
Abstract
Pangolins are scale-covered mammals, containing eight endangered species. Maintaining pangolins in captivity is a significant challenge, in part because little is known about their genetics. Here we provide the first large-scale sequencing of the critically endangered Manis javanica transcriptomes from eight different organs using Illumina HiSeq technology, yielding ~75 Giga bases and 89,754 unigenes. We found some unigenes involved in the insect hormone biosynthesis pathway and also 747 lipids metabolism-related unigenes that may be insightful to understand the lipid metabolism system in pangolins. Comparative analysis between M. javanica and other mammals revealed many pangolin-specific genes significantly over-represented in stress-related processes, cell proliferation and external stimulus, probably reflecting the traits and adaptations of the analyzed pregnant female M. javanica. Our study provides an invaluable resource for future functional works that may be highly relevant for the conservation of pangolins.
Similar content being viewed by others


Introduction
The Malayan pangolin (Manis javanica) is a remarkable and distinctive mammal species belonging the order Pholidota, a sister taxon of Carnivora within the superclade Laurasiatheria. Pholidota pangolins are not closely related to other anteaters in South America (Xenarthra) although there are several morphological and adaptive similarities, some due to convergence but others likely inherited from the insectivore precursors of placental mammals that coexisted among the dinosaurs some 100MY ago1,2,3,4. Also being referred to as ‘scaly or spiny anteaters’, pangolins have a peculiar anatomy with keratinized scales made up of agglutinated hairs that overlap with one another and covering most of their body. Pangolins also lack teeth in adults (edentulism), an adaptation for the specialized diet made up of ants and termites (insect-eating - myrmecophagy)1 and possess ‘incomplete zygomatic arches’ and a ‘extremely reduced, bladelike mandible’ with each denture having a single bony protrusion5,6.
The number of Malayan pangolins in the wild is dramatically declining for several reasons and the species is characterized as being critically endangered in The International Union for Conservation of Nature and Natural Resources (IUCN) Red List of Threatened Species7,8. One of the major threats for its declining numbers is the rapid loss and deterioration of their natural habitat due to deforestation activities and human agricultural expansion8,9. Malayan pangolins (and pangolins in general) are also heavily hunted for their meat, skin, and scales, as illegal trade in live animals has become a severe threat to pangolins10,11,12. In China, pangolin meat is consumed as an exotic delicacy while the scales are used for traditional medicinal purposes such as skin diseases and cancer remedies, among other illnesses8. There has also been an attempt to relocate and breed the species in captivity while imitating its natural habitat, but with little success as pangolins do not survive and breed well in captivity13.
Genetic studies of endangered species have become widespread during the last several decades but more recently, the genomes and transcriptomes of endangered species have been sequenced14,15,16. With the emergence of high-throughput Next-Generation Sequencing (NGS) technologies, more molecular information about endangered species can be acquired and studied in-depth. Here, we present the sequencing of the first M. javanica transcriptomes, an important resource for the detailed study of this species that may assist the future management and conservation of this critically endangered mammal. We used short read Illumina HiSeq technology to sequence eight transcriptomes representing the following M. javanica organs: cerebellum, cerebrum, heart, kidney, liver, lung, spleen and thymus. The high quality transcriptomes generated were used for downstream analyses, which provided insights into functional and phylogenetic aspects of pangolin biology. The RNA-Seq reads are accessible at the Sequence Read Archive (SRA) at the National Center for Biotechnology Information (NCBI) under the accession number SRP064341. Genome raw reads and assemblies are also available for download at our repository http://pangolin-genome.um.edu.my or http://www.genomesolutions.com.my/iparc/pgd.
Results
RNA isolation and whole-transcriptome sequencing
To catalog a representative pangolin transcriptome, we sequenced eight RNA samples derived from a pregnant female M. javanica representing the following different organs provided by the Department of Wildlife and National Parks: cerebrum, cerebellum, thymus, liver, kidney, lung, spleen and heart. The quality of all extracted RNAs was evaluated using the Agilent Bioanalyzer 2100 (Agilent Technologies, Palo Alto, CA) before library preparation and sequencing. All samples had sufficient yields of RNAs with very good RNA integrity number (RIN) values (>9.00), indicating the high integrity of RNA samples for sequencing (Supplementary Figure 1). We generated approximately 373 million reads of 100 bp paired-end RNA-Seq data for the eight samples with the number of paired-end reads per sample ranging from 41,532,741 to 52,722,994 (Supplementary Table 1). The quality of sequencing reads was assessed based on the Phred scoring system17. More than 96% of reads for each organ had a read quality higher than the standard threshold (Q30), indicating good quality of the sequencing reads.
Construction of the pangolin transcriptome
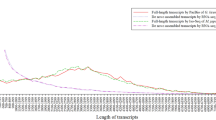
To generate a comprehensive and representative transcriptome of M. javanica, a total of 373,303,322 Illumina HiSeq reads were pooled from the eight different tissues. After normalization of in silico reads using a Perl script housed in the Trinity software package18, the reduced reads were assembled following the steps described in Fig. 1, using three different assemblers: Trinity19, SOAPdenovo-Trans20, and Velvet21. The consensus set of unigenes of the three different methods was used as the final representative transcriptome assembly, which resulted in 89,754 unigenes with a N50 of 3,741 bps (Supplementary Table 2). The length distribution of M. javanica assembled unigenes is displayed in Supplementary Figure 2 with comparison to the orthologous dog genes from ENSEMBL and the human genes from both ENSEMBL and RefSeq databases.

The final M. javanica transcriptome was represented by unigenes, which are consensus assembled transcripts from three different assembly methods.
Transcriptome assembly quality assessment
Although several pipelines to assess the quality of transcriptome assemblies have been recently developed and studied22,23, there are no established standard metrics for quantifying a transcriptome assembly, which is particularly difficult for a de novo non-model transcriptome assembly24. Therefore, we assessed the reconstructed M. javanica transcriptome using the following three in silico methods: (1) transcript sequence quality quantified by RNA-Seq by Expectation-Maximization (RSEM)25; (2) percentage of reads used to recover the unigene set as well as their coverage; and (3) sequence completeness and its contiguity.
All sequencing reads were filtered based on a Phred score of 30 and each assembled transcript was supported by a minimum Fragments per kilo base of transcript per million reads (FPKM) value of 1.00 (Supplementary Figure 3). When working with transcriptomes, read count is a way of quantifying the transcripts by abundance, and also determines whether or not the genes or transcripts produced are highly dependent on length and library size. Taking the FPKM values into consideration in generating the assemblies promotes normalization of the data, as well as minimizing the dependence of transcript selection based on the two variables, thereby producing high quality transcriptomes. RSEM outputs were used for transcript abundance estimation. Secondly, the normalized reads were mapped against the representative sequences. We observed that approximately 70% of the normalized paired reads were mapped back to the M. javanica unigenes with the mean mapping coverage per base of 27. The relatively high percentage indicates that a high amount of transcripts were recovered and retained for analyses after undergoing several stringent filtering steps.
Similarity search by Basic Local Alignment Search Tool (BLAST) against RefSeq database showed that 52.5% of M. javanica unigenes had significant matches (e-value <1e−6) with human and 40.1% with mouse. These results showed that most of M. javanica unigenes are homologous to its closely related and well-annotated mammalian (human and mouse) known genes, supporting that these unigenes are well-assembled.
To examine the completeness of the M. javanica unigenes, we used the TransDecoder program18. Our results showed that 57,214 sequences (63.7%) were protein-coding unigenes, with 38,168 unigenes predicted to have complete coding sequences (CDSs). Furthermore, 4,859 sequences were 3′ partial CDS while 12,486 were 5′ partial CDS. Only 1,701 sequences were internal sequences, which were defined as unknown. These results indicate a relatively high level of completeness of M. javanica unigenes based on the observations that most mammals have 20,000~30,000 gene loci in their genome26,27,28,29 and assuming that this pangolin species did not experience any lineage-specific whole-genome duplication30.
Conservation among mammals
To the best of our knowledge, the M. javanica transcriptome reported in this study is the first Pholidota transcriptome ever sequenced. Because there was no reference pangolin genome available at the start of our study we compared the M. javanica unigenes against the NCBI non-redundant nucleotide database containing sequences for major organisms (from microorganisms to vertebrates) using BLASTX. The total number of BLAST top hit sequences was 69,565 with 55,568 sequences assigned to known species. As anticipated, the top 20 known species with the most abundant BLAST top hits are all derived from mammals with Homo sapiens (15.1%) being the highest, followed by the Equus caballus (12.3%), Ailuropoda melanoleuca (12.1%), Canis lupus (10.4%), and Sus scrofa (7.4%), supporting the view that the list of the assembled pangolin genes are highly similar to known mammalian genes.
Unigene validation
To further determine the quality and accuracy of M. javanica assembled unigenes, we performed Polymerase Chain Reaction (PCR) and Sanger sequencing in addition to in silico quality screening. A total of 10 unigenes were randomly selected for PCR and sequencing. The details of these sequences and the primer sequences used are described in Supplementary Table 3. Pooled RNA samples from the eight tissues were subjected to cDNA (complementary DNA) synthesis and PCR analysis. PCR products of the 10 genes matched the expected sizes (Fig. 2). To confirm the accuracy of each unigene, each PCR product was sequenced and its sequence was compared with the assembled unigenes. Our alignment results showed that the sequences from Sanger sequencing are almost perfectly aligned to our assembled unigenes with an average sequence identity of 99.6% and an average sequence completeness of 99.2% (Supplementary Table 3). These results further support the high quality of our M. javanica assembly.

The size of the PCR products of the validated unigenes meets well with the expected size. “N” is a negative control.
Gene Ontology (GO) and KEGG pathway analysis
To characterize the functional properties of the M. javanica transcriptome, unigene sequences were annotated using Blast2GO31. As a result, a total of 46,720 (52.1%) unigenes were assigned with GO terms (Supplementary Figure 5). As anticipated, functional annotation of M. javanica unigenes revealed high homology with known genes responsible for various biological roles. 50.4% of the unigenes were assigned to biological processes, 23.3% to molecular functions, and 26.3% to cellular components. For biological process, the most highly represented terms were cellular process (16.1%), metabolic process (14.2%) and single-organism process (13.1%). The fourth top represented term was biological regulation (12.0%), followed by response to stimulus (8.8%), cellular component organization (7.1%), developmental process (6.6%), signaling (6.4%), multicellular organismal process (5.9%), localization (5.4%), reproduction (2.1%), multi-organism process (1.1%), and growth (1.1%). The terms associated with reproduction, developmental processes, cellular component organization, and growth may be indicative of the involvement of the M. javanica transcriptome in various growth and developmental activities, which is common for an organism in a gestation period (the female pangolin used in this study was pregnant).
For molecular functions, the sequences were mainly assigned to binding (52.4%) and catalytic activity (25.9%), with the rest of the other terms distributed at 0.1–5.0%. As anticipated, cell (37.6%) and organelle (28.4%) are the most predominant terms assigned to the pangolin transcriptome in cellular components, followed by membrane (10.5%), macromolecular complex (10.2%), membrane-enclosed lumen (8.2%), extracellular region (4.1%), and lastly extracellular matrix (1.0%). Overall, these results are indicative of the broad range of biological activities related with the expressed pangolin transcriptome, representing a pooled collection of the multiple tissues sequenced.
To identify the pathways in which the unigenes are involved, we mapped the unigenes of M. javanica on the known Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. We found a total of 19,165 (21.4%) M. javanica unigenes were associated with 138 unique KEGG pathways, with a total of 133 representing metabolic pathways, 3 environmental information processing pathways (signal transduction), 1 pathway in genetic information passing, and 1 pathway in organismal systems (immune systems) (Supplementary Table 4). The highest represented pathways in M. javanica unigenes included purine (2,784 unigenes) and thiamine (1,963 unigenes) metabolism, followed by aminobenzoate degradation (683), T cell receptor signaling pathway (582), and pyrimidine metabolism (489). For instance, we found that M. javanica unigenes were involved in the phosphatidylinositol signaling pathway (Fig. 3). Phosphatidylinositol 3-Kinase/Protein Kinase B signaling pathway (PI3K/Akt) involved in the phosphatidylinositol signaling pathway, is crucial for host survival (glycolysis/gluconeogenesis) and plays important roles in cell growth, proliferation (cell cycle) and survival signals (e.g. apoptosis)32,33. Interestingly, we also observed the involvement of M. javanica unigenes in the insect hormone biosynthesis pathway (KEGG map 00981). The genes that are associated with this pathway are MJU195075_c0_seq20, MJU195075_c0_seq3, MJU195075_c0_seq30, and MJU195075_c0_seq5. The biosynthesis of insect hormone belongs to the higher class of terpenoids and polyketides, and in vertebrates, is similar to cholesterol-driven pathway as shown in KEGG map 00981, which could be related to ant-eating traits. The future characterization of the pangolin digestive system transcriptome would be important to test the hypothesis that these genes are related to a specialized myrmecophagy phenotype34.

Phosphatidylinositol signaling pathway in the M. javanica transcriptome.
Lipid metabolism
Termites and insect larvae, a staple diet of pangolins, are rich in nutrition, especially fatty acids in the form of palmitic acid, oleic acid and linolenic acid35. In order to use them as an energy source, several important metabolic pathways are involved, namely the linoleic and alpha-linolenic acid metabolism pathways (KEGG:00591 & KEGG:00592), glycerolipid metabolism (KEGG:00561) and fatty acid degradation (KEGG:00071). A total of 747 unigenes were identified in the transcriptome that may participate in these pathways. These unigenes are involved in different pathways and some of them may overlap in different pathways (Supplementary Figure 6). These results provide an important resource in further understanding the potentially evolved lipid metabolism system of pangolins and improving their feeding management in captivity36. Likewise, the future characterization of the pangolin digestive transcriptome would be needed directly investigate the link between these lipid metabolism-related genes and the pangolin diet.
Comparative analysis
We compared pangolin unigenes with the well-annotated genes of H. sapiens and the closest relatives of pangolin from Carnivora, Canis lupus familiaris (dog) (CanFam 3.1, Ensembl Release 67) and Felis catus (cat) (Felis_catus-6.2, Ensembl Release 67) using OrthoMCL (Supplementary Table 5)37. We identified a large number of genes (5,506) specific to M. javanica, suggesting pangolins are divergent compared to their closest relatives (Fig. 4a). We used the Gene Ontology (GO) annotations to find the GO terms for which the M. javanica specific unigenes are enriched by running Fisher’s exact test (0.05 FDR) on a total of M. javanica specific unigenes. We identified nine significantly enriched GO terms in biological processes that include response to external stimulus, signal transduction, response to stress, cell proliferation, and response to biotic stimulus (Fig. 4b). Interestingly, most of these terms are well associated with cell interactions and response to stimulus, indicating that the M. javanica unigenes may be actively involved in response to stimulus or stress. At the molecular function level, the pangolin-specific genes are significantly enriched in functions such as cytoskeletal protein binding, receptor binding, receptor activity, RNA binding and protein kinase activity. Interestingly, the proteins involving in the cytoskeletal protein binding are known to interact selectively with any protein components of any cytoskeleton which includes actin, microtubule and intermediate filament cytoskeleton38,39. Pangolins have strong and sophisticated musculoskeletal system for digging and tree climbing7,40. Besides that, pangolins have a unique protecting mechanism allowing them to quickly roll themselves into visually impenetrable balls defending them against enemies, which may likely required the evolution of a sophisticated musculoskeletal system. Moreover, pangolin scales are structurally similar to human nails and hairs that are made of keratin41. The intermediate filaments could be transformed into keratin-like stretchable muscular-structure filaments, forming durable and strong scale structures. We hypothesize that the enriched pangolin-specific unigenes in cytoskeletal protein binding may be required for these unique features of pangolins (the development of pangolin keratinised scales and highly sophisticated musculoskeletal system for fossoriality or arboreality). However, further studies on these pangolin-specific genes may be required to insightfully understand the unique traits or adaptation of pangolins compared to other mammals.

(A) Venn diagram showing comparison between the genes of pangolin and other mammals. (B) Functional enrichment analysis of pangolin-specific genes. Significant functional categories were shown. GO terms highlighted in light red are biological processes, whereas GO terms highlighted in yellow are molecular functions.
Repetitive elements discovery
Repetitive elements can usually be classified into two major categories: (1) transposon-derived interspersed repeats and (2) simple sequence repeats. These two repeat types can be further classified according to the pattern of repeating nucleotide base, mode of repeat expansion, and sequence homology with the consensus repeats family. These consensus repeat families are the core sequence from the repeat database RepBase, which is used by RepeatMasker42 to detect homologous repeats present in the transcripts. To identify the repeats in the M. javanica transcriptome, we screened the repetitive elements in the 89,754 unigenes using the RepeatMasker software42. For instance, we found many predicted repeats, covering a considerable portion (13.90% or 31,832,508 bp) of the total genomic length (229,081,942 bp) of all unigenes. The most represented repetitive elements were Long Interspersed Nuclear Elements (LINEs), where LINE1 family consists of 28,578 elements (4.67%), whereas Short Interspersed Elements (SINEs) have 33,009 elements (1.93%) in its Mammalian-wide interspersed repeats (MIRs) family (Table 1). The relatively high number of expressed repeats may be due to frequent exonization of transposable elements43 in pangolins. They may play an important role in pangolin transcriptome diversity as the repeats could introduce novel splice sites resulting in alternative splicing44.
Simple Sequence Repeats (SSR), also known as Short Tandem Repeats (STR) or microsatellites, are another major class of repetitive elements. Most of the SSRs found in the transcriptome or the protein-coding regions of the genome are trinucleotide repeats, since they do not cause frameshift mutations and retain protein integrity45. Trinucleotide repeats are known to affect the chemical and physical properties of certain proteins46 and length changes of triplet repeats are related to more than 40 neurological diseases found in humans47. The microsatellite searching tool MISA48 was used to find microsatellites present in the unigenes. In total, 45,223 Short Sequence Repeats (SSR) were identified from 89,754 non-redundant unigenes including their 5′- and 3′ UTR regions (Supplementary Table 6). Mono-nucleotides SSRs were the most abundant motif (20,705 copies), followed by di-nucleotide SSRs (20,064 repeats), tri- (4,001 repeats), tetra- (432 repeats) and penta-nucleotides (21 repeats) SSRs. The top mono-nucleotide repeat motifs and di-nucleotide repeat motifs included A/T (19,402 repeats or 42.9%) and AC/GT (10,776 repeats or 23.8%), respectively. Most of the SSRs found in the protein-coding regions of the genome are trinucleotide repeats (759 repeats). The major trinucleotide sequences found in the pangolin transcriptome coding regions are the AGG/CCT and CCG/CGG type. These putative SSRs could be useful as genetic markers for further investigations on genetic variation of the associated expressed genes.
Pairwise comparisons of different transcriptomic profiles
To examine the similarity of each organ transcriptome, we performed statistical correlation analysis for each pair of organs using log10 (FPKM + 1) to normalize the plots (Fig. 5). Our data showed that the thymus and lung transcriptomes have the most similar expression profiles (the coefficient of determination, R2 = 0.88), followed by cerebellum and cerebrum (R2 = 0.77). The cerebellum and liver have the least similar transcriptomic profiles with R2 = 0.40, reflecting the highly different complexity between the two organs49 As anticipated, the lung and thymus or the cerebellum and cerebrum showed the highest inter-tissue correlations, which is consistent with a previous study based on human tissues using gene expression profiles50. These results also fit into the biological origin of the tissues since they represent the immune system and brain, respectively.

Correlation between any two organs resulting from pairwise comparison using expression values in log10 (FPKM+1) quantified by RSEM.
Discussion and conclusion
Pangolins are considered as one of the world’s most bizarre mammal. Belonging to the Pholidota, all eight species of extant pangolins are classified within the single family Manidae. Unfortunately, all the eight pangolin species are currently categorized from vulnerable to critically endangered, with M. javanica being one of the critically endangered species7. Many efforts have been made to maintain the population, but without success, in large part because pangolins do not survive and breed well in captivity13. Here we have successfully generated transcriptomic data from eight different organs of M. javanica. We have also assembled and reported the first comprehensive and representative catalog of M. javanica genes and their expression profiles across different organs as a starting platform to study the genomic and molecular basis of this lesser-known unique mammalian species.
Functional annotation of the M. javanica unigenes revealed the involvement of the species in various essential KEGG pathways such as T-cell receptor pathway and lipid metabolism. The lipid metabolism pathway, on the other hand, may support the myrmecophagus feeding habits of this mammalian species1,35.
Comparative analysis between M. javanica and its closest relatives, the Carnivora, revealed a large number of genes unique to pangolins (Fig. 4). These genes were significantly over-represented in biological processes such as response to external stimulus, response to stress and signal transduction. The enriched genes in these processes might be associated with the fact that the pangolin used here was pregnant. Another possible explanation is that the pangolin might be under stress after being caught or kept in captivity. Furthermore, we also cannot rule out the possibility that pangolins might have evolved to have different mechanisms to deal with stress and external stimulus compared to the closely related dogs and cats or even more distant humans. Studying and comparing the transcriptomes of more pangolins at different conditions may provide clearer insights into each possibility in the future.
In conclusion, we believe the high quality M. javanica transcriptome datasets serve as the first step towards understanding the uniquely specialized evolution of pangolins and a valuable genomic resource for functional studies on pangolins that may be important to unveil the mysteries of the biology and evolution of this rare and unique mammal.
Methods
Ethics statement
Veterinary officers conducted all procedures involving animals and experts at the Department of Wildlife and National Parks (DWNP), Malaysia, following internationally recognized guidelines and approved by the University of Malaya Institutional Animal Care and Use Committee (UM IACUC) [reference number of the approval: DRTU/11/10/2013/RH (R)].
Biological sample
Briefly, a female pregnant pangolin sample weighing 2.73 kg was provided by the DWNP. Organ tissues (cerebellum, cerebrum, heart, kidney, liver, lungs, skin, spleen, and thymus) were harvested by DWNP veterinary officers and stored at −80 °C.
RNA isolation and sequencing
Frozen tissues (~30 mg) were weighed and ruptured using TissueRuptor (Qiagen); followed by extraction using RNAeasy mini kit (Qiagen) following manufacturer’s manual. RNA concentrations were measured using Nanodrop® ND-1000 Spectrophotometer (Thermo Scientific, Wilmington, DE). Quality of the isolated RNA was assessed using Agilent Bioanalyzer 2100 (Agilent Technologies, Palo Alto, CA), prior sequencing. Eight organs RNA sample (cerebellum, cerebrum, heart, kidney, liver, lungs, spleen, and thymus) of average insert size of 200 bps were sent for preprocessing. DNA contaminants were further removed using DNase enzyme digestion, followed by rRNA removal, then cDNA synthesis and PCR amplified into complete cDNA library sent for sequencing using Illumina HiSeq™ 2000 platform (2 × 100 bp strategy). The cDNA libraries were constructed according to Illumina TruSeq™ Stranded mRNA Sample Preparation Guide (Rev E, October 2013) for Illumina Paired-End Sequencing service provided by BGI, Hong Kong.
De novo assembly strategies
Clean reads from Illumina HiSeq RNA-Seq were obtained after filtering out reads with adaptors, unknown nucleotides larger than 5%, and reads with low quality (more than 20% of the bases’ qualities are less than 10 in a read). A quality check was performed on the reads using the FastQC (version 0.10.1) software (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/).
De novo assembly of each individual tissue sample was performed using Trinity with default parameters previously described. To generate the whole transcriptome of Malayan pangolin, we assembled the pooled reads from eight samples using three different assemblers: Trinity18, SOAPdenovo-Trans20, and Velvet21. Total reads were first normalized by k-mer coverage using Trinity software. The assembled transcripts from three different assemblers were then individually filtered to remove poorly supported transcripts using the filtering criteria fragments per kilo base per million transcripts, FPKM, equals to 1.00.
Filtered transcripts from the three assemblers were then selected for overlaps and merged together; before being further clustered using CD-Hit-EST program51 using clustering threshold of 98% identity, to reduce redundancy. The singletons (unclustered transcripts), and the longest sequence representatives in each clustered transcripts were selected and retained, and classified as the unigenes; the sequences that cannot be extended on either ends. Figure 1 summarizes the workflow of de novo assembly to generate the Malayan pangolin unigenes. The unigenes then underwent foreign contamination screening (FCS) using BLAST with 98% identity cut-off to screen for any foreign organisms’ chromosome, mitochondria DNA, vectors, and sequencing adaptors.
Functional annotation with GO and KEGG pathway analysis
To further validate and annotate the Malayan pangolin transcriptome, the assembled unigenes were subjected to sequence similarity search by BLASTX against NCBI’s non-redundant (nr) protein database with the configuration of E-value = 1e-3 and HSP length cut-off of 33. Results were exported into Blast2GO program to undergo mapping and functional annotation to retrieve GO terms associated with biological processes, cellular components, and molecular functions using the e-value hit-filter of 1e-6. Blast2GO also annotate the unigenes following KEGG database.
Correlation between any two pangolin tissue transcriptomes
To examine the close-relatedness of pangolin tissue transcriptomes, the expression values of the unigenes (FPKM) in the transcriptomes of each tissue are manipulated. By utilizing the tool ‘RSEM-calculate-expression’ in the RSEM pipeline, the reads of each tissue were mapped to the unigenes25. Gene expression values, expressed as log10 (FPKM + 1) for each tissue transcriptome were plotted against one another producing the scatter plots. R2 values were then calculated from the scatter plots to estimate the correlation between any two pangolin transcriptomes.
Additional Information
How to cite this article: Yusoff, A. M. et al. De novo sequencing, assembly and analysis of eight different transcriptomes from the Malayan pangolin. Sci. Rep. 6, 28199; doi: 10.1038/srep28199 (2016).
References
Reiss, K. Z. Using Phylogenies to Study Convergence: The Case of the Ant-Eating Mammals. Integrative and Comparative Biology 41, 507–525, doi: 10.1093/icb/41.3.507 (2001).
Delsuc, F. et al. Molecular phylogeny of living xenarthrans and the impact of character and taxon sampling on the placental tree rooting. Molecular biology and evolution 19, 1656–1671 (2002).
Honeycutt, R. L. & Adkins, R. M. Higher Level Systematics of Eutherian Mammals: An Assessment of Molecular Characters and Phylogenetic Hypotheses. Annual Review of Ecology and Systematics 24, 279–305, doi: 10.1146/annurev.es.24.110193.001431 (1993).
Murphy, W. J. et al. Molecular phylogenetics and the origins of placental mammals. Nature 409, 614–618, doi: 10.1038/35054550 (2001).
Nowak, R. M. Walker’s Mammals of the World 6th Edition., (Johns Hopkins University Press, 1999).
Davit-Béal, T., Tucker, A. S. & Sire, J.-Y. Loss of teeth and enamel in tetrapods: fossil record, genetic data and morphological adaptations. Journal of Anatomy 214, 477–501, doi: 10.1111/j.1469-7580.2009.01060.x (2009).
IUCN. The IUCN Red List of Threatened Species. Version 2014.3., http://www.iucnredlist.org (2014).
Challender, D. et al. Manis javanica. The IUCN Red List of Threatened Species. Version 2014.3, http://www.iucnredlist.org (2014).
CITES. Prop. 11.13. Manis crassicaudata, Manis pentadactyla, Manis javanica. Transfer from Appendix II to Appendix I (India, Nepal, Sri Lanka, United States). Available at: http://www.cites.org/eng/cop/11/prop/13.pdf, 2000).
Challender, D. W. S. Asian pangolins: Increasing affluence driving hunting pressure. TRAFFIC Bulletin 23, 92–93 (2011).
Pantel, S. & Chin, S. Y. Proceedings of the Workshop on Trade and Conservation of Pangolins native to South and Southeast Asia. TRAFFIC Southeast Asia (2009).
Pantel, S. & Anak, N. A. A preliminary assessment of the pangolin trade in Sabah. TRAFFIC Southeast Asia (2010).
Yang, C. W. et al. History and dietary husbandry of pangolins in captivity. Zoo Biology 26, 223–230, doi: 10.1002/zoo.20134 (2007).
Li, R. et al. The sequence and de novo assembly of the giant panda genome. Nature 463, 311–317, doi: 10.1038/nature08696 (2009).
Liu, Z. et al. De novo Assembly of the Indo-Pacific Humpback Dolphin Leucocyte Transcriptome to Identify Putative Genes Involved in the Aquatic Adaptation and Immune Response. PLoS ONE 8, e72417, doi: 10.1371/journal.pone.0072417 (2013).
Dobrynin, P. et al. Genomic legacy of the African cheetah, Acinonyx jubatus. Genome biology 16, 277, doi: 10.1186/s13059-015-0837-4 (2015).
Schmieder, R. & Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864, doi: 10.1093/bioinformatics/btr026 (2011).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature Protocols 8, 1494–1512, doi: 10.1038/nprot.2013.084 (2013).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology 29, 644–652, doi: 10.1038/nbt.1883 (2011).
Xie, Y. et al. SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 30, 1660–1666, doi: 10.1093/bioinformatics/btu077 (2014).
Zerbino, D. R. & Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Research 18, 821–829, doi: 10.1101/gr.074492.107 (2008).
Martin, J. et al. Rnnotator: an automated de novo transcriptome assembly pipeline from stranded RNA-Seq reads. BMC Genomics 11, 663, doi: 10.1186/1471-2164-11-663 (2010).
Li, B. et al. Evaluation of de novo transcriptome assemblies from RNA-seq data, doi: 10.1101/006338 (2014).
Martin, J. A. & Wang, Z. Next-generation transcriptome assembly. Nature Reviews Genetics 12, 671–682, doi: 10.1038/nrg3068 (2011).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. Bmc Bioinformatics 12, doi: 10.1186/1471-2105-12-323 (2011).
Chinwalla, A. T. et al. Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562, doi: 10.1038/nature01262 (2002).
Okazaki, Y. et al. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 420, 563–573, doi: 10.1038/nature01266 (2002).
Versteeg, R. The Human Transcriptome Map Reveals Extremes in Gene Density, Intron Length, GC Content, and Repeat Pattern for Domains of Highly and Weakly Expressed Genes. Genome Research 13, 1998–2004, doi: 10.1101/gr.1649303 (2003).
Lander, E. S. et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921, doi: 10.1038/35057062 (2001).
Lespinet, O. The Role of Lineage-Specific Gene Family Expansion in the Evolution of Eukaryotes. Genome Research 12, 1048–1059, doi: 10.1101/gr.174302 (2002).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676, doi: 10.1093/bioinformatics/bti610 (2005).
Riley, J. K., Carayannopoulos, M. O., Wyman, A. H., Chi, M. & Moley, K. H. Phosphatidylinositol 3-kinase activity is critical for glucose metabolism and embryo survival in murine Blastocysts. J Biol Chem 281, 6010–6019, doi: 10.1074/jbc.M506982200 (2006).
Nicholson, K. M. & Anderson, N. G. The protein kinase B/Akt signalling pathway in human malignancy. Cellular signalling 14, 381–395 (2002).
Reiss, K. Z. Using Phylogenies to Study Convergence: The Case of the Ant-Eating Mammals. Integrative and Comparative Biology 41, 507–525, doi: doi: 10.1093/icb/41.3.507 (2001).
Tomotake, H., Katagiri, M. & Yamato, M. Silkworm pupae (Bombyx mori) are new sources of high quality protein and lipid. Journal of nutritional science and vitaminology 56, 446–448 (2010).
Lin, M. F., Chang, C. Y., Yang, C. W. & Dierenfeld, E. S. Aspects of digestive anatomy, feed intake and digestion in the Chinese pangolin (Manis pentadactyla) at Taipei zoo. Zoo biology 34, 262–270, doi: 10.1002/zoo.21212 (2015).
Li, L., Stoeckert, C. J., Jr. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13, 2178–2189, doi: 10.1101/gr.1224503 (2003).
dos Remedios, C. G. et al. Actin binding proteins: regulation of cytoskeletal microfilaments. Physiological reviews 83, 433–473, doi: 10.1152/physrev.00026.2002 (2003).
Erickson, H. P. Evolution of the cytoskeleton. BioEssays : news and reviews in molecular, cellular and developmental biology 29, 668–677, doi: 10.1002/bies.20601 (2007).
Endo, H. et al. The functional anatomy of the masticatory muscles of the Malayan pangolin, Manis javanica. Mammal Study 23, 1–8 (1998).
Tong, J., Ma, Y.-H., Ren, L.-Q. & Li, J.-Q. Tribological characteristics of pangolin scales in dry sliding. Journal of materials science letters 19, 569–572 (2000).
A.F.A., S., R., H. & P., G. RepeatMasker Open-4.0, http://www.repeatmasker.org (2015).
Zhang, W. et al. Inferring the expression variability of human transposable element-derived exons by linear model analysis of deep RNA sequencing data. BMC genomics 14, 1 (2013).
Nekrutenko, A. & Li, W.-H. Transposable elements are found in a large number of human protein-coding genes. TRENDS in Genetics 17, 619–621 (2001).
Sutherland, G. R. & Richards, R. I. Simple tandem DNA repeats and human genetic disease. Proceedings of the National Academy of Sciences 92, 3636–3641, doi: 10.1073/pnas.92.9.3636 (1995).
Hancock, J. M. & Simon, M. Simple sequence repeats in proteins and their significance for network evolution. Gene 345, 113–118, doi: 10.1016/j.gene.2004.11.023 (2005).
Pearson, C. E., Nichol Edamura, K. & Cleary, J. D. Repeat instability: mechanisms of dynamic mutations. Nature reviews. Genetics 6, 729–742, doi: 10.1038/nrg1689 (2005).
Thiel, T., Michalek, W., Varshney, R. K. & Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). TAG. Theoretical and applied genetics. Theoretische und angewandte Genetik 106, 411–422, doi: 10.1007/s00122-002-1031-0 (2003).
Ramsköld, D., Wang, E. T., Burge, C. B. & Sandberg, R. An Abundance of Ubiquitously Expressed Genes Revealed by Tissue Transcriptome Sequence Data. PLoS Computational Biology 5, e1000598, doi: 10.1371/journal.pcbi.1000598 (2009).
Shmueli, O. et al. GeneNote: whole genome expression profiles in normal human tissues. Comptes rendus biologies 326, 1067–1072 (2003).
Huang, Y., Niu, B., Gao, Y., Fu, L. & Li, W. CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 26, 680–682, doi: 10.1093/bioinformatics/btq003 (2010).
Acknowledgements
We would like to thank the members of Genome Informatics Research Laboratory and the International Pangolin Research Consortium (IPaRC), University of Malaya for their IT and bioinformatics assistance and inputs in this study. Furthermore, we would like to appreciate SJO supported as PI by Russian Ministry of Science Mega-grant no.11.G34.31.0068. This project was supported by University of Malaya and Ministry of Education, Malaysia under the High Impact Research (HIR) grant UM.C/625/HIR/MOHE/CHAN-08.
Author information
Authors and Affiliations
Contributions
S.W.C., G.J.W., and A.M.Y. conceived and coordinated this project. S.W.C., K.V.K, A.M.Y., W.G.J., J.R.-R.J, R.H. and F.T.S. performed animal handling, sampling and tagging. A.M.Y. performed RNA extraction. S.W.C. and A.M.Y. led and designed library construction and RNA-Seq experiments. S.W.C. and A.M.Y. designed primers, PCR and Sanger sequencing validation experiments. A.M.Y., T.K.T., R.H., W.Y.W., A.A. and K.P.K. performed data analyses, data interpretation and oversaw various analyses. A.M.Y., S.W.C., T.K.T., K.P.K., A.A., R.H., L.L, S.J.O., W.C.W. and W.G.J. wrote manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Mohamed Yusoff, A., Tan, T., Hari, R. et al. De novo sequencing, assembly and analysis of eight different transcriptomes from the Malayan pangolin. Sci Rep 6, 28199 (2016). https://doi.org/10.1038/srep28199
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep28199
This article is cited by
-
New horizons in the reproductive biology of Chinese pangolin (Manis pentadactyla) using the gonadal hormonal profile
Scientific Reports (2023)
-
Research-ready data: the C-Surv data model
European Journal of Epidemiology (2023)
-
Development and characterization of 21 SNP markers in Manis javanica based on high-throughput sequencing
Conservation Genetics Resources (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
