
Abstract
The major human pathogen Streptococcus pneumoniae is a leading cause of disease and death worldwide. Pneumococcal biofilm formation within the nasopharynx leads to long-term colonization and persistence within the host. We have previously demonstrated that the capsular surface-associated pneumococcal serine rich repeat protein (PsrP), key factor for biofilm formation, binds to keratin-10 (KRT10) through its microbial surface component recognizing adhesive matrix molecule (MSCRAMM)-related globular binding region domain (BR187–385). Here, we show that BR187–385 also binds to DNA, as demonstrated by electrophoretic mobility shift assays and size exclusion chromatography. Further, heterologous expression of BR187–378 or the longer BR120–378 construct on the surface of a Gram-positive model host bacterium resulted in the formation of cellular aggregates that was significantly enhanced in the presence of DNA. Crystal structure analyses revealed the formation of BR187–385 homo-dimers via an intermolecular β-sheet, resulting in a positively charged concave surface, shaped to accommodate the acidic helical DNA structure. Furthermore, small angle X-ray scattering and circular dichroism studies indicate that the aggregate-enhancing N-terminal region of BR120–166 adopts an extended, non-globular structure. Altogether, our results suggest that PsrP adheres to extracellular DNA in the biofilm matrix and thus promotes pneumococcal biofilm formation.
Similar content being viewed by others

Introduction
The human adapted, Gram-positive commensal bacterium Streptococcus pneumoniae (pneumococcus) colonizes the upper respiratory tract in about 10% of healthy adults and up to 60% of children without necessarily causing any symptoms1. The organization of bacterial communities into complex and dynamic biofilm structures allows for growth in a hostile environment, providing protection against anti-microbial agents2,3. Pneumococcal colonization is the precursor for otitis media and sinusitis, as well as severe diseases including pneumonia, septicemia and meningitis4. Pneumococcal biofilms are found on the surface of adenoid and mucosal epithelial tissues in children with recurrent middle-ear infections and otitis media with effusion, highlighting the role of microbial biofilms in disease development5.
Recently, studies using in vitro and in vivo pneumococcal biofilm models have begun to unveil the biogenesis, regulation, structure and composition of pneumococcal biofilms6,7,8. The exact composition of the pneumococcal biofilm extracellular polymer matrix (EPM) depends largely on the species and environmental conditions5,9. Most often, the EPM is composed of exo-polysaccharides, proteins, nucleic acids and lipids, and its overall structure is stabilized through intermolecular interaction networks, often involving non-catalytic carbohydrate- or eDNA-binding proteins such as lectins and pilins8,10. In pneumococcus, the amount of capsular polysaccharides is highly regulated and significantly reduced during colonization compared to invasion1. Non-encapsulated strains are hyper-adhesive to epithelial cells and form biofilms more efficiently than encapsulated strains. On the other hand, they are also less virulent5,11,12,13. The increased adhesive phenotype in non-encapsulated strains can possibly be attributed to an increased presentation of adhesion proteins13,14.
The pneumococcal serine rich repeat protein (PsrP), present in 60% of strains capable of causing pneumonia in children, has been identified as a key factor for biofilm formation using in vitro and in vivo models7,15. A pneumococcal ΔPsrP deletion mutant strain was unable to form biofilm aggregates in an in vivo mice colonization model and elicited a significantly enhanced immune response compared to the wild-type strain7. PsrP belongs to the serine rich repeat protein (SRRP) family found in Gram-positive bacteria. SRRPs are attached to the capsular surface via a cell wall anchoring domain and display a long, highly repetitive and glycosylated C-terminal serine rich-repeat (SRR) region. The SRR region varies between 400 and 4000 residues in length, extending the functional binding region (BR) domain out of the capsule15,16,17,18 (Fig. 1A). SRRP BR domains which bind to a broad range of targets including glyco-conjugates and keratins, are variable in sequence and organized into modular domains17,18. Fold topologies similar to the microbial surface component recognizing adhesive molecule (MSCRAMM) CnaA from Staphylococcus aureus were determined for domains of the SRRPs GspB and Fap1 from Streptococcus parasanguinis and Streptococcus gordonii, respectively, and also predicted for domains of four other SRRPs16,17,19. Although the fold topology of the BR domain of PsrP compared to the other BR domains could not be predicted due to very low sequence similarity17, the recently determined crystal structure of the KRT10-binding domain of PsrP (BR187–385) revealed a topology distantly related to the CnaA fold20. Interestingly, GspB, Fap1 and PsrP, among several other SRRPs, were demonstrated to mediate intra-species interactions during biofilm formation15,17,18,21,22. A molecular model for the oligomerization of the helical subdomain NRα of Fap1 has also been proposed based on higher oligomers found in the crystal lattice and predicted as stable in solution21. The suggested molecular model is one of only a few known examples of biofilm-associated protein oligomerizations for which high-resolution structural data exist23. Recently, the crystal structures of the Staphylococcus aureus surface protein G (SasG) and of the accumulation-associated protein (Aap) from Staphylococcus epidermidis, both essential in biofilm formation, have been determined24,25,26,27. Like PsrP, these homologous multi-domain proteins are covalently attached to the peptidoglycan via their LPxTG motifs. Dimerization of the Aap B-repeat domains requires upstream proteolytic cleavage and a similar mechanism has also been suggested for SasG24,26. Crystal structures of self-oligomerizing B-repeat regions of both proteins revealed remarkable “free-standing” β-sheet topologies25,27. Based on the Aap B-domain crystal structure, a model for the Zn2+-dependent dimerization of the entire B-repeat domains was suggested according to which two molecules are wrapped around each other like entangled fibers in a rope27. Although a molecular understanding of the Zn2+-dependent dimerization of the MSCRAMM Fibronectin-Binding Protein A (FnBPA) of S. aureus is still lacking, atomic force microscopy (AFM) experiments elegantly demonstrated that cellular aggregation can be induced through multiple low-affinity homophilic bonds between copies of a single protein28.

(A) PsrP is organized into five domains: the N-terminal signal sequence (S) for extracellular translocation of PsrP, two putatively glycosylated serine rich repeat regions (SRR1 and SRR2), the binding region (BR) domain and the cell wall domain (CW). BR harbors two distinct sub-regions for KRT10 binding (black bar, residues 273–341) and self-oligomerization (black bar, residues 122–166), respectively. The sequence K164RRKR168 is recognized by the human Furin protease. The three constructs BR120–395, BR187–385 and BR187–378 used within this study are displayed below the overall schematic representation of PsrP. In the mutated version of BR120–395 (BR*120–395), KRRKR was substituted to KSRKS. (B) A protease cleavage assay confirmed that furin recognized the KRRKR motif, but did not cleave in the presence of the furin protease inhibitor or when the KRRKR sequence was substituted to KSRKS. Cleavage was performed at room temperature (RT) and 37 °C for the indicated incubation times. The inhibitor was used at 100 μM and 250 μM, shown as plus signs in regular and bold format, respectively. (C) Distinct populations of BR187–385 were eluted using Superdex 200 HiLoad 16/600 at elution column volumes (CV) of 89, 78, 71 and 66 mL corresponding to apparent molecular weights of 22, 55 and 90, 150 kDa, as well as higher oligomers. In this study, we show that the irreversibly associated dimer of BR187–385 is formed through a domain swap mechanism (PDB: 5JUI, Fig. S2) and that a low-affinity β-sheet dimer is created between two symmetry-related molecules in the previously determined crystal structure of the BR187–385 monomer (PDB: 3ZGH, Fig. 3). (D) BR*120–395 was eluted from the same column at CV of 79 and 69 mL corresponding to apparent MW of 50 and 115 kDa. (C,D) When the isolated monomer and oligomer populations of BR187–385 and BR*120–395 were re-applied on analytical Superdex 200 HR10/30 columns, the monomer and oligomer populations were stable and did not interconvert between each other.
The self-oligomerization property of PsrP has previously been suggested to occur through residues 122–166 (BR122–166), preceding the KRT10-binding region (BR187–385)15. However, a molecular mechanism underlying PsrP oligomerization has so far not been provided. In this study, crystal structure analysis and atomic force microscopy experiments revealed homophilic low affinity interactions, through the formation of an intermolecular β-sheet between two BR187–385 molecules. Electrophoretic mobility shift assays demonstrated that BR187–385 binds efficiently to DNA. Importantly, heterologous display of BR187–385 on the surface of Staphylococcus carnosus demonstrated the capacity of BR187–385 to promote eDNA-dependent cellular aggregation. Small-angle X-ray scattering and circular dichroism experiments revealed a disordered and non-globular structure in the region corresponding to BR122–166. We here demonstrate that BR122–166 is released from PsrP through proteolytic cleavage by the human furin protease, a known maturation factor of other toxins and virulence factors. The discovered molecular mechanisms provide novel structural and mechanistic insights into the role of PsrP during pneumococcal biofilm formation.
Results and Discussion
The BR domain comprises a furin protease recognition site and forms irreversibly associated dimers only during heterologous expression
Sequence analysis and biochemical protease cleavage assays revealed that the sequence motif K164RRKR168, localized between the KRT10-binding region20 and the predicted non-globular N-terminal extension BR120–166, is cleaved by the human furin protease (Fig. 1A,B). While wild-type BR120–395 was specifically cleaved by the human furin protease and prone to degradation during purification, a BR*120–395 construct with a mutated furin site was not cleaved and significantly more stable compared to wild-type, with no degradation product observed during purification (data not shown). Furin-like proteases activate a large number of pro-protein substrates as well as bacterial and viral pathogenic agents29,30. Thus, we hypothesized that the N-terminal fragment comprising BR120–166 and SRR1 could be released following proteolysis by furin, similarly to the previously described shedding of the glycoprotein Flo11p from Saccharomyces cerevisiae cells into the extracellular matrix of yeast mats31.
We have recently determined the crystal structure of the KRT10-binding domain of PsrP (BR187–385), revealing a novel MSCRAMM-related DEv-IgG fold resembling a compressed barrel20. The crystals were obtained from a monomeric preparation of BR187–385 that did not form higher oligomers in solution as assessed by analytical ultracentrifugation (AUC) and small angle X-ray scattering (SAXS) experiments at different concentrations20. However, during purification of the heterologously produced BR187–385, higher oligomer states were formed in the final size exclusion chromatography (SEC) step (Fig. 1C). Interestingly, these isolated oligomer populations were stable in solution but not in steady-state equilibrium with the monomer, as demonstrated using AUC and SEC (Figs 1C and S1A, Table S1). Similarly, the larger BR120–395 construct produced stable higher oligomer species, but at a significantly reduced ratio (Figs 1D and S1B, Table S1).
Since stable oligomer formation of PsrP could be relevant for pneumococcal biofilm formation, crystallization trials were set up for the isolated BR187–385 dimer in order to obtain a molecular understanding for stable dimer formation. However, crystals were hard to reproduce and those obtained diffracted poorly. Therefore, a shorter construct was designed by deleting the seven C-terminal residues in BR187–385 that were not built in the previously determined crystal structure due to missing electron density20. Well-diffracting crystals were obtained of the isolated irreversibly associated BR187–378 dimer. Analysis of the crystal structure solved by molecular replacement revealed that the residue stretches L202-G315 (region I) and Y316-S377 (region II) are reciprocally exchanged between two separate chains (Fig. S2, Table S2). This reciprocal exchange of entire protein segments is termed 3D-domain swapping and has been first described for diphtheria toxin32,33. A common feature of domain-swapped oligomers is the requirement to pass a high-energy barrier between the non-swapped and swapped states. Such inter-conversions usually do not occur spontaneously, but require harsh and denaturing conditions32. Thus, it should be noted that the irreversibly associated dimers were only obtained following heterologous production of the isolated BR protein constructs, and that, at this stage, we remain uncertain about their formation in vivo.
The putative self-oligomerization region BR122–166 is non-globular and does not promote self-aggregation
In order to obtain structural information about the longer BR*120–395 construct in solution, comparative small angle X-ray scattering (SAXS) data were collected for both monomeric BR187–385 and BR*120–395 (Fig. 2A,B and Table S1). The directly accessible and sample-characteristic radii of gyration (Rg) and maximal dimensions (Dmax) of BR187–385 and BR120–395 were 20 ± 1 Å and 29 ± 2 Å as well as 78 ± 8 Å and 125 ± 12 Å, respectively, indicating a more extended structure for BR120–395 (Fig. S3A,B). Indeed, molecular models obtained from the ensemble optimization method (EOM) support the presence of a single globular domain common to BR187–385 and BR*120–395, but with a longer highly mobile structural extension for BR120–395 (Fig. 2C,D). In dimensionless Kratky plots, the curve of BR187–385 closely resembles the bell-shaped curve expected for a globular protein, while the curve of BR120–395 is clearly shifted towards the shape of a molecule that comprises a more disordered, random chain (Fig. S3C). This is in line with our interpretation that the longer BR120–395 construct comprises an extended, highly flexible section in solution compared to BR187–385. Additional structural information was obtained from circular dichroism spectra of BR187–385 and BR*120–395 that also indicated the presence of disordered and helical regions for the additional residues in BR*120–395 (Fig. S3D). Combining the structural information obtained from AUC, SAXS, and CD, we conclude that the N-terminal region in BR*120–395 does not promote aggregation and corresponds to a non-globular, largely disordered structure.

(A,B) Processed solution scattering data (black dots) from (A) BR187–385 and (B) BR*120–395 fit to the theoretical scattering curves (red lines) from ensembles of 50 monomer models obtained from the ensemble optimization method (EOM) with χ-values of 0.9 and 1.7. (C,D) The histograms of the radii of gyration distribution of the pool of 10 000 structures generated by EOM (dotted lines) and the Rg distribution of the selected ensembles (solid lines). (C) The ensemble of 50 BR187–385 molecules selected by EOM was separated into two groups. The first group had an Rg value of around 20 Å and is represented by the molecular model with the extension colored in cyan. The second had an extended Rg value of 22.5 Å and is represented by the molecular model with the extension colored in blue. (D) The ensemble of 50 BR*120–395 molecules had a similar distribution of Rg values as compared the large pool of molecules with random conformations of the extensions. The two illustrated models displayed in blue and cyan have Rg values of 52 and 26 Å, respectively.
BR187–385 forms low-affinity homo-dimers through intermolecular β-sheets
Considering that the described domain swap mechanism is unlikely to occur spontaneously in vivo, and that the previously suggested self-aggregating BR120–166 region does not promote self-oligomerization in our experiments, we looked back into the crystal structure of the isolated BR187–385 monomer20 for hints of a more probable oligomerization mechanism. Indeed, we discovered intermolecular β-sheets between symmetry-related monomeric molecules in the P4122 crystal form (PDB: 3ZGH) and in the P43212 crystal form (PDB: 3ZGI) forming dimers with interface surface areas of 600 Å2 and 440 Å2, respectively (Figs 3 and S4A). Moreover, intermolecular β-sheets are also observed between symmetry-related domain-swapped dimers with similar interfaces and hydrogen bond patterns (Fig. S4B,C). However, although the formation of such edge-to-edge β-sheets is regarded as a fundamental feature underlying molecular recognition in protein-protein interactions34, a predicted weak interaction of this interface could prevent its detection in the performed SEC, SAXS and AUC experiments (Figs 1C and 2A and S1B)20.

Intermolecular β-sheets are formed between symmetry mates of BR187–385 in the P4122 (PDB: 3ZGH) crystal structure with a total interface surface area (ISA) of 600 Å2. The sheet is stabilized by eight hydrogen bonds between the backbones of the two A1 strands. The molecules forming intermolecular β-sheets are colored grey and wheat. The view of the right panel was rotated by ~90° compared to the left panel. Hydrogen bond interactions are represented as dashed red lines.
Single-cell and single-molecule atomic force microscopy have recently been applied to demonstrate that low-affinity (15 mM) homophilic interactions of the fibronectin binding protein A (FnBPA) of Staphylococcus aureus promote cellular aggregation28. Therefore, we also applied single-molecule force spectroscopy to test the capacity of BR187–378 and BR*120–395 to mediate similar low-affinity homophilic bonds. Indeed, homophilic binding events were measured for BR187–378 and BR*120–395 with mean adhesion forces of 70 ± 18 pN and 68 ± 14 pN, respectively (Fig. S5A–C). And as expected for specific bimolecular bonds35, the adhesion forces increased linearly with the logarithm of the loading rate (Fig. S5D).
BR187–385 binds to DNA and forms a positively charged saddle that snuggly fits the acidic helical structure of double-stranded DNA
Although the measured weak homophilic binding forces could be sufficient for cellular aggregation, we hypothesized that the highly basic BR187–385 could also bind to acidic biofilm-associated extracellular DNA. Indeed, electromobility shift assays (EMSA) demonstrated binding of a randomly chosen 276 bp long DNA (DNA276bp) molecule to BR187–385 with an apparent affinity in the lower micromolar range (Fig. 4A). After incubation of BR187–378 with DNA276bp at a 1:750 molar ratio, the purified high-molecular weight DNA population also comprised bound BR187–385 molecules (Fig. 4B,C). Interestingly, the DNA-bound BR187–385 ran at a higher molecular weight in SDS-PAGE, indicating that the DNA was not completely removed even by lauryl-dodecyl sulfate detergent treatment. Based on our previously suggested ligand-binding model, it is tempting to speculate that the negatively charged DNA binds to the extended highly basic groove of the intermolecular β-sheet dimer of BR187–385 (Fig. 4D). Molecular dynamic (MD) simulations also suggested that the docked DNA:BR187–385 complex is flexible but most of the non-nucleotide specific protein-DNA contacts are preserved over a 50 ns simulation period (Movie S1).

(A) BR187–385 concentration-dependent complex formation with a non-specific 270 bp DNA molecule (DNA270bp) was detected as retarded DNA bands in an electrophoretic mobility shift assay (EMSA). The apparent affinity of the interaction was estimated to around 10 μM. (B) After incubation of BR187–385 with DNA270bp, the area of the BR187–385 monomer peak, obtained at a retention volume of around 17 mL and an absorption wavelength of 280 nm, was slightly reduced. The high-molecular weight population was detected at a retention volume of about 8 mL with an estimated MW of about >1 MDa. DNA270bp alone would elute at the same retention volume (not shown). (C) The putative BR187–385-DNA complex was isolated from SEC and run on SDS-PAGE (left) and DNA-Agarose (right) gels. In SDS-PAGE the protein band was detected as a slightly larger complex, possibly retarded by the bound negatively charged DNA. The DNA270bp that was isolated from SEC was detected at the expected size, and was not retarded due to the low amount of bound BR187–385. (D) The intermolecular β-sheet dimer resembles a saddle-like structure with an extended intermolecular β-sheet. The electrostatic potentials are plotted in kbT ec−1 with the Boltzmann’s constant kb, the charge of an electron ec at a temperature T of 298 K. The saddle-like structure snuggly fits an acidic helical non-specific DNA molecule, as predicted in a molecular model of the BR187–385-DNA complex. (E) Similar to the BR187–385 dimer, the TATA-box binding protein (TBP, PDB: 1YTB) adopts a saddle-like structure. TBP was superimposed onto the BR187–387 dimer via shape similarity and represented as white and blue ribbons.
Strikingly, the structure of the BR187–385 dimer resembles the molecular saddle of the transcriptional pre-initiation complex-associated TATA-box binding protein (TBP) (Fig. 4E). While the concave under-surface of the TBP saddle is also highly basic, the specificity of the protein:TATATAAA interaction is primarily mediated through hydrophobic interactions36,37. However, it is well established that the specificity of protein-DNA interactions is manifested entirely in the non-electrostatic interaction component, while the electrostatic component typically contributes to the majority of the affinity38. Similar to the interaction of TBP with non-related DNA, the apparent affinity measured for binding of BR187–385 to DNA was in the low micromolar range. Thus, although we cannot exclude the possibility that PsrP recognizes a specific DNA sequence, we here hypothesize that PsrP binds non-specifically to DNA.
BR187–385 promotes DNA-dependent bacterial aggregation
Display of heterologously produced proteins on the surface of the Gram-positive bacterium Staphylococcus carnosus represents an excellent model system to study the functions of surface-associated proteins, e.g. the biofilm-promoting function of the SasC protein of Staphylococcus aureus39,40. In order to assess the capacity of each segment of the BR domain to promote cellular aggregation, BR187–378 and BR*120–378 were fused separately to the SasC-derived DUF-domain linker followed by a C-terminal LPxTG motif that becomes covalently attached to the staphylococcal cell wall through Sortase-mediated enzymatic linkage (Fig. 5A). The DUF domain was chosen to substitute the role of the SRR domain of native PsrP, which is to extend the BR region away from the cellular surface for functional accessibility. Dotblot assays of 3C-protease-cleaved surface-released proteins using polyclonal antisera against BR143–156 and BR187–378 confirmed that both BR*120–378 and BR187–378 were produced (Fig. 5B). The poly-His tags were detected for all constructs including the DUF control strain.

(A) The domain organization for surface display of BR187–378 and BR*120–378 on the surface of S. carnosus. PP - propeptide from S. hyicus lipase; His6 – hexahistidine tag; 3C - recognition sequence for protease 3C; X and M - regions from the SpA gene for covalent anchoring of the proteins to the peptidoglycan cell wall. (B) Supernatants of 3C-protease incubated staphylococcal cells were spotted on nitrocellulose membranes. Staining using polyclonal anti-serum against BR187–385 confirmed the expression of both BR*120–378 and BR187–378, and the absence and presence of signal for the BR143–156 region allowed for an unambiguous discrimination between BR*120–378- and BR187–378-expressing cells, respectively. As expected, signals from HRP-coupled anti-His antibodies were detected for all three constructs. (C) The BR187–378 and BR*120–378 displaying cells were stained using fluorescently labeled secondary antibodies directed against primary polyclonals against BR187–385. Singlets, duplets and higher aggregates are highlighted in white, bronze and pink, respectively. (D) Phase-like contrast microscopy images revealed the formation of cellular aggregates of S. carnosus displaying BR187–378 and BR*120–378, but not of cells displaying the DUF domain alone. (E) The two-dimensional aggregates sizes of staphylococcal cells were quantitatively analyzed from an area of about 0.05 mm2, which corresponds to approximately 64 times the area shown in panel D. The derived particle areas are plotted as normalized density histogram distributions, with the Scar-DUF control and the corresponding samples in black and red, respectively. The histogram represents the particle distribution obtained from a single experiment. The results from three independent experiments are summarized in the q-q plot shown in panel G. (F) Incubation of Scar-BR187–378 with DNaseI reduced the formation of aggregates, compared to the same sample not incubated with DNaseI (panel D). (G) The mean particle area sizes of quantiles with cut-off values at 25%, 50%, 75%, 85%, and 95% were calculated from three independent experiments, and summarized in a quantile-quantile plot (q-q plot). After incubation with DNaseI, both q-q plot curves of Scar-BR187–378 and Scar-BR120–378 are downward-shifted (light and dark green, respectively).
While bacteria displaying the DUF linker domain alone (Scar-DUF) were mainly observed as single cells or in doublets, cells displaying either BR*120–378 (Scar-BR*120–378) or BR187–378 (Scar-BR187–378) were heavily aggregated (Fig. 5C,D). Using fluorescence phase-contrast microscopy, single bacteria of Scar-BR187–378 and -BR*120–378 could be easily discriminated even within aggregates, while this was more difficult for Scar-DUF that appeared mostly in duplets (Fig. 5C). The origin of the slightly altered morphology of Scar-DUF bacteria under phase-contrast compared to the Scar-BR constructs is not known, but could be related to the absence and presence of the positively charged BR proteins, respectively. While Scar-DUF was not stained when incubated with fluorescently labeled secondary antibodies directed against primary polyclonals against BR187–385, both Scar-BR*120–378 and Scar-BR187–378 bacteria were stained (Fig. 5C).
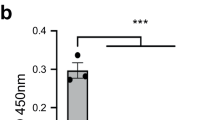
Quantitative analysis of the collected microscopy images confirmed our initial qualitative estimation that Scar-BR187–378 and Scar-BR*120–378 were significantly more aggregated compared to Scar-DUF, which is apparent from the particle size histogram plots with median particle sizes (q50) of 11 ± 2 μm2, 11 ± 2 μm2 and 3.7 ± 0.2 μm2, respectively (Fig. 5D,E). Since the staphylococcal cells were grown in terrific broth (TB) medium comprising yeast extract, we reasoned that the DNA extracted from yeast contributed to the observed cellular aggregates. Indeed, following incubation with the DNA-degrading enzyme DNaseI, aggregates of Scar-BR*120–378 and Scar-BR187–378 were reduced by almost 40% over all listed quantile values (Fig. 5F,G). A quantile is the fraction at which x% percent of the data fall below and (100 − x)% fall above that value. While two identical populations give a straight line (Fig. 5G, Scar-DUF in black), upward-shifted curves reveal increased particle sizes for Scar-BR187–378 and Scar-BR120–378 (Fig. 5G, Scar-BR187–378 and Scar-BR120–378 in red and blue, respectively). Similar results were obtained when Scar-DUF, Scar-BR187–378 and Scar-BR120–378 were grown in M9 minimal medium in the presence and absence of added sheared eDNA derived from herring sperm (Fig. S6). While presence of eDNA did not have any effect on Scar-DUF, both Scar-BR187–378 and Scar-BR120–378 were significantly more aggregated, albeit at a slightly reduced extent compared to bacteria grown in TB. We hypothesize that the observed slight reduction can be attributed to the restricted sequence length of the added eDNA between 15 and 50 bp. From these experiments we conclude that the globular BR187–378 domain of PsrP also promotes eDNA-dependent cellular aggregation within pneumococcal biofilms. Future experiments will address the exact contribution of each of the two factors, namely BR dimerization and binding to eDNA for biofilm formation.
Conclusions
PsrP has previously been suggested to promote pneumococcal biofilm formation through residues 122–166 (BR122–166), preceding the globular KRT10-binding MSCRAMM-related BR187–385 domain. In this study, small angle X-ray scattering and circular dichroism spectroscopy analysis revealed a non-globular, disordered structure for the BR122–166 region that does not promote self-aggregation. Importantly, surface-display of BR187–385 demonstrated the capacity of the globular domain to promote eDNA-dependent bacterial aggregation. Structural studies indicate that the saddle-like structure of the BR187–385 β-sheet dimer snuggly fits the acidic helical double-stranded DNA structure.
Materials and Methods
Cloning, expression and purification of protein constructs
Expression constructs comprising residues 120–395 of PsrP (BR120–395), 187–385 (BR187–385), and 187–378 (BR187–378) all with a N-terminal poly-His (HHHHHH) tag, were cloned into the pET21d (Novagen) expression vector as described previously20. The mutated expression construct of BR120–395 (BR*120–395) with R165S and R168S substitutions was generated following previously described protocols41. All coding sequences of the protein-expression constructs were confirmed by DNA sequencing and are listed in the supplemental information.
Heterologous protein expression in E. coli (T7 express, New England Biolabs) was induced at OD 0.4–0.7 using 400 μM IPTG and performed over night at 25 °C. The poly-Histidine-tagged proteins were purified using immobilized metal affinity (IMAC), cation exchange chromatography (CEC) (HisTrap FF and HiTrap SPFF, GE-Healthcare) and size exclusion chromatography (SEC) on Superdex 75 or 200 columns (GE Healthcare).
Furin cleavage assay
Two units of Furin protease (New England Biolabs, P8077) were added to a total assay volume of 100 μL with a concentration of 0.7 mg/mL BR120–395 and mutated BR*120–395 in 20 mM HEPES, 500 mM NaCl, 10% glycerol, 1 mM CaCl2, pH 7.5. Hexa-D-arginine amide (Sigma: SCP0148) was used as furin inhibitor at concentrations of 100 μM and 250 μM. The cleavage assays were carried out at room temperature and at 37 °C. Samples for SDS-PAGE were taken at after 0 h, 2 h, 4 h and overnight incubation.
Small angle X-ray scattering
Sample preparation and data collection
Data were collected at beamline BM29 at the European Synchrotron Radiation Facility (ESRF, Grenoble)42. Using a Pilatus 1 M detector at a sample to detector distance of 2.9 m and a wavelength of λ = 0.9919 Å, the range of momentum transfer 0.006 < s < 0.45 Å−1 was covered (s = 4π sin θ/λ, where 2θ is the scattering angle). Prior to data collection, proteins were buffer exchanged into phosphate buffered saline (PBS) with 5% glycerol, pH 7.4. Samples were measured at concentrations between 0.3 and 3 mg/ml using a continuous flow cell capillary.
Data analysis
The forward scattering I(0), the radius of gyration Rg along with the pair distribution function of the particle p(r) and the maximum dimension Dmax were derived using the automated SAXS data analysis pipeline43. Using Ensemble Optimization Method (EOM)44 analysis of BR187–385 and BR120–395, a pool of 10,000 models comprising a rigid domain (residues K206-E375) and flexible N- and C-termini were generated. A subset of the pool was selected using a genetic algorithm such that the calculated averaged scattering of the selected models agreed with the experimental data. The Rg distributions of the selected ensembles were obtained by repeating the selection process multiple times.
Structural analysis
All figures were created using PyMOL version 1.3.045. Interfaces of macromolecular assemblies in the crystal structures were investigated using PBDePISA46. The molecular model of the BR187–385-DNA complex was predicted using the non-specific DNA-rigid protein-docking algorithm ParaDock47, and the electrostatic surface was visualized using APBS48,49. The structure of TBP (PDB: 1YTB) was superimposed onto the BR187–387 dimer using the shape similarity algorithm of subcomp in which three-dimensional models are represented as simplified ensembles of points36,50.
Electrophoretic mobility shift assay (EMSA) and isolation of BR187–378-DNA276bp complex using SEC
The 276 bp DNA molecule (DNA276 sequence listed in supplemental information) was produced and isolated by PCR amplification and gel extraction (QIAEXII gel extraction kit, Qiagen). BR187–385 was titrated to DNA at a final concentration of 100 nM in TAE buffer (40 mM Tris, 20 mM acetic acid, and 1 mM EDTA) supplemented with 300 mM NaCl. After an hour incubation time, the samples were mixed with sample loading buffer and run on a 1.5% (w/V) GelGreen-stained (Biotium) agarose gel in TAE buffer.
For SEC analysis, BR187–385 and DNA276 were incubated at concentrations of 30 μM and 440 nM, respectively. Before mixing, BR187–385 and DNA276 were prepared in 20 mM Hepes, 200 mM NaCl, 10% glycerol, pH 7.5 and 10 mM Tris at pH 8.5, respectively. The mixed samples were loaded on a Superdex S200 10/300 GL (GE healthcare) equilibrated in 20 mM Hepes, 200 mM NaCl, 10% glycerol, pH 7.5.
Surface display of BR on the surface of Staphylococcus carnosus
Cellular aggregation assay
The PCR-amplified PsrP constructs and the gene-synthesized (Eurofins Genomics, Germany) DUF1542 repeat domain of SasC (Uniprot ID: C7BUR8) were cloned into the staphylococcal display vector ‘pHis3C’ using a sequence and ligation independent cloning (SLIC) method51,52. Coding sequences of the protein-display constructs were confirmed by DNA sequencing and are listed in the supplemental information. Transformed S. carnosus cells were cultured in TB medium with chloramphenicol (10 ug/mL). OD-adjusted cells treated or non-treated with DNaseI were gently re-suspended in PBS, filled into wells of a 96-μ-well plate (Ibidi, Germanu) and images were taken using the ZOE Fluorescent Cell Imager (Biorad, USA) at 20x magnification. Bacteria were also grown in M9 derived minimal medium53 comprising 1x M9 salts, 2 mM MgSO4, 0.1 mM CaCl2, 1% glucose, 1% casaminoacids, 1 mM Thiamine-HCl and 0.05 mM nicotinamide, with or without the addition of herring sperm derived degraded DNA (D3159, Sigma Aldrich) at a concentration of 10 ug/mL that corresponds to approximately 1 μM of 15 bp oligonucleotide fragments. The bacteria were adjusted in OD and imaged in the same way as described previously. Images were analyzed using the software cell profiler (www.cellprofiler.org), the particle parameter values were imported into the R software package for statistical analysis and visualization using density histogram and q-q plots54,55.
Dotblot assays and fluorescence imaging
Polyclonal antibodies against the TEV-cleaved purified BR187–385 monomer and a synthesized BR143–156 (RKKPASDYVASVTN) peptide were raised in rabbits and the obtained sera yielded titers of about 75000 and 6000, respectively (Innovagen, Sweden). For fluorescent staining, bacteria were incubated with affinity-purified anti-BR187–385 antibodies and detected using fluorescein isothiocyanate (FITC)-labeled goat anti-rabbit IgG (life technologies) secondary antibody. Bacteria were fixed in 4% paraformaldehyde, washed, and visualized using a fluorescence microscope (Leica Leitz DMRBE). For dotblot assays, surface-displayed proteins were cleaved as previously described52 and applied on nitrocellulose filter membranes. Proteins were detected using polyclonal rabbit-antisera against BR187–385, BR143–156 as well as HRP-coupled anti-His antibodies (ab1187; Abcam, UK). The anti-BR antibody stained samples were incubated using HRP-coupled monoclonal anti-rabbit IgGγ (A1949, Sigma Aldrich). Membranes were developed using Pierce ECL western blotting substrate (Thermo Scientific). Further details are given in the supplementary material.
Additional Information
Accession codes: Coordinates and structure factors have been deposited in the Protein Data Bank with accession number 5JUI. SAXS data have been deposited in the Small Angle Scattering Biological Databank (sasbdb) with accession numbers SASDAC6 and SASDAE6.
How to cite this article: Schulte, T. et al. The BR domain of PsrP interacts with extracellular DNA to promote bacterial aggregation; structural insights into pneumococcal biofilm formation. Sci. Rep. 6, 32371; doi: 10.1038/srep32371 (2016).
Accession codes
References
van der Poll, T. & Opal, S. M. Pathogenesis, treatment, and prevention of pneumococcal pneumonia. Lancet 374, 1543–1556 (2009).
Costerton, J. W., Stewart, P. S. & Greenberg, E. P. Bacterial Biofilms: A Common Cause of Persistent Infections. Science 284, 1318–1322 (1999).
Hall-Stoodley, L., Costerton, J. W. & Stoodley, P. Bacterial biofilms: from the Natural environment to infectious diseases. Nat. Rev. Microbiol. 2, 95–108 (2004).
Simell, B. et al. The fundamental link between pneumococcal carriage and disease. Expert Rev. Vaccines 11, 841–855 (2012).
Domenech, M., Garcia, E. & Moscoso, M. Biofilm formation in Streptococcus pneumoniae . Microb. Biotechnol. 5, 455–465 (2012).
Domenech, M., García, E., Prieto, A. & Moscoso, M. Insight into the composition of the intercellular matrix of Streptococcus pneumoniae biofilms. Environ. Microbiol. 15, 502–516 (2013).
Blanchette-Cain, K. et al. Streptococcus pneumoniae Biofilm Formation is Strain Dependent, Multifactorial, and Associated with Reduced Invasiveness and Immunoreactivity during Colonization. mBio 4, e00745-13 (2013).
Hobley, L., Harkins, C., MacPhee, C. E. & Stanley-Wall, N. R. Giving structure to the biofilm matrix: an overview of individual strategies and emerging common themes. FEMS Microbiol. Rev. fuv015, 10.1093/femsre/fuv015 (2015).
Flemming, H.-C. & Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 8, 623–633 (2010).
van Schaik, E. J. et al. DNA binding: a novel function of Pseudomonas aeruginosa type IV pili. J. Bacteriol. 187, 1455–1464 (2005).
Camilli, R., Pantosti, A. & Baldassarri, L. Contribution of serotype and genetic background to biofilm formation by Streptococcus pneumoniae . Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. Eur. Soc. Clin. Microbiol. 30, 97–102 (2011).
Hall-Stoodley, L. et al. Characterization of biofilm matrix, degradation by DNase treatment and evidence of capsule downregulation in Streptococcus pneumoniae clinical isolates. BMC Microbiol. 8, 173 (2008).
Sanchez, C. J. et al. Streptococcus pneumoniae in Biofilms Are Unable to Cause Invasive Disease Due to Altered Virulence Determinant Production. Plos One 6, e28738 (2011).
Muñoz-Elías, E. J., Marcano, J. & Camilli, A. Isolation of Streptococcus pneumoniae Biofilm Mutants and Their Characterization during Nasopharyngeal Colonization. Infect. Immun. 76, 5049–5061 (2008).
Sanchez, C. J. et al. The pneumococcal serine-rich repeat protein is an intra-species bacterial adhesin that promotes bacterial aggregation in vivo and in biofilms. Plos Pathog. 6 (2010).
Ramboarina, S. et al. Structural insights into serine-rich fimbriae from gram-positive bacteria. J. Biol. Chem. 285, 32446–32457 (2010).
Pyburn, T. M. et al. A Structural Model for Binding of the Serine-Rich Repeat Adhesin GspB to Host Carbohydrate Receptors. Plos Pathog. 7, e1002112 (2011).
Lizcano, A., Sanchez, C. J. & Orihuela, C. J. A role for glycosylated serine-rich repeat proteins in Gram-positive bacterial pathogenesis. Mol. Oral Microbiol. 27, 257–269 (2012).
Shivshankar, P., Sanchez, C., Rose, L. F. & Orihuela, C. J. The Streptococcus pneumoniae adhesin PsrP binds to Keratin 10 on lung cells. Mol. Microbiol. 73, 663–679 (2009).
Schulte, T. et al. The basic keratin 10-binding domain of the virulence-associated pneumococcal serine-rich protein PsrP adopts a novel MSCRAMM fold. Open Biol. 4, 130090 (2014).
Garnett, J. A. et al. Structural insight into the role of Streptococcus parasanguinis Fap1 within oral biofilm formation. Biochem. Biophys. Res. Commun. 417, 421–426 (2012).
Wu, H., Zeng, M. & Fives-Taylor, P. The glycan moieties and the N-terminal polypeptide backbone of a fimbria-associated adhesin, Fap1, play distinct roles in the biofilm development of Streptococcus parasanguinis . Infect. Immun. 75, 2181–2188 (2007).
Garnett, J. A. & Matthews, S. Interactions in Bacterial Biofilm Development: A Structural Perspective. Curr. Protein Pept. Sci. 13, 739–755 (2012).
Corrigan, R. M., Rigby, D., Handley, P. & Foster, T. J. The role of Staphylococcus aureus surface protein SasG in adherence and biofilm formation. Microbiology 153, 2435–2446 (2007).
Gruszka, D. T. et al. Staphylococcal Biofilm-Forming Protein Has a Contiguous Rod-Like Structure. Proc. Natl. Acad. Sci. 109, E1011–E1018 (2012).
Rohde, H. et al. Induction of Staphylococcus epidermidis biofilm formation via proteolytic processing of the accumulation-associated protein by staphylococcal and host proteases. Mol. Microbiol. 55, 1883–1895 (2005).
Conrady, D. G., Wilson, J. J. & Herr, A. B. Structural basis for Zn2+-dependent intercellular adhesion in staphylococcal biofilms. Proc. Natl. Acad. Sci. USA 110, E202–211 (2013).
Herman-Bausier, P., El-Kirat-Chatel, S., Foster, T. J., Geoghegan, J. A. & Dufrêne, Y. F. Staphylococcus aureus Fibronectin-Binding Protein A Mediates Cell-Cell Adhesion through Low-Affinity Homophilic Bonds. mBio 6 (2015).
Klimpel, K. R., Molloy, S. S., Thomas, G. & Leppla, S. H. Anthrax toxin protective antigen is activated by a cell surface protease with the sequence specificity and catalytic properties of furin. Proc. Natl. Acad. Sci. 89, 10277–10281 (1992).
Thomas, G. Furin at the cutting edge: From protein traffic to embryogenesis and disease. Nat. Rev. Mol. Cell Biol. 3, 753–766 (2002).
Karunanithi, S. et al. Shedding of the Mucin-Like Flocculin Flo11p Reveals a New Aspect of Fungal Adhesion Regulation. Curr. Biol. 20, 1389–1395 (2010).
Gronenborn, A. M. Protein acrobatics in pairs–dimerization via domain swapping. Curr. Opin. Struct. Biol. 19, 39–49 (2009).
Bennett, M. J. & Eisenberg, D. Refined structure of monomeric diphtheria toxin at 2.3 A resolution. Protein Sci. Publ. Protein Soc. 3, 1464–1475 (1994).
Nowick, J. S. & Chung, D. M. Sequence-Selective Molecular Recognition between β Sheets. Angew. Chem. Int. Ed. 42, 1765–1768 (2003).
Hinterdorfer, P. & Dufrêne, Y. F. Detection and localization of single molecular recognition events using atomic force microscopy. Nat. Methods 3, 347–355 (2006).
Kim, Y., Geiger, J. H., Hahn, S. & Sigler, P. B. Crystal structure of a yeast TBP/TATA-box complex. Nature 365, 512–520 (1993).
Hahn, S., Buratowski, S., Sharp, P. A. & Guarente, L. Yeast TATA-binding protein TFIID binds to TATA elements with both consensus and nonconsensus DNA sequences. Proc. Natl. Acad. Sci. USA 86, 5718–5722 (1989).
Privalov, P. L., Dragan, A. I. & Crane-Robinson, C. Interpreting protein/DNA interactions: distinguishing specific from non-specific and electrostatic from non-electrostatic components. Nucleic Acids Res. 39, 2483–2491 (2011).
Samuelson, P., Gunneriusson, E., Nygren, P. A. & Ståhl, S. Display of proteins on bacteria. J. Biotechnol. 96, 129–154 (2002).
Schroeder, K. et al. Molecular Characterization of a Novel Staphylococcus Aureus Surface Protein (SasC) Involved in Cell Aggregation and Biofilm Accumulation. Plos One 4, e7567 (2009).
Li, J. et al. Site-directed mutagenesis by combination of homologous recombination and DpnI digestion of the plasmid template in Escherichia coli . Anal. Biochem. 373, 389–391 (2008).
Pernot, P. et al. Upgraded ESRF BM29 beamline for SAXS on macromolecules in solution. J. Synchrotron Radiat. 20, 660–664 (2013).
Franke, D., Kikhney, A. G. & Svergun, D. I. Automated acquisition and analysis of small angle X-ray scattering data. Nucl. Instrum. Methods Phys. Res. Sect. Accel. Spectrometers Detect. Assoc. Equip. 689, 52–59 (2012).
Bernadó, P., Mylonas, E., Petoukhov, M. V., Blackledge, M. & Svergun, D. I. Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 129, 5656–5664 (2007).
Schrödinger, L. PyMOL Molecular Graphics System. (2010).
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007).
Banitt, I. & Wolfson, H. J. ParaDock: a flexible non-specific DNA—rigid protein docking algorithm. Nucleic Acids Res. 39, e135 (2011).
Dolinsky, T. J., Nielsen, J. E., McCammon, J. A. & Baker, N. A. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–7 (2004).
Baker, N. A., Sept, D., Joseph, S., Holst, M. J. & McCammon, J. A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 98, 10037–41 (2001).
Kozin, M. B. & Svergun, D. I. Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 34, 33–41 (2001).
Li, M. Z. & Elledge, S. J. Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat. Methods 4, 251–256 (2007).
Kronqvist, N., Löfblom, J., Severa, D., Ståhl, S. & Wernérus, H. Simplified characterization through site-specific protease-mediated release of affinity proteins selected by staphylococcal display. FEMS Microbiol. Lett. 278, 128–136 (2008).
Reed, P. et al. Staphylococcus aureus Survives with a Minimal Peptidoglycan Synthesis Machine but Sacrifices Virulence and Antibiotic Resistance. Plos Pathog. 11, e1004891 (2015).
Jones, T. R. et al. CellProfiler Analyst: data exploration and analysis software for complex image-based screens. BMC Bioinformatics 9, 482 (2008).
R Core Team. R: A language and environment for statistical computing. (R Foundation for Statistical Computing) (2011).
Acknowledgements
We gratefully acknowledge fruitful discussions with Tatyana Sandalova as well as access to synchrotron beamlines BM29 and ID23-1 at ESRF (Grenoble, France). This work was facilitated by the Protein Science Facility at Karolinska Institute/SciLifeLab (http://psf.ki.se). We thank John Löfblom (KTH, Stockholm) and Simon Elsässer (MBB, Karolinska Institute, Stockholm) for providing the S. carnosus cells and access to the ZOE Fluorescent Cell Imager, respectively. This work was supported by grants from the Swedish Research Council (AA, BHN), the Knut and Alice Wallenberg Foundation (AA, BHN), the Swedish foundation for strategic research (AA and BHN), the Wenner Gren Foundation and (TS), the European Community’s Seventh Framework Programme (FP7/2007–2013) under BioStruct-X (grant agreement No. 283570). We gratefully acknowledge access to synchrotron beamlines BM29 and ID23-1 at ESRF (Grenoble, France). We acknowledge the platforms of the Grenoble Instruct center (ISBG; UMS 3518 CNRS-CEA-UGA-EMBL) supported by the French Infrastructure for Integrated Structural Biology Initiative FRISBI (ANR-10-INSB-05-02) and by the Grenoble Alliance for Integrated Structural Cell Biology GRAL (ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology (PSB). Work at the Université catholique de Louvain was supported by the National Fund for Scientific Research (FNRS), the FNRS-WELBIO under Grant no. WELBIO-CR-2015A-05, the Federal Office for Scientific, Technical and Cultural Affairs (Interuniversity Poles of Attraction Programme), and the Research Department of the Communauté française de Belgique (Concerted Research Action). Y.F.D. is a Research Director at the FNRS. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Author information
Authors and Affiliations
Contributions
T.S. and A.A. wrote the initial draft that was complemented by contributions from C.M., A.B., A.K., A.P., C.E., F.F., B.H.-N., Y.F.D., D.S., P.-Å.N. and A.A.; T.S., C.M., J.L., M.D. and S.W. performed the cloning work for heterologous expression in E. coli and S. carnosus; T.S. and C.M. produced the BR constructs and performed the furin cleavage assays as well SEC analysis; C.M. collected the SAXS data that were analyzed by A.K., T.S. and C.M.; T.S. performed and analyzed the in vitro DNA binding of BR, F.F. performed and analyzed the M.D. simulations, A.B. collected the AFM data that were analyzed by A.B. and Y.F.D.; A.P. took the fluorescence images; C.E. collected and analyzed the AUC data; M.D., S.W. and T.S. performed and analyzed the heterologous surface display experiments.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Schulte, T., Mikaelsson, C., Beaussart, A. et al. The BR domain of PsrP interacts with extracellular DNA to promote bacterial aggregation; structural insights into pneumococcal biofilm formation. Sci Rep 6, 32371 (2016). https://doi.org/10.1038/srep32371
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep32371
This article is cited by
-
Formation of a biofilm matrix network shapes polymicrobial interactions
The ISME Journal (2023)
-
Assigned NMR backbone resonances of the ligand-binding region domain of the pneumococcal serine-rich repeat protein (PsrP-BR) reveal a rigid monomer in solution
Biomolecular NMR Assignments (2020)
-
Detecting ligand interactions in real time on living bacterial cells
Applied Microbiology and Biotechnology (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
