
Abstract
Coffee is one of the most consumed beverages world-wide and one of the primary sources of caffeine intake. Given its important health and economic impact, the underlying genetics of its consumption has been widely studied. Despite these efforts, much has still to be uncovered. In particular, the use of non-additive genetic models may uncover new information about the genetic variants driving coffee consumption. We have conducted a genome-wide association study in two Italian populations using additive, recessive and dominant models for analysis. This has uncovered a significant association in the PDSS2 gene under the recessive model that has been replicated in an independent cohort from the Netherlands (ERF). The identified gene has been shown to negatively regulate the expression of the caffeine metabolism genes and can thus be linked to coffee consumption. Further bioinformatics analysis of eQTL and histone marks from Roadmap data has evidenced a possible role of the identified SNPs in regulating PDSS2 gene expression through enhancers present in its intron. Our results highlight a novel gene which regulates coffee consumption by regulating the expression of the genes linked to caffeine metabolism. Further studies will be needed to clarify the biological mechanism which links PDSS2 and coffee consumption.
Similar content being viewed by others

Introduction
Coffee is one of the most widely drunk beverages, being second only to water and tea1. Given that it contains many different physiologically active compounds such as caffeine, polyphenols (e.g., chlorogenic acids), niacin, N-methylpyridinium ion and others2, many studies have investigated its impact on health, finding associations with common disorders. For example, coffee consumption has been linked to protective effects on various common pathologies such as cardiovascular diseases3, hypertension4,5, Alzheimer’s and Parkinson’s diseases6,7, type 2 diabetes8,9,10, some types of cancer11,12 and hearing functions13, while it may predispose to others such as sleep disturbances14,15. The earliest studies on the genetic bases of coffee consumption can be dated back as far as the 1960’s with the first description of its heritability in Italy16, which has been estimated to range between 0.36 and 0.58 in twin studies. Different independent genome-wide association studies (GWAS) carried out mostly in Northern European populations have highlighted associations between coffee or caffeine consumption and several genes: CYP1A1-CYP1A217,18, AHR17 NRCAM and ULK318 while moderate association has been seen with the adenosine receptor A2, which is actually one of the effector proteins of caffeine17. More recently, a very large study comprising more than 120 thousand people has confirmed some of these (CYP1A1-CYP1A2 and AHR) while identifying 6 novel ones19. Despite these successes much of the heritability of coffee consumption still remains unexplained, part of which could be explained by the fact that the additive genetic model is almost always used in association studies, whereas, in fact, genes can exhibit recessive or dominant effects. It is in fact very well known that miss-specifying the genetic model will result in a considerable loss of power, especially under certain conditions20. For this reason we have decided do conduct a genome-wide association study looking for non-additive effects on two isolated populations of Italy in order to verify whether genes are affecting coffee consumption under these genetic models.
Materials and Methods
Study Populations and study design
For the discovery phase we used the samples from two isolated Italian populations participating to the INGI consortium. In particular, our study used 370 individuals from INGI-CARL, a population coming from Carlantino, a small village located in Puglia (Southern Italy), and 843 defined as INGI-FVG, making reference to 6 villages situated in the Friuli Venezia Region in North-Eastern Italy for a total of 1207 samples. For replication we used the samples coming from the Erasmus Rucphen Family (ERF) study, a cross-sectional cohort including 3,000 living descendants of 22 couples who had at least 6 children baptized in the community church around 1850–1900; 1731 samples were used from this study.
Phenotype and sample selection
Coffee consumption was assessed in the INGI cohorts through field interviews while it was self-reported in the ERF cohort. In all cohorts coffee consumption was measured as number of coffee cups per day. Given that the distribution of the trait was extremely skewed we transformed this to log10(Ncoffee cups + 1). In order to remove outliers, we excluded people who drank more than 9 cups of coffee per day in the Italian cohorts and more than 20 in ERF. This difference was due to the very different coffee consumption distribution observed in the Italian vs. the Dutch populations. Finally, all people under hypertensive medication were excluded from the analysis since those are usually advised to reduce or avoid coffee consumption.
Genotyping and Imputation
Genotyping was carried out as previously described21,22. Briefly, INGI-CARL, and INGI-FVG have been genotyped with the Illumina 370k high density SNP array. Genotype imputation on the INGI cohorts was conducted after standard QC using SHAPEIT223 for the phasing step and IMPUTE224 for the imputation using the1000 Genomes phase I v3 reference set25. ERF has been genotyped with different genotyping platforms: Illumina 318 k, 350 k, 610 k and Affymetrix 200 k. Genotypes were pooled together after QC, phased and imputed to the 1000 Genomes dataset phase I v326 using MaCH and minimac27. After imputation SNPs with MAF <0.01 or Info score <0.4 we excluded from the statistical analyses for each of the populations but ERF, for which R2 < 0.3 was used instead.
Association analysis
Association analysis was conducted using mixed model linear regression where the standardized food liking was used as the dependent variable and the SNP dosages as the independent variable. Sex and age were used as covariates. The kinship matrix based on all available genotyped SNPs was used as the random effect. For ERF the kinship matrix was estimated on 14.4 k SNPs common to all different genotyping platforms used. Association analysis was conducted using the FASTA method28 as implemented in the GenABEL 1.7–229 R package in order to eliminate the effect of familial relatedness from the trait. MixABEL30 was used for the actual association of the imputed SNPs. SNPs that did not pass quality control for more than one population were discarded. It is common practice in GWAS to run association presuming the genetic variants have an additive effect on the phenotype, however this is not necessarily true. Therefore, we decided to also use non-additive genetic models, in particular the dominant and recessive models. For the discovery step, association analysis was conducted separately for each INGI cohort and results were pooled using the inverse-variance weighting method. Given that no meta-analysis software supports non-additive genetic models we developed custom R scripts. After meta-analysis in the INGI cohorts, genomic control was used to eliminate any residual stratification and all significant SNPs where taken forward to the replication step in the ERF cohort. The significance threshold was estimated considering the number of equivalent tests performed when running multiple genetic models, which has been estimated as being 2.231, and was thus set at 2.27 × 10−8. In order to avoid false positive results due to rare categories, in each GWAS we eliminated those SNPs in which the coded category was either <0.01 or >0.99. This resulted in a different number of SNPs depending on the model used. Supplementary Table S2 summarizes the number of SNPs used for each cohort under each model.
Comparison of the association pattern of coffee consumption with eQTL results
In order to verify if the observed SNP association pattern could be traced back to an expression quantitative locus (eQTL), we have decided to compare it with that coming from the cis-eQTL of PDSS2 gene available from 14 different tissues from the GTEx database32. We have downloaded the full dataset of results and extracted all SNPs tested against the PDSS2 gene which fell inside the two recombination hotspots delimiting our association signal (chr6:107400000-107900000). If the two association signals can be traced back to each other we would expect that the strengths of the association should resemble each other and the relationships between the direction of their effect should be consistent across all SNPs examined and in particular in those showing the lowest p-values. In order to give more weight to the SNPs with the lowest p-values and to keep the directionality of the effect, we assigned to each SNP a score equal to −log10(p-value) × 1 if the direction of the effect was positive and log10(p-value) otherwise. Given that the two association patterns do not have a normal distribution we used the Kendal  and correlation test to verify if the two patterns resembled each other and if the resulting correlation was significant.
and correlation test to verify if the two patterns resembled each other and if the resulting correlation was significant.
Functional annotation of the discovered SNPs
In order to understand the possible functional role of the 5 replicated SNPs we annotated them using Haploreg v4.133. Briefly, Haploreg allows one to annotate a list of SNPs by reporting the associated chromatin stated on a 15-state model from the using the Roadmap epigenomics project34 and Encode project35. Moreover it checks if the SNPs have been previously associated to any eQTL, QTL or metabolite levels according to various databases. Finally, it allows one to set an LD threshold based on which all SNPs with r2 > threshold will be included in the annotation. For this study we limited the analysis to those SNPs that have r2 > 0.8 with our 5 SNPs. The results from this analysis can be found in Supplementary Table S4.
Ethical statement
All studies adhered to the tenets of the Declaration of Helsinki. The ERF study was approved by the Medical Ethics Committee of the Erasmus Medical Center in Rotterdam. Informed consent was obtained after explanation of the nature and possible consequences of the study.
All subjects in the INGI-CARL and INGI-FVG studies provided written informed consent before participation. Approval for the research protocol was obtained from the ethical committee of IRCCS-Burlo Garofolo Hospital.
Results
Supplementary Table S1 summarizes the population characteristics for each of the studies used. The Italian populations had comparable composition and distribution of coffee consumption. In ERF we detected a much higher coffee consumption with a mean more than double (5.6 cups per day vs 1.9 in INGI-CARL and 2.3 in INGI-FVG) that of the other populations and a much higher standard deviation (3.6 vs 1.6 in both INGI populations).
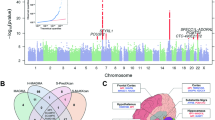
None of the discovery step genome-wide associations revealed evidence of residual stratification either in each separate cohort or after meta-analysis (genomic control λ between 0.99 and 1.02, see Table S2 for details). Meta-analysis between the results from the INGI cohorts revealed 21 SNPs under the recessive model to be genome-wide significant, with the top hit rs6568479 showing a p-value = 8.9 × 10−10 and an effect of 0.086 corresponding to a difference of 1.2 cups of coffee per day for the people homozygous for the G allele. No association were found using the dominant genetic model. Table 1 summarizes the results of the association analysis and replication for the 21 significant SNPs. Effect sizes in the two populations were extremely similar (CARL 0.0864, FVG 0.0861). Some of the SNPs showed significant results also under the additive model although p-values were higher. For this reason, we consider the recessive model to be the true genetic model. Figure 1 shows the Manhattan plot from the recessive model meta-analysis. As can be seen in Fig. 2 all SNPs fell in the same locus in chromosome 6 all inside the PDSS2 gene which codes for the coenzyme Q10 (Fig. 2 shows the regional plot for the same SNPs). Replication of the results from the 21 significant SNPs in the ERF cohort revealed that 5 out of 21 SNP had a p < 0.05 with a concordant although attenuated effect. All 5 SNPs were significant after pooling the results from the three cohorts with a p-value lower than the one observed in the discovery step. None of the identified SNPs are coding, however we sought to verify if any of these could be associated with an existing e-QTL of PDSS2. For this reason we downloaded the full results from the Gtex Database32 for all cis-eQTL SNPs tested against PDSS2 falling inside the two recombination hot-spots delimiting the association signal. Comparing the patterns of association for all the SNPs (see Materials and Methods for details) between the two recombination hot spots that show significant correlation with all the association patterns for the tested tissues shows correlation values varying between −0.27 for Adipose Subcutaneous tissue and −0.61 for Esophagus Mucosa tissue. In all cases correlation was negative indicating that people with a higher consumption of coffee have a lower expression of PDSS2. Supplementary Table S3 reports the Kendall’s  values for each tissue. The annotation with Haploreg showed 7 further SNPs in strong LD with the 5 replicated ones which were not in included in the association analysis. Although none of the 12 SNPs resulted to be coding, 6 out of 12 are located inside enhancers regions active in numerous tissues (Table S4). Finally consistently with the analysis on the Gtex Database all SNPs showed significant association with tissue specific expression of PDSS2.
values for each tissue. The annotation with Haploreg showed 7 further SNPs in strong LD with the 5 replicated ones which were not in included in the association analysis. Although none of the 12 SNPs resulted to be coding, 6 out of 12 are located inside enhancers regions active in numerous tissues (Table S4). Finally consistently with the analysis on the Gtex Database all SNPs showed significant association with tissue specific expression of PDSS2.

Manhattan plot of the genome-wide association analysis under the recessive model.

Regional plot of the results from the association analysis around the most significant SNPs.
Discussion
In this study we have highlighted a novel association between coffee consumption and the PDSS2 gene. Moreover we have shown that the association pattern correlates very strongly with the expression QTLs in different tissues. Finally the bioinformatics analysis shows that several of the identified SNPs are either in or in LD with enhancers sequences active in various tissues. All these results would suggest that the SNPs would act on coffee consumption by regulating the expression PDSS2 suggesting a possible functional explanation. The gene PDSS2 codes for an enzyme responsible for the synthesis of the prenyl side chain of coenzyme Q10. Its mutation in mice leads to the insurgence of a renal phenotype (kd) which is characterized by tubulointerstitial nephritis, dilated tubules, and proteinuria36. In humans only one case of a PDSS2 compound heterozygous mutant has been described in a child with Leigh syndrome, a very serious condition characterized by neonatal pneumonia, hypotonia, nephrotic syndrome and blindness37, although neither of the parents presented any symptoms at all. Further studies have shown that although the reduction in Q10 due to the PDSS2 mutation is ubiquitous among the different tissues the observed phenotype is due to an augmented production of reactive oxygen species (ROS) specific to the affected tissues probably to the use of alternative mechanisms in the non-affected tissues38.
Conditional knockout of PDSS2 in the liver has been shown to increase the expression of the genes of the caffeine metabolism pathway (GSEA normalized enrichment score 1.59, FDRp = 0.03) while on knock-out mice treated with probucol (which restores the knock-out phenotype) a strong inhibition was observed when compared to knockout mice (GSEA normalized enrichment score −1.87, FDR p = 0.02)39. We have shown through the comparison of our results with those of PDSS2 eQTLs that in all examined tissues there is an inverse correlation between PDSS2 expression and coffee consumption. Assuming that this is true also for the liver we may hypothesize that higher PDSS2 expression would inhibit the expression of the genes in the caffeine metabolism pathway thus inhibiting caffeine degradation. The hypothesis of an eQTL in the liver is strengthened by the presence of one of our associated SNPs in an enhancer active in this specific tissue. This would fit very well with previous results that have associated speed in metabolizing caffeine and coffee intake through the CYP1A2 genotype18,19. Unfortunately, liver eQTLs for PDSS2 data on 1000G are not available yet and these results will need to be confirmed.
The difference in strength of effect between the Italian and the Dutch cohorts could be explained by differences in the way coffee is prepared in the two countries. In fact, while in Italy moka or espresso are the preferred way of drinking coffee, in the Netherlands filtered coffee is preferred. Although the concentrations of caffeine in the different preparation methods are similar, given the differences between cup sizes, Dutch intake of caffeine per cup is almost three times higher than Italians (average 173.25 mg/cup for filtered coffee vs. 67.18 and 60.95 for moka and espresso, respectively)40. It is thus possible that while CYP1A2 genotype is important in determining coffee consumption at higher caffeine intakes, PDSS2 may have a role at lower levels. According to this view we detected only an extremely mild association with previously reported CYP1A2 SNPs in the Italian populations (rs2472297, p = 0.027). Of course further functional studies are needed to clarify this hypothesis.
Our results have highlighted a novel gene associated with coffee consumption adding new information towards understanding the genetic drivers of coffee consumption. The fact that this gene was not identified in previous much larger studies could be due to the use of a much denser SNP map (1000G) or to the fact that we have specified a different model of inheritance. Although further studies on larger cohorts will be needed to confirm our findings we believe to have added an important piece to the understanding of the genetic basis of coffee consumption and potentially to the mechanisms regulating caffeine metabolism.
Additional Information
How to cite this article: Pirastu, N. et al. Non-additive genome-wide association scan reveals a new gene associated with habitual coffee consumption. Sci. Rep. 6, 31590; doi: 10.1038/srep31590 (2016).
References
Grigg, D. The worlds of tea and coffee: Patterns of consumption. GeoJournal 57, 283–294 (2002).
Farah, A. In Coffee: Emerging Health Effects and Disease Prevention (ed. Chu, Y.-F. ) 1–20 (Wiley-Blackwell) doi: 9780470958780 (2012).
Kleemola, P., Jousilahti, P., Pietinen, P., Vartiainen, E. & Tuomilehto, J. Coffee consumption and the risk of coronary heart disease and death. Arch. Intern. Med. 160, 3393–3400 (2000).
Robertson, D. et al. Caffeine and hypertension. Am. J. Med. 77, 54–60 (1984).
Umemura, T. et al. Effects of acute administration of caffeine on vascular function. Am. J. Cardiol. 98, 1538–1541 (2006).
J. Lindsay, P.-H Carmichael, E. & Kroeger, D. L. In Emerging Health Effects and Disease Prevention (ed. Chu, Y.-F. ) 97–100 (Wiley-Blackwell) doi: 10.1002/9781119949893.ch5 (2012).
Lim, J.-W. & Tan, E.-K. In Emerging Health Effects and Disease Prevention (ed. Chu, Y.-F. ) 111–122 (Wiley-Blackwell) doi: 10.1002/9781119949893.ch6 (2012)
van Dam, R. M. & Feskens, E. J. M. Coffee consumption and risk of type 2 diabetes mellitus. Lancet 360, 1477–1478 (2002).
Tuomilehto, J., Hu, G., Bidel, S., Lindström, J. & Jousilahti, P. Coffee consumption and risk of type 2 diabetes mellitus among middle-aged Finnish men and women. JAMA 291, 1213–1219 (2004).
Carlsson, S., Hammar, N., Grill, V. & Kaprio, J. Coffee consumption and risk of type 2 diabetes in Finnish twins. Int. J. Epidemiol. 33, 616–617 (2004).
Dórea, J. G. & da Costa, T. H. M. Is coffee a functional food? Br. J. Nutr. 93, 773–782 (2005).
Nkondjock, A. In Coffee: Emerging Health Effects and Disease Prevention (ed. Chu, Y.-F. ) 197–209 (Wiley-Blackwell, 2012).
Dragana, Vuckovic, Ginevra, Biino, Francesco, Panu, Mario, Pirastu & Gasparini Paolo, G. G. Lifestyle and normal hearing function in Italy and Central Asia: a significant role of coffee. Hear. Balanc. Commun. in press, (2013).
Goldstein, A., Warren, R. & Kaizer, S. Psychotropic effects of caffeine in man. i. individual differences in sensitivity to caffeine-induced wakefulness. J. Pharmacol. Exp. Ther. 149, 156–159 (1965).
Heath, A. C., Eaves, L. J., Kirk, K. M. & Martin, N. G. Effects of lifestyle, personality, symptoms of anxiety and depression, and genetic predisposition on subjective sleep disturbance and sleep pattern. Twin Res. 1, 176–188 (1998).
Conterio, F. & Chiarelli, B. Study of the inheritance of some daily life habits. Heredity (Edinb). 17, 347–359 (1962).
Cornelis, M. C. et al. Genome-wide meta-analysis identifies regions on 7p21 (AHR) and 15q24 (CYP1A2) as determinants of habitual caffeine consumption. PLoS Genet. 7, e1002033 (2011).
Amin, N. et al. Genome-wide association analysis of coffee drinking suggests association with CYP1A1/CYP1A2 and NRCAM. Mol. Psychiatry 17, 1116–1129 (2012).
Cornelis, M. C. et al. Genome-wide meta-analysis identifies six novel loci associated with habitual coffee consumption. Mol. Psychiatry 20, 647–656 (2015).
Lettre, G., Lange, C. & Hirschhorn, J. N. Genetic model testing and statistical power in population-based association studies of quantitative traits. Genet. Epidemiol. 31, 358–362 (2007).
Girotto, G. et al. Hearing function and thresholds: a genome-wide association study in European isolated populations identifies new loci and pathways. J. Med. Genet. 48, 369–374 (2011).
Aulchenko, Y. S. et al. Linkage disequilibrium in young genetically isolated Dutch population. Eur. J. Hum. Genet. 12, 527–534 (2004).
Delaneau, O., Zagury, J.-F. & Marchini, J. Improved whole-chromosome phasing for disease and population genetic studies. Nat. Methods 10, 5–6 (2013).
Howie, B. N., Donnelly, P. & Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 5, e1000529 (2009).
Abecasis, G. R. et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012).
van Leeuwen, E. M. et al. Population-specific genotype imputations using minimac or IMPUTE2. Nat. Protoc. 10, 1285–1296 (2015).
Howie, B., Fuchsberger, C., Stephens, M., Marchini, J. & Abecasis, G. R. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat. Genet. 44, 955–959 (2012).
Chen, W.-M. & Abecasis, G. R. Family-based association tests for genomewide association scans. Am. J. Hum. Genet. 81, 913–926 (2007).
Aulchenko, Y. S., Ripke, S., Isaacs, A. & van Duijn, C. M. GenABEL: an R library for genome-wide association analysis. Bioinformatics 23, 1294–1296 (2007).
Karssen, L. C. et al. The GenABEL Project for statistical genomics. F1000Research 5, 914 (2016).
González, J. R. et al. Maximizing association statistics over genetic models. Genet. Epidemiol. 32, 246–254 (2008).
Lonsdale, J. et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585 (2013).
Ward, L. D. & Kellis, M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 40, D930–D934 (2012).
Jin, F. et al. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature 503, 290 (2013).
Dunham, I. et al. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Peng, M. et al. Mutant prenyltransferase-like mitochondrial protein (PLMP) and mitochondrial abnormalities in kd/kd mice. Kidney Int. 66, 20–28 (2004).
López, L. C. et al. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am. J. Hum. Genet. 79, 1125–1129 (2006).
Quinzii, C. M. et al. Tissue-specific oxidative stress and loss of mitochondria in CoQ-deficient Pdss2 mutant mice. FASEB J. 27, 612–621 (2013).
Falk, M. J. et al. Probucol ameliorates renal and metabolic sequelae of primary CoQ deficiency in Pdss2 mutant mice. EMBO Mol. Med. 3, 410–427 (2011).
Caporaso, N., Genovese, A., Canela, M. D., Civitella, A. & Sacchi, R. Neapolitan coffee brew chemical analysis in comparison to espresso, moka and American brews. Food Res. Int. 61, 152–160 (2014).
Acknowledgements
We would like to thank all the participants in the various studies for their contribution and support. The INGI-FVG study was founded through the Italian Ministry of health. The ERF study as a part of EUROSPAN (European Special Populations Research Network) was supported by European Commission FP6 STRP grant number 018947 (LSHG-CT-2006-01947) and also received funding from the European Community’s Seventh Framework Programme (FP7/2007-2013)/grant agreement HEALTH-F4-2007-201413 by the European Commission under the programme “Quality of Life and Management of the Living Resources” of 5th Framework Programme (no. QLG2-CT-2002-01254). High-throughput analysis of the ERF data was supported by joint grant from Netherlands Organisation for Scientific Research and the Russian Foundation for Basic Research (NWO-RFBR 047.017.043). We are grateful to all study participants and their relatives, general practitioners and neurologists for their contributions and to P. Veraart for her help in genealogy, J. Vergeer for the supervision of the laboratory work and P. Snijders for his help in data collection. Statistical analyses were partly carried out on the Genetic Cluster Computer (http://www.geneticcluster.org) which is financially supported by the Netherlands Scientific Organization (NWO 480-05-003 PI: Posthuma) along with a supplement from the Dutch Brain Foundation and the VU University Amsterdam. This research was financially supported by BBMRI-NL, a Research Infrastructure financed by the Dutch Government (NWO 184.021.007). The work of LCK was partially funded by the FP7 projects MIMOmics (grant no. 305280) and Pain-Omics (grant no. 602736).
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: N.P. and P.G. Performed the experiments: N.P., L.C.K. and A.R. Analyzed the data: N.P., M.K. and A.V.D.S. Contributed reagents/materials/analysis tools: C.V.D., P.G. and N.A. Wrote the paper: N.P., C.V.D. and P.G. All authors contributed to the review of the manuscript and agreed to the final content.
Corresponding author
Ethics declarations
Competing interests
L. Navarini is an employee of Illy s.p.a. This does not alter in any way our adherence to all the Scientific Reports policies on sharing data and materials.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Pirastu, N., Kooyman, M., Robino, A. et al. Non-additive genome-wide association scan reveals a new gene associated with habitual coffee consumption. Sci Rep 6, 31590 (2016). https://doi.org/10.1038/srep31590
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep31590
This article is cited by
-
Genetic determinants of liking and intake of coffee and other bitter foods and beverages
Scientific Reports (2021)
-
A genome-wide association study in Japanese identified one variant associated with a preference for a Japanese dietary pattern
European Journal of Clinical Nutrition (2021)
-
A genome-wide association study in the Japanese population identifies the 12q24 locus for habitual coffee consumption: The J-MICC Study
Scientific Reports (2018)
-
Acute caffeine ingestion reduces insulin sensitivity in healthy subjects: a systematic review and meta-analysis
Nutrition Journal (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
