
Abstract
We report identification and genetic characterization of a rare skeletal disorder that remained unidentified for decades in a village of Jammu and Kashmir, India. The population residing in this region is highly consanguineous and a lack of understanding of the disorder has hindered clinical management and genetic counseling for the many affected individuals in the region. We collected familial information and identified two large extended multiplex pedigrees displaying apparent autosomal recessive inheritance of an uncharacterized skeletal dysplasia. Whole exome sequencing (WES) in members of one pedigree revealed a rare mutation in WISP3:c.156C > A (NP_003871.1:p.Cys52Ter), that perfectly segregated with the disease in the family. To our surprise, Sanger sequencing the WISP3 gene in the second family identified a distinct, novel splice site mutation c.643 + 1G > A, that perfectly segregated with the disease. Combining our next generation sequencing data with careful clinical documentation (familial histories, genetic data, clinical and radiological findings), we have diagnosed the families with Progressive Pseudorheumatoid Dysplasia (PPD). Our results underscore the utility of WES in arriving at definitive diagnoses for rare skeletal dysplasias. This genetic characterization will aid in genetic counseling and management, critically required to curb this rare disorder in the families.
Similar content being viewed by others

Introduction
Rare disorders are very often poorly understood in part due to their low prevalence and in the absence of specialized diagnostic tools and medical facilities. Such rare disorders may remain uncharacterized or misdiagnosed for years, even generations. This scenario is not uncommon in developing countries. We report identification and genetic characterization of a very rare skeletal disorder that remained undiagnosed for many generations, in a village of the Jammu and Kashmir region of India. In recent years the prevalence of the disorder has reportedly increased in the village, while the absence of any medical intervention and severity of the progressive skeletal disease has rendered affected individuals physically challenged and disabled for life. The village itself is located deep in the lower Himalayan mountains (approximate geographic coordinates: 33°47′17.9′N and 74°16′50.4′E) that until recently were inaccessible by modern transportation. The population residing in the region is highly endogamous and consanguineous with a size of approximately 4372 individuals (as per records from Ministry of Drinking Water and Sanitation, Government of India). Studies in the past have indicated that consanguineous marriages lead to increased chance of autosomal recessive disorders (reviewed in Hamamy 20121) as also reported in the present study.
Report
We collected familial information for the affected individuals and generated two highly extended multiplex pedigrees (supplementary Fig. 1a,b). The analyses of both pedigrees indicated very high consanguinity and an autosomal recessive mode of inheritance. We examined the phenotypic features of the disorder segregating within the two pedigrees. Affected individuals were reported to be normal at birth had normal intelligence, normal appearance and no symptoms of joint effusion or inflammation, with onset of disease symptoms between 4–6 years of age and intensifying with age. The disorder was reported to initiate with general physical symptoms including enlarged ankles, knees and elbow joints, knobby appearance of proximal interphalangeal joints of hand and mild involvement of hip joints. By ten years of age, in addition to the above symptoms, the affected appeared to develop a knobby appearance of metacarpophalangeal and distal interphalangeal joints of the hand and a slightly abnormal spine. In early adolescence, some affected individuals displayed gait disturbances due to knee varus, tilted pelvis and/or other spine deformities e.g. scoliosis. Beyond adolescence, affected individuals developed stiffness in the enlarged joints, comptodactyly in the fingers and toes, involvement of the shoulders, shortened stature by 10–18 cm due to dorsal kyphosis, a highly affected pelvis, and flexion contracture of the elbows and knees. Most individuals also presented with a chest deformity due to rib and spine involvement and they were unable to stand and extend their joints. The musculoskeletal features of the disorder warranted radiological screenings but facilities for this were lacking in the local village. We ultimately succeeded in transporting and an affected male (AR-11 of age 32 years) from the first family (Supplementary Fig. 1a) and two affected sisters (AR-17 of age 9 and AR-16 of age 26 years) and two unaffected members (IV-12, carrier father of age 60 and V-18, unaffected brother age 22 years) from second family (Supplementary Fig. 1b). In addition to a radiological examination the family members were evaluated by blood tests including: haemoglobin(Hb), erythrocyte sedimentation rate (ESR), blood sugar, calcium and alkaline phosphatase levels, presence of rheumatoid (RA) factor and anti nuclear antibodies (ANA). The results indicated values within normal ranges for all individuals. Radiological findings in the affected 9 year old sister showed enlarged epiphyses and metaphyses of the metacarpals and phalanges; enlarged femoral necks with reduced articular space of the hip joints; flexion of knees and reduced articular space in the knee joints; and reduced articular space in the feet (Fig. 1a). The older individuals (26 years, radiographs in Fig. 1a and 32 years, radiographs in Fig. 1b) exhibited additional radiological features including enlarged carpal bones with reduced interosseous space and comptodactyly; enlarged and flat femoral heads with complete loss of articular space in hip joints, significant osteopenia; and pelvic tilt due to abnormal curvature of the spine, complete loss of articular space in the knee joints and various spinal abnormalities (fused cervical vertebrae, narrowing of intervertebral disc spaces, kyphoscoliosis, platyspondyly, erosion of end plates and narrowing of intervertebral disc spaces) (Fig. 1a,b). Collectively these parameters indicate an inherited skeletal dysplasia with Mendelian recessive Inheritance. However, these clinical details were consistent with various diagnoses, making it difficult to precisely characterize the dysplasia.

(1a) Radiographs of Affected Sibs of Age 9 years and 26 years and Unaffected of Age 22 years. Panels show (a) Hand (b) Radial and Ulnar joints at Elbow (c) Feet (d) Knee Joint (lateral view) (e) Tibia and Fibula at knee joint (Anterior View) (f) Pelvis. (1b) Radiographs of affected individual of 32 years from 2nd family. Panels show (a) Head and Neck (lateral view) (b) Spine Thoracic region (posterior view) (c) Radial and Ulnar joints at Elbow (d) hand (posterior view) (e) Spine (posterior View) (f) Spine (lateral view) (g) Pelvis (h) Knee Joint (lateral view) (i) Foot (lateral view).
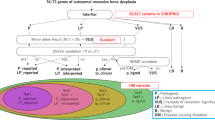
Next generation sequencing (NGS), specifically whole exome sequencing (WES), has proven quite powerful for hastening the identification and characterization of rare Mendelian disorders2,3. Combining comprehensive WES, appropriate data filtering, and pedigree mapping can be sufficiently statistically informative to identify the single gene and causal mutation with as few as 2–4 family members members with very rare Mendelian disease4,5. We opted to carry out WES in the affected siblings from family 1 (Supplementary Fig. 1). We expected to observe a relatively high proportion of variants shared between these two individuals due to the consanguinity within the family rather than the disease, making it more difficult to distinguish true candidate variants. To partially overcome this issue and filter out false positives that would not segregate with the disease in the family we selected a distant cousin (AR-15 also a carrier) of the parents in family 1 (Fig. 2). We outsourced exome capture and sequencing to SciGenom Labs Private Limited. Blood samples were collected during the visit to the village and transported to lab in ice packs. DNA was isolated using Qiagen DNeasy Blood & Tissue Kit and approximately 2ug of genomic DNA and Agilent sureselect v5 kit for exome capture was used. Paired End (PE-100 × 2) Sequencing was done on Illumina HiSeq 2000 with a targeted mean coverage of 100X. A bioinformatics pipeline spanning from alignment variant calling to variant annotation was used (Details in SUPPLEMENTARY FILE Whole Exome Sequencing Methodology). The sequencing of the samples provided us over 90 million reads for each sample. More than 99% of the total reads aligned to the reference genome (GRCh37/HG19) for each sample. After filtering the alignment for mapping quality, 98% of the passed mapped reads have Q60 Phred score reflecting very good alignment quality. Variant calling was performed using BROAD Institute’s GATK-Toolkit6 and were called using the complete Human Reference Genome. Exome sequencing produced 26382 called variants that were shared in the affected individuals. After variant filtering for rare (<1%) conserved in evolution, functional homozygous recessive mutations, we identified the most promising causative variant in WISP3; NM_003880.3:c.156C > A NP_003871.1:p.Cys52*, which is listed in the USA National Institutes of Health ClinVar database7 (Fig. 2). This variation has been previously reported8,9 and is predicted to cause premature termination of the WISP3 protein. The mutation is diagnostic of Progressive Pseudorheumatoid Dysplasia (PPD), Progressive Pseudorheumatoid Arthropathy of Childhood (PPAC) or Spondyloepiphyseal Dysplasia Tarda with Progressive Arthropathy (SEDT-PA) (OMIM:208230). PPD is a very rare disorder with incidence estimated to be one case per million people in the UK10. The disorder has also been reported in India but primarily in Southern India11. We noticed, interestingly, the presence of another nonsynonymous variation WISP3; NM_003880.3:c.248G > A, NP_003871.1:p.Gly83Glu in the same exon. Both variations were checked for quality, and Sanger sequencing of the exon confirmed the finding (Supplementary Fig. 2a,b). Sanger sequencing was also performed on exon 2 of the WISP3 gene in all the collected samples, with both variants showing perfect segregation with the disease in family 1 (Supplementary Fig. 3). However neither variant was detected by Sanger sequencing exon 2 in affected members of family 2. We further sequenced all the exons, exon-intron junctions and 5′-3′ UTRs of WISP3 in family 2 and detected a novel splice site variant, NM_003880.3:c.643 + 1 G > A, at the junction of exon 4 and intron 4. This variant showed perfect segregation with the disease in family 2 (Supplementary Fig. 4). In-silico evaluation of the variant12 revealed that the site is highly conserved across the vertebrates (from humans to zebrafish). This change is predicted to alter the Exon 4 splice donor site (Supplementary Fig. 5), may be leading to either exon skipping/alternative splicing in the mRNA or premature termination of the protein, which needs to be experimentally validated. WISP3 is a member of the CCN gene family that encodes cysteine-rich secreted proteins with roles in cell growth and differentiation. It has been reported to modulate bone morphogenic protein (BMP) and the canonical WNT signaling pathway13. Earlier reports also note that homozygous or compound heterozygous mutations in the WISP3 gene also cause PPD [reviewed in Garcia Segarra et.al.14].

Whole Exome Sequencing and Variant Filtering Strategy adapted to narrow down to most promising causative mutation in Family 1.
In summary, the combined evidence of the segregation of WISP3 functional variations with the disease, autosomal recessive transmission, and clinical and radiological features supported a diagnosis of PPD in both families. This identification and genetic characterization will aid in carrier screening and genetic counseling in both families. The ability to definitively identify PPD carriers and affected individuals may also help in managing the disorder, which ordinarily has a prevalence of one case per million people in the UK10 but thousands of fold higher prevalence in this isolated village. It was intriguing and highly unexpected to identify distinct causal mutations in the two families. Our results suggest that further screening of all affected individuals within the village and other members of these multiplex families is warranted to identify possible compound heterozygotes for these mutations. Further detailed medical evaluations are also needed to identify possible correlations between genotype and phenotype. It is critical for our future perspective to elucidate the functional consequence of the novel splice site variation we found (c.643 + 1 G > A) and its effect on the WISP3 protein to understand the mechanism of causation of the disease. This effort demonstrates the first genetic study exploiting WES to understand a disorder in the state of Jammu and Kashmir, India. WES may be an effective approach in understanding rare disorders in regions where such disorders exist in abundance but remain undiagnosed/misdiagnosed due to lack of specialized clinical resources.
Ethics approval
Institutional Ethical Review Board (IERB) SMVDU Katra, J&K, India approved the study and study was according to the approved guidelines. Informed consent for the study and genetic analyses was obtained from all individuals or their parents.
Additional Information
Accession Codes: Next generation sequencing data (WES) will be available at SRA (NCBI) through Bioprojects; Project ids: PRJNA291671, PRJNA291678, PRJNA291679.
How to cite this article: Rai, E. et al. Whole Exome Screening Identifies Novel and Recurrent WISP3 Mutations Causing Progressive Pseudorheumatoid Dysplasia in Jammu and Kashmir-India. Sci. Rep. 6, 27684; doi: 10.1038/srep27684 (2016).
References
Hamamy, H. Consanguineous marriages: Preconception consultation in primary health care settings. J Community Genet 3, 185–192, doi: 10.1007/s12687-011-0072-y (2012).
Gilissen, C., Hoischen, A., Brunner, H. G. & Veltman, J. A. Unlocking Mendelian disease using exome sequencing. Genome biology 12, 228, doi: 10.1186/gb-2011-12-9-228 (2011).
Zhu, X. et al. Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios. Genet Med 17, 774–781, doi: 10.1038/gim.2014.191 (2015).
Bamshad, M. J. et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nature reviews. Genetics 12, 745–755, doi: 10.1038/nrg3031 (2011).
Boycott, K. M., Vanstone, M. R., Bulman, D. E. & MacKenzie, A. E. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nature reviews. Genetics 14, 681–691, doi: 10.1038/nrg3555 (2013).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature genetics 43, 491–498, doi: 10.1038/ng.806 (2011).
Landrum, M. J. et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic acids research 42, D980–985, doi: 10.1093/nar/gkt1113 (2014).
Dalal, A. et al. Analysis of the WISP3 gene in Indian families with progressive pseudorheumatoid dysplasia. American journal of medical genetics. Part A 158A, 2820–2828, doi: 10.1002/ajmg.a.35620 (2012).
Hurvitz, J. R. et al. Mutations in the CCN gene family member WISP3 cause progressive pseudorheumatoid dysplasia. Nature genetics 23, 94–98, doi: 10.1038/12699 (1999).
Wynne-Davies, R., Hall, C. & Ansell, B. M. Spondylo-epiphysial dysplasia tarda with progressive arthropathy. A “new” disorder of autosomal recessive inheritance. The Journal of bone and joint surgery. British volume 64, 442–445 (1982).
Ekbote, A. V. et al. A descriptive analysis of 14 cases of progressive-psuedorheumatoid-arthropathy of childhood from south India: review of literature in comparison with juvenile idiopathic arthritis. Seminars in arthritis and rheumatism 42, 582–589, doi: 10.1016/j.semarthrit.2012.09.001 (2013).
Desmet, F. O. et al. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic acids research 37, e67, doi: 10.1093/nar/gkp215 (2009).
Nakamura, Y. et al. The CCN family member Wisp3, mutant in progressive pseudorheumatoid dysplasia, modulates BMP and Wnt signaling. The Journal of clinical investigation 117, 3075–3086, doi: 10.1172/JCI32001 (2007).
Garcia Segarra, N. et al. The diagnostic challenge of progressive pseudorheumatoid dysplasia (PPRD): a review of clinical features, radiographic features, and WISP3 mutations in 63 affected individuals. American journal of medical genetics. Part C, Seminars in medical genetics 160C, 217–229, doi: 10.1002/ajmg.c.31333 (2012).
Acknowledgements
We thank all the Villagers who helped us by all possible means to conduct this study in difficult terrain. Especially, we are indebted to Moulvi Fareed, Mr. Shabir Bhat, Dr. Puneet Kar, Mr. Sumesh Raina, Mr. Qamar Latif and Mr. Mohd Hamid for their assistance in communication with the people in the local language and their assistance in the collection of familial information and samples, which were key factors in identification of the disorder. We also thank Chief Medical Officer (CMO) Poonch and Director, Health Services, Jammu (Government of J&K) for their consistent support through out the study. We also thank Dr. Sanjana Kaul (SBT, University of Jammu) for facilitating sequencing of the samples. Authors are highly thankful to Ms. Liza Nowlin (TSRH, USA) and Ms. Marjorie Ehrmann (TSRH, USA) for their valuable edit to improve the linguistic quality of the Manuscript.
Author information
Authors and Affiliations
Contributions
E.R., S.S., K.K.P. and P.K. planned the study. E.R., S.S. and K.K.P. mainly wrote the paper. E.R., A.M., P.K. and A.A. assisted in collection of samples, DNA isolation and Sanger Sequencing. E.R. and S.S. analyzed the WES data. C.W., S.I., K.T. and M.K.D. contributed in study design and helped in understanding the genetics and evaluation of pedigrees. K.K.P. and S.R. contributed in clinical evaluation of the affected individuals in person and S.I. helped in understanding clinical and radiological findings. All the authors approve the content of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Rai, E., Mahajan, A., Kumar, P. et al. Whole Exome Screening Identifies Novel and Recurrent WISP3 Mutations Causing Progressive Pseudorheumatoid Dysplasia in Jammu and Kashmir-India. Sci Rep 6, 27684 (2016). https://doi.org/10.1038/srep27684
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep27684
This article is cited by
-
The combined prevalence of classified rare rheumatic diseases is almost double that of ankylosing spondylitis
Orphanet Journal of Rare Diseases (2021)
-
Giant axonal neuropathy with novel GAN pathogenic variant in a patient of consanguineous origin from Poonch Jammu and Kashmir-India
Molecular Biology Reports (2021)
-
Progressive pseudorheumatoid dysplasia: a rare childhood disease
Rheumatology International (2019)
-
Progressive pseudorheumatoid dysplasia with new-found gene mutation of Wntl inducible signaling pathway protein 3
Pediatric Rheumatology (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
