Dekker, R. J. et al. KLF2 provokes a gene expression pattern that establishes functional quiescent differentiation of the endothelium. Blood 107, 4354–4363, 10.1182/blood-2005-08-3465 (2006).
Lin, Z. et al. Kruppel-like factor 2 (KLF2) regulates endothelial thrombotic function. Circ Res 96, e48–57, 10.1161/01.RES.0000159707.05637.a1 (2005).
Savage, B., Sixma, J. J. & Ruggeri, Z. M. Functional self-association of von Willebrand factor during platelet adhesion under flow. Proceedings of the National Academy of Sciences of the United States of America 99, 425–430, 10.1073/pnas.012459599 (2002).
Zhang, X., Halvorsen, K., Zhang, C. Z., Wong, W. P. & Springer, T. A. Mechanoenzymatic cleavage of the ultralarge vascular protein von Willebrand factor. Science. New York, N.Y 324, 1330–1334 (2009).
Rieger, M. et al. Relation between ADAMTS13 activity and ADAMTS13 antigen levels in healthy donors and patients with thrombotic microangiopathies (TMA). Thromb Haemost 95, 212–220, 10.1160/TH05-08-0550 (2006).
Thyberg, J. & Moskalewski, S. Microtubules and the organization of the Golgi complex. Exp Cell Res 159, 1–16 (1985).
Yoshida, T., Kamiya, T., Imanaka-Yoshida, K. & Sakakura, T. Low cytoplasmic pH causes fragmentation and dispersal of the Golgi apparatus in human hepatoma cells. International journal of experimental pathology 80, 51–57 (1999).
Lin, Z., Hamik, A., Jain, R., Kumar, A. & Jain, M. K. Kruppel-like factor 2 inhibits protease activated receptor-1 expression and thrombin-mediated endothelial activation. Arteriosclerosis, thrombosis, and vascular biology 26, 1185–1189, 10.1161/01.ATV.0000215638.53414.99 (2006).
Minogue, S. et al. Relationship between phosphatidylinositol 4-phosphate synthesis, membrane organization, and lateral diffusion of PI4KIIalpha at the trans-Golgi network. Journal of lipid research 51, 2314–2324, 10.1194/jlr.M005751 (2010).
Fish, R. J. et al. Fluvastatin inhibits regulated secretion of endothelial cell von Willebrand factor in response to diverse secretagogues. Biochem J 405, 597–604, 10.1042/BJ20070404 (2007).
Yamakuchi, M. et al. HMG-CoA reductase inhibitors inhibit endothelial exocytosis and decrease myocardial infarct size. Circ Res 96, 1185–1192, 10.1161/01.RES.0000170229.49776.81 (2005).
Leoncini, M., Toso, A., Maioli, M., Tropeano, F. & Bellandi, F. Statin treatment before percutaneous cononary intervention. Journal of thoracic disease 5, 335–342, 10.3978/j.issn.2072-1439.2013.05.09 (2013).
Patti, G. et al. Atorvastatin pretreatment improves outcomes in patients with acute coronary syndromes undergoing early percutaneous coronary intervention: results of the ARMYDA-ACS randomized trial. Journal of the American College of Cardiology 49, 1272–1278, 10.1016/j.jacc.2007.02.025 (2007).
Chia, J. et al. RNAi screening reveals a large signaling network controlling the Golgi apparatus in human cells. Mol Syst Biol 8, 629, 10.1038/msb.2012.59 (2012).
Hewlett, L. et al. Temperature-dependence of Weibel-Palade body exocytosis and cell surface dispersal of von Willebrand factor and its propolypeptide. PLoS One 6, e27314, 10.1371/journal.pone.0027314 (2011).
Acknowledgements
We are grateful to Tom Mckinnon and Mike Laffan (Imperial College London) for providing purified plasma vWF. We thank the members of the Cutler lab for providing useful feedback on the manuscript. Daniel F. Cutler (MC-UU-12018/2 grant) and Robin Ketteler are funded by the UK Medical Research Council. Hwee Kuan Lee is supported by the Biomedical Research Council of A*STAR (Agency for Science, Technology and Research), Singapore.
Author information
Authors and Affiliations
Contributions
F.F. and D.F.C. designed the study. F.F. and M.L.d.S. performed experiments. R.K. provided technical support and essential instruments. F.F., M.L.d.S. and J.K.-V. analysed and interpreted data. W.G. and H.K.L. developed methods and W.G. analysed data. F.F. and D.F.C. wrote the manuscript. M.L.d.S., J.K.-V. and R.K. revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Ferraro, F., da Silva, M., Grimes, W. et al. Weibel-Palade body size modulates the adhesive activity of its von Willebrand Factor cargo in cultured endothelial cells. Sci Rep 6, 32473 (2016). https://doi.org/10.1038/srep32473
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep32473
This article is cited by
-
Structure modeling hints at a granular organization of the Golgi ribbon
BMC Biology (2022)
-
Pharmacological dissection of the cellular mechanisms associated to the spontaneous and the mechanically stimulated ATP release by mesentery endothelial cells: roles of thrombin and TRPV
Purinergic Signalling (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
