
Abstract
Multidrug resistance 3 (MDR3), encoded by the ATP-binding cassette, subfamily B, member 4 gene (ABCB4), localizes to the canalicular membrane of hepatocytes and translocates phosphatidylcholine from the inner leaflet to the outer leaflet of the canalicular membrane. Progressive familial intrahepatic cholestasis type 3 (PFIC3) is a rare hepatic disease caused by genetic mutations of ABCB4. In this study, we characterized 8 ABCB4 mutations found in PFIC3 patients, using in vitro molecular assays. First, we examined the transport activity of each mutant by measuring its ATPase activity using paclitaxel or phosphatidylcholine. Then, the pathogenic mechanisms by which these mutations affect MDR3 were examined through immunoblotting, cell surface biotinylation, and immunofluorescence. As a result, three ABCB4 mutants showed significantly reduced transport activity. Among these mutants, one mutation A364V, located in intracellular domains, markedly decreased MDR3 expression on the plasma membrane, while the others did not affect the expression. The expression of MDR3 on the plasma membrane and transport activity of A364V was rescued by a pharmacological chaperone, cyclosporin A. Our study provides the molecular mechanisms of ABCB4 mutations and may contribute to the understanding of PFIC3 pathogenesis and the development of a mutation-specific targeted treatment for PFIC3.
Similar content being viewed by others


Introduction
The liver is the one of the major organs responsible for the elimination of drugs and other xenobiotics from the body1. The ATP-binding cassette, subfamily B, member 4 gene (ABCB4), codes for human multidrug resistance 3 (MDR3)2. MDR3 is mainly expressed on the canalicular membrane of hepatocytes and plays an important role in the protection of the liver through translocation of phosphatidylcholine from the inner leaflet to the outer leaflet of the canalicular membrane2,3. Progressive familial intrahepatic cholestasis type 3 (PFIC3) is caused by genetic mutations in ABCB44. PFIC denotes a group of rare hepatic diseases, and onset varies from the neonatal period to early adulthood, depending on the specific type of disease. PFIC patients are usually candidates for liver transplantation before adulthood, due to complications such as hepatic failure, liver cirrhosis, and hepatocellular carcinoma5. Recently, new approaches for the treatment of hepatic diseases resulting from canalicular transport defects, as occurs in PFIC, have been suggested, including inducing an increase in the expression of functional protein by using synthetic farnesoid X receptor ligands or pharmacological chaperones6.
To date, several studies have been conducted to identify and functionally characterize the ABCB4 mutations found in PFIC3 patients, however, they do not include all the identified mutations7,8,9,10,11,12. A missense mutation found in PFIC3 patients, I541F, was shown to decrease transport activity through reduction of membrane MDR3 expression, which could be functionally rescued by low temperature or cyclosporin A8,9. Some genetic variations of ABCB4 are associated with other hepatobiliary diseases such as obstetric cholestasis, cholelithiasis, drug-induced liver injury, and primary biliary cirrhosis13,14,15,16,17,18. Recently, we performed functional characterization of the ABCB4 promoter variants and found that two common ABCB4 promoter haplotypes led to significantly decreased promoter activity. In addition, we identified nuclear factor-Y (NF-Y) as a possible transcriptional factor involved in the regulation of ABCB4 transcription19.
Previously, ABCB4 mutations were newly identified through direct sequencing, using genomic DNA samples from 68 PFIC3 patients7. The authors found 29 mutations in the coding region, including 23 missense, four nonsense, and two short insertion mutations. To evaluate the effect of these mutations on protein function, they mapped each variant on the predicted tertiary structure of MDR3, and found that 10 out of 29 mutations were located on transmembrane domains (TMs). In particular, TM 7, which may be required for the translocation of phosphatidylcholine, was most commonly affected. However, these studies did not perform a functional characterization of mutations.
In the present study, we selected 8 missense mutations of ABCB4 that were first reported by Degiorgio et al.7 and investigated the effect of each mutant on MDR3 transport activity or expression by using a range of in vitro assays such as a membrane vesicular adenosine triphosphatase (ATPase) assay, immunoblotting, surface biotinylation assay, and immunofluorescence. Our study provides the molecular pathogenic mechanisms of ABCB4 mutations, and our findings may contribute to the development of a new drug for the treatment of PFIC3 or other MDR3-deficiency related diseases.
Results
ABCB4 mutations examined in this study
We selected 8 missense mutations that were first reported by Degiorgio et al.7. Among the 29 ABCB4 mutations identified by Degiorgio et al.7, we excluded nonsense mutations and mutations that were also found in other MDR3-deficient phenotypes or those that affect amino acid residues, which are conserved in other ABC transporters, in particular, the MDR1 transporter, as these had already been investigated20,21,22. The 8 missense ABCB4 mutations examined in this study are listed in Table 1.
ABCB4 mutations affect transporter activity
To characterize the functional effects of the ABCB4 mutations, we examined the transport activity of each mutant by measuring the ATPase activity using paclitaxel or phosphatidylcholine, which are known MDR3 substrates23,24 (Fig. 1). To exclude ATPase activity by other endogenous ABC transporters, such as MDR1, values for transport activity were obtained by subtracting the uptake in empty-vector-transfected cells from that in cells transfected with ABCB4 wild type or mutant-bearing vectors, at each paclitaxel or phosphatidylcholine concentration. Inhibition of transport by verapamil, a known inhibitor of MDR323, confirmed that the transport was MDR3-mediated (Supplementary Fig. S1). As a result, the paclitaxel-induced ATPase activity was significantly reduced in membrane vesicles, which expressed three ABCB4 mutations: A364V, A737V, and A1193T (Fig. 1a). Table 2 shows the paclitaxel Vmax and Km values for the ABCB4 wild type or mutants. We observed that the average value of Vmax/Km for three ABCB4 mutants was significantly reduced compared to that of the ABCB4 wild type. This resulted from a reduced Vmax. MDR3 shows phosphatidylcholine-induced ATPase activity25; therefore, we additionally performed the ATPase assay using phosphatidylcholine, for the three mutants: A364V, A737V, and A1193T. We observed a significant decrease in their ATPase activities and this is consistent with the results obtained using paclitaxel (Fig. 1b).

Inside-out membrane vesicles were prepared after transfection of ABCB4 wild type or mutant-bearing plasmids into HEK-293T cells, and the ATPase activity induced by either paclitaxel (a) or phosphatidylcholine (b) was measured. The X-axis represents paclitaxel or phosphatidylcholine concentration. The Y-axis represents the amount of inorganic phosphate that was produced by the ATPase activity of MDR3. Data shown represent mean ± SD from five independent experiments and analyzed by one-way analysis of variance followed by Dunnett’s two-tailed test. *P < 0.05, **P < 0.01, ***P < 0.001 vs. wild type (WT).
The effect of mutations on the expression level of MDR3
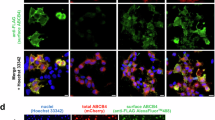
To examine the mechanisms through which ABCB4 mutations alter the transport activity, we investigated MDR3 expression levels of the mutants on the plasma membrane by immunofluorescence and cell surface biotinylation. Upon overexpression of wild type and mutant ABCB4 in HEK-293T cells, among the three ABCB4 mutants that showed decreased transport activities, A737V and A1193T was mainly expressed on the plasma membrane like ABCB4 wild type (Fig. 2). However, we observed that the MDR3 expression of A364V on the plasma membrane was decreased and a large of fraction of MDR3 was present in the endoplasmic reticulum or Golgi (Fig. 2). In addition, cell surface biotinylation demonstrated that A364V mutant had significantly decreased MDR3 expression on the plasma membrane by 30% as compared to that in the wild type vector transfected cells (Fig. 3). The surface expression level of A737V was comparable with that of the wild type, and that of A1193T seemed to decrease, but it was not statistically significant (Fig. 3), even though their transport activities were decreased. Based on these results, we suspected that A364V mutant had folding or trafficking defects. Proteins with trafficking defects usually undergo degradation by proteasome or lysosome, we examined this for A364V. We observed that MDR3 expression of A364V was significantly increased by 116% and 115%, after treatment with an inhibitor of proteasomal proteolysis, MG132 (Fig. 4a), and an inhibitor of lysosomal degradation, bafilomycin A1, respectively (Fig. 4b). While, MDR3 expression of the wild type vector transfected cells was not as much as that for A364V, expression was increased by 46% and 18% after treatment with MG132 and bafilomycin A1, respectively (Fig. 4). These data suggest that A364V is vulnerable to intracellular degradation, and the decreased MDR3 expression of this mutant might be a result of proteasomal or lysosomal degradation. For the five mutations that showed similar transport activities as compared to the wild type, their MDR3 expression levels were comparable with those of the wild type (Supplementary Fig. S2).

HEK-293T cells were transfected with ABCB4 wild type or mutant plasmids. Cells were fixed and permeabilized with methanol, and immunostained with anti-MDR3, and an endoplasmic reticulum maker BiP or a Golgi marker Gigantin antibodies. Scale bars, 10 μm.

(a) HEK-293T cells transfected with ABCB4 wild type or mutant plasmids, and surface biotinylated. CsA was treated at 10 μM for 24 hours. Absence of the cytosolic protein aldolase A in the biotinylated fraction confirms cell surface protein-specific labeling in each experiment. (b) Quantification of surface expression of MDR3. Bar graphs represent band density of surface biotinylated MDR3 compared to the wild type ABCB4 and error bars indicate the SD for three independent experiments. *P < 0.05, Student’s t test.

MDR3 expression was investigated after transfection with ABCB4 wild type or A364V mutant plasmids. Immunoblotting was performed after treatment with MG132 (a) or bafilomycin A1 (b). Data shown represent mean ± SD from three independent experiments and analyzed by one-way analysis of variance followed by Dunnett’s two-tailed test. **P < 0.01 vs. expression of A364V without MG132 or bafilomycin A1 treatment.
The effect of cyclosporin A on the expression and function of a trafficking-defective mutant
A previous study by Gautherot et al.9 showed that a trafficking-defective mutation of ABCB4 was functionally rescued by cyclosporin A. In their study, cyclosporin A produced a dose-dependent increase in MDR3 expression. Therefore, we tested whether a trafficking-defective mutant of ABCB4, A364V could be rescued by cyclosporin A. To examine it, we performed cell surface biotinylation after cells were treated with 10 μM cyclosporine A. As a result, we observed that cyclosporin A significantly increased MDR3 expression levels of A364V (Fig. 3). Our finding was similar to those of a previous study9. Then, to examine the effect of cyclosporin A on the transport activity of A364V, membrane vesicular ATPase assays were performed after treatment with cyclosporin A. We found that cyclosporin A increased the transport activity of this mutant significantly in the ATPase assays using paclitaxel or phosphatidylcholine (Fig. 5).

Membrane vesicular ATPase assays were performed using paclitaxel (a) or phosphatidylcholine (b) after treatment with CsA. Data shown represent mean ± SD from three independent experiments, and the P values for comparison between the results obtained before and after CsA treatment, were calculated using Student’s t test. *P < 0.05, ***P < 0.001 vs. transport activity of A364V without CsA treatment.
Discussion
Recently, screening of genomic DNA samples from 68 PFIC3 patients identified 29 mutations, 25 of which were novel7. In this study, authors examined the effect of each mutation on the protein tertiary structure using a three-dimensional model of MDR3. MDR3 consists of twelve TMs, six intracellular domains (ICDs), six extracellular loops (ECs), and a linker peptide connecting the N-terminal to the C-terminal transmembrane domain-nucleotide binding domain (TMD-NBD). Most mutations were located in the NBDs, TMs, or ICDs, which may be involved in substrate binding and translocation, coupling the energy from ATP hydrolysis to substrate transport, and the conformational change involved in substrate extrusion. Ten mutations were located in TMs, nine in ICDs, and eight in NBDs. However, functional characterization of each mutant was not performed in this previous study.
The present study was conducted to functionally characterize the ABCB4 mutations reported in the previous study by Degiorgio et al.7. Among 29 ABCB4 mutations, we selected 8 missense mutations. Then, we measured the transport activity of each mutant indirectly by ATPase assays using paclitaxel or phosphatidylcholine. we found that three mutants showed significantly decreased transport activities, whereas the other five mutants had similar transport activities compared to that of the wild type. These results are unexpected, however, two mutants, A250P and F357L, were initially identified as in cis with M630V and T775M, respectively7, suggesting that F357L or A250P is probably not sufficient to cause loss-of-function of MDR3 transport activity by itself. In addition, the pathogenicity of A286V, V475A and T715I may depend on the trans-associated mutation, as shown for NPHS2 R229Q26. In other words, these alleles lead to a disease phenotype only when it is associated specifically with certain ABCB4 mutations. For three mutants which exhibited decreased transport activities, we found that the reduced expression for A364V, located in the ICDs, might be due to proteasomal and lysosomal degradation. In the case of A737V and A1193T, the results from the cell surface biotinylation, immunoblotting, and immunofluorescence experiments indicated that decreased transport activity of these mutants did not result from the reduced MDR3 expression on the plasma membrane. Recently Gautherot et al.27 reported that two ABCB4 mutants, T34M and R47G, resulted in abnormal phosphorylation of its N-terminal domain, leading to decreased secretion of phosphatidylcholine while the targeting to the plasma membrane of those mutants was comparable with the wild type ABCB4. Further in vitro study would be necessary to elucidate the cause of decreased transport activity of A737V and A1193T.
Most of the ABC transporters are expressed on the plasma membrane of cells, where they transport various substrates. Serious human disease can occur if the transporters are not expressed on the cell surface properly due to folding defects28. The correction of folding defects caused by specific mutations, through alteration of intracellular environment or addition of pharmacological compounds, is a valuable approach for the treatment of some genetic diseases. For example, ∆F508 of cystic fibrosis conducting regulator (CFTR), the most common mutation in patients with cystic fibrosis, impairs CFTR folding29. Many studies reported that folding and trafficking defects of ∆F508 could be partially rescued by various modulators such as thapsigargin, sodium 4-phenylbutyrate, and aminoarylthiazoles or by overexpression of heat shock protein 70 or Golgi reassembly stacking proteins9,29,30,31. In the case of the other ABC transporters, a few approaches have focused on correcting trafficking-defective mutations. For example, a processing mutant of ABCB1 could also be rescued by substrates or modulators of MDR12,29. In particular, it has been shown that the pharmacological chaperone cyclosporin A, a known inhibitor of MDR1, increases the levels of total and matured protein of MDR1 by stabilizing the mutant protein32. Recently, two separate studies reported that an ABCB4 trafficking-defective mutation, I541F, could be rescued by low temperature or addition of cyclosporin A, which could correct the folding defects introduced by this mutation8,9. Similarly, in this study, we found that the trafficking defect induced by the mutation A364V could be rescued by cyclosporin A. We also found that the transport activity of this mutant was restored with cyclosporin A.
In some PFIC3 patients with mild symptoms, therapy with ursodeoxycholic acid could be effective5. However, most PFIC3 patients are ultimately candidates for liver transplantation. Although solid organ transplantation, including liver transplantation, is regarded as one of the most significant advances in medical science, it still faces various challenges such as organ unavailability, postoperative complications, and high costs33,34. In the case of pediatric liver transplantation, there are additional problems such as growth retardation due to prolonged steroid exposure, lower physical and psychosocial function after liver transplantation, graft loss, and preoperative malnutrition35,36. The identification of successful mechanism-based drugs for the treatment of PFIC3 is necessary to improve the quality of life in pediatric PFIC3 patients.
The present study has a limitation in that the transport activity or phosphatidylcholine flippase activity was not measured directly. ABC transporters, including MDR3, use ATP as an energy source to transport their substrates37; we measured this ATPase activity and thus, indirectly evaluated the transporter activities of wild type and mutant ABCB4. However, functional study of transporters using the ATPase assay has been reported previously38,39. In particular, Kwak et al.39 investigated the transport activity of MDR1 using two different methods, ATPase assays and direct transport assays using rhodamine 123 and reported that the results from the ATPase assays were comparable with those from the direct assays.
In conclusion, we have characterized a few ABCB4 mutations found in PFIC3 patients and found that the A364V, A737V, and A1193T mutants show significantly decreased transport activities, as measured indirectly using the ATPase assay. The mechanisms by which these ABCB4 mutants may decrease transport activities were determined in this study: A364V, a mutant in the ICDs, was trafficking-defective, while others did not affect MDR3 expression on the plasma membrane. We reported that both the expression of MDR3 on the plasma membrane and the transport activity of this trafficking-defective mutant could be rescued by a cyclosporin A. To our knowledge, this is the first study to functionally characterize a large number of ABCB4 mutations. The functional characterization of ABCB4 mutations could enable development of mutation-specific treatment regimens for PFIC3 patients in the future.
Methods
Construction of ABCB4 plasmids
To construct a plasmid containing a wild type ABCB4 gene, the BC_042531 vector was purchased (Thermo Fisher Scientific Inc., Waltham, MA, USA) and subcloned into the pcDNA3.1(+) vector. Mutant-bearing plasmids were produced using QuikChange® II Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA, USA). Nucleotide location numbers were assigned according to the ABCB4 mRNA sequence (GenBank accession number: NM_018849.2). Supplementary Table S1 lists primers used in this study.
Membrane vesicle preparation
Membrane vesicles were prepared according to a previously described method40. ABCB4 wild type or mutant-bearing plasmids were transfected into human embryonic kidney-293T (HEK-293T) cells using the Calcium Phosphate Transfection Kit (Life Technologies Corporation, Carlsbad, CA, USA). Forty-eight hours later, cells were harvested and subjected to nitrogen cavitation at 350 pounds per square inch, for 15 min. After transfer into a tube containing 0.5 M EDTA, centrifugation was performed twice at 1,900 × g, for 10 min each time, to obtain the supernatant. The membrane vesicle fractions were collected by sucrose density gradient centrifugation at 1,000,000 × g for 90 min and suspended in buffer containing 250 mM sucrose and 50 mM Tris.
ATPase assay
The vanadate-sensitive ATPase activity was measured using the SensoLyte® MG Phosphate Assay Kit (AnaSpec, Fremont, CA, USA) for paclitaxel or phosphatidylcholine transport, according to a previously described method39. First, membrane vesicles (4 μg/well) were prepared in 4 mM MgCl2, 5 mM 3-(N-morpholino)-propanesulfonic acid-Tris (pH 7.0), 4 mM ATP, and various concentrations of paclitaxel or phosphatidylcholine. For the inhibition assay, 150 or 400 μM verapamil was added to each well. Finally, absorbance was measured at 620 nm, using a microplate reader. ATPase activities were determined as the inorganic phosphate liberation, normalized by subtracting the activities of the mock vesicles, without the transfection of ABCB4 wild type or mutant-bearing plasmids.
Immunoblotting
The ABCB4 wild type or mutant-bearing plasmids were transfected into HEK-293T cells by using the Lipofectamine LTX and Plus reagents (Life Technologies). To examine the effect of MG132 or bafilomycin A1 on ABCB4 mutants, cells were treated with 10 μM MG132 or 0.1 mg/ml bafilomycin A1 24 h after transfection. To investigate the effect of cyclosporin A, cells were treated with 10 μM cyclosporin A, 6 h after transfection. Thirty or forty-eight hours after transfection, immunoblotting was performed using the primary antibodies mouse anti-MDR3 antibody (Abcam, Cambridge, UK) or goat anti-β-actin antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The intensity of each band was measured using ImageJ (National Institutes of Health, Bethesda, MD, USA).
Surface biotinylation assay
Biotinylation experiments were conducted using a Cell Surface Protein Isolation Kit (Thermo Fisher Scientific Inc.), performed according to the manufacturer’s protocol, using the cells obtained from transfection of the ABCB4 wild type or mutant-bearing plasmids into HEK-293T cells. To examine the effect of cyclosporin A on ABCB4 mutants, cells were treated with 10 μM cyclosporin A, 6 h after transfection. A rabbit polyclonal anti-Na+/K+ ATPase α-1 (EMD Millipore, Billerica, MA, USA) and a goat polyclonal anti-Aldolase A (Santa Cruz Biotechnology) antibodies were used as internal standards.
Immunofluorescence
For immunofluorescence, HEK-293T cells were grown on cover glasses, in a 24-well plate, and the ABCB4 wild type or mutant-bearing plasmids were transfected into HEK-293T cells. Twenty-four hours after transfection, cells were fixed and permeabilized with cold methanol for 10 min at −20 °C, following which blocking was performed. To detect MDR3, cells were incubated overnight at 4 °C with the anti-MDR3 and anti-BiP (Abcam) or anti-gagantin (Abcam) antibodies. After washing with PBS, Alexa Fluor® 488 rabbit anti-mouse IgG and Alexa Fluor® 594 goat anti-rabbit IgG (Life Technologies) were used as fluorophore-tagged secondary antibodies, and DAPI was used for nucleic acid staining. Confocal images were captured using a confocal laser scanning microscope, and the digital images were analyzed using an LSM Image Examiner (Carl Zeiss, Oberkochen, Germany).
Statistical analysis
Statistical analysis was performed using the GraphPad Prism 4.0 software package (GraphPad Software Inc., San Diego, CA, USA). The data shown in the ATPase assay, immunoblotting, and surface biotinylation assay represent mean ± SD from more than three separate experiments. P values for comparison between the results obtained before and after cyclosporin A treatment were calculated using Student’s t test. The other P values were calculated using one-way analysis of variance followed by Dunnett’s two-tailed test, and P values of less than 0.05 were considered statistically significant.
Additional Information
How to cite this article: Park, H. J. et al. Functional characterization of ABCB4 mutations found in progressive familial intrahepatic cholestasis type 3. Sci. Rep. 6, 26872; doi: 10.1038/srep26872 (2016).
References
Shitara, Y., Horie, T. & Sugiyama, Y. Transporters as a determinant of drug clearance and tissue distribution. European journal of pharmaceutical sciences: official journal of the European Federation for Pharmaceutical Sciences 27, 425–446, 10.1016/j.ejps.2005.12.003 (2006).
Oude Elferink, R. P. & Paulusma, C. C. Function and pathophysiological importance of ABCB4 (MDR3 P-glycoprotein). Pflugers Archiv: European journal of physiology 453, 601–610, 10.1007/s00424-006-0062-9 (2007).
Ruetz, S. & Gros, P. Phosphatidylcholine translocase: a physiological role for the mdr2 gene. Cell 77, 1071–1081 (1994).
Deleuze, J. F. et al. Defect of multidrug-resistance 3 gene expression in a subtype of progressive familial intrahepatic cholestasis. Hepatology 23, 904–908, 10.1002/hep.510230435 (1996).
Davit-Spraul, A., Gonzales, E., Baussan, C. & Jacquemin, E. Progressive familial intrahepatic cholestasis. Orphanet journal of rare diseases 4, 1, 10.1186/1750-1172-4-1 (2009).
Stapelbroek, J. M., van Erpecum, K. J., Klomp, L. W. & Houwen, R. H. Liver disease associated with canalicular transport defects: current and future therapies. Journal of hepatology 52, 258–271, 10.1016/j.jhep.2009.11.012 (2010).
Degiorgio, D. et al. Molecular characterization and structural implications of 25 new ABCB4 mutations in progressive familial intrahepatic cholestasis type 3 (PFIC3). European journal of human genetics: EJHG 15, 1230–1238, 10.1038/sj.ejhg.5201908 (2007).
Delaunay, J. L. et al. A missense mutation in ABCB4 gene involved in progressive familial intrahepatic cholestasis type 3 leads to a folding defect that can be rescued by low temperature. Hepatology 49, 1218–1227, 10.1002/hep.22775 (2009).
Gautherot, J. et al. Effects of cellular, chemical, and pharmacological chaperones on the rescue of a trafficking-defective mutant of the ATP-binding cassette transporter proteins ABCB1/ABCB4 . The Journal of biological chemistry 287, 5070–5078, 10.1074/jbc.M111.275438 (2012).
Gordo-Gilart, R. et al. Heterozygous ABCB4 mutations in children with cholestatic liver disease. Liver international: official journal of the International Association for the Study of the Liver 36, 258–267, 10.1111/liv.12910 (2016).
Delaunay, J. L. et al. A functional classification of ABCB4 variations causing progressive familial intrahepatic cholestasis type 3. Hepatology, 10.1002/hep.28300 (2015).
Degiorgio, D. et al. Two ABCB4 point mutations of strategic NBD-motifs do not prevent protein targeting to the plasma membrane but promote MDR3 dysfunction. European journal of human genetics: EJHG 22, 633–639, 10.1038/ejhg.2013.214 (2014).
Eloranta, M. L. et al. Multidrug resistance 3 gene mutation 1712delT and estrogen receptor alpha gene polymorphisms in Finnish women with obstetric cholestasis. European journal of obstetrics, gynecology, and reproductive biology 105, 132–135 (2002).
Rosmorduc, O. et al. ABCB4 gene mutation-associated cholelithiasis in adults. Gastroenterology 125, 452–459 (2003).
Pauli-Magnus, C. et al. Sequence analysis of bile salt export pump (ABCB11) and multidrug resistance p-glycoprotein 3 (ABCB4, MDR3) in patients with intrahepatic cholestasis of pregnancy. Pharmacogenetics 14, 91–102 (2004).
Lang, C. et al. Mutations and polymorphisms in the bile salt export pump and the multidrug resistance protein 3 associated with drug-induced liver injury. Pharmacogenetics and genomics 17, 47–60, 10.1097/01.fpc.0000230418.28091.76 (2007).
Ohishi, Y. et al. Single-nucleotide polymorphism analysis of the multidrug resistance protein 3 gene for the detection of clinical progression in Japanese patients with primary biliary cirrhosis. Hepatology 48, 853–862, 10.1002/hep.22382 (2008).
Yoshikado, T. et al. Itraconazole-induced cholestasis: involvement of the inhibition of bile canalicular phospholipid translocator MDR3/ABCB4 . Molecular pharmacology 79, 241–250, 10.1124/mol.110.067256 (2011).
Jang, G. H., Kim, T. H., Choe, Y., Ham, A. & Choi, J. H. Functional characterization of genetic variations in the MDR3 promoter. Biochemical and biophysical research communications 430, 1312–1318, 10.1016/j.bbrc.2012.12.041 (2013).
Loo, T. W., Bartlett, M. C. & Clarke, D. M. Processing mutations located throughout the human multidrug resistance P-glycoprotein disrupt interactions between the nucleotide binding domains. The Journal of biological chemistry 279, 38395–38401, 10.1074/jbc.M405623200 (2004).
Loo, T. W. & Clarke, D. M. Identification of residues in the drug-binding domain of human P-glycoprotein. Analysis of transmembrane segment 11 by cysteine-scanning mutagenesis and inhibition by dibromobimane. The Journal of biological chemistry 274, 35388–35392 (1999).
Loo, T. W. & Clarke, D. M. Location of the rhodamine-binding site in the human multidrug resistance P-glycoprotein. The Journal of biological chemistry 277, 44332–44338, 10.1074/jbc.M208433200 (2002).
Smith, A. J. et al. MDR3 P-glycoprotein, a phosphatidylcholine translocase, transports several cytotoxic drugs and directly interacts with drugs as judged by interference with nucleotide trapping. The Journal of biological chemistry 275, 23530–23539, 10.1074/jbc.M909002199 (2000).
Andress, E. J. et al. Molecular mechanistic explanation for the spectrum of cholestatic disease caused by the S320F variant of ABCB4 . Hepatology 59, 1921–1931,10.1002/hep.26970 (2014).
Ellinger, P., Kluth, M., Stindt, J., Smits, S. H. & Schmitt, L. Detergent screening and purification of the human liver ABC transporters BSEP (ABCB11) and MDR3 (ABCB4) expressed in the yeast Pichia pastoris. PloS one 8, e60620, 10.1371/journal.pone.0060620 (2013).
Tory, K. et al. Mutation-dependent recessive inheritance of NPHS2-associated steroid-resistant nephrotic syndrome. Nature genetics 46, 299–304, 10.1038/ng.2898 (2014).
Gautherot, J. et al. Phosphorylation of ABCB4 impacts its function: insights from disease-causing mutations. Hepatology 60, 610–621, 10.1002/hep.27170 (2014).
Loo, T. W., Bartlett, M. C. & Clarke, D. M. Rescue of folding defects in ABC transporters using pharmacological chaperones. Journal of bioenergetics and biomembranes 37, 501–507, 10.1007/s10863-005-9499-3 (2005).
Lukacs, G. L. & Verkman, A. S. CFTR: folding, misfolding and correcting the DeltaF508 conformational defect. Trends in molecular medicine 18, 81–91, 10.1016/j.molmed.2011.10.003 (2012).
Gee, H. Y., Noh, S. H., Tang, B. L., Kim, K. H. & Lee, M. G. Rescue of DeltaF508-CFTR trafficking via a GRASP-dependent unconventional secretion pathway. Cell 146, 746–760, 10.1016/j.cell.2011.07.021 (2011).
Okiyoneda, T. et al. Mechanism-based corrector combination restores DeltaF508-CFTR folding and function. Nature chemical biology 9, 444–454, 10.1038/nchembio.1253 (2013).
Loo, T. W. & Clarke, D. M. Correction of defective protein kinesis of human P-glycoprotein mutants by substrates and modulators. The Journal of biological chemistry 272, 709–712 (1997).
Soler, X., Myo Bui, C. C., Aronson, L. A. & Saied, A. S. Current issues in pediatric liver transplantation. International anesthesiology clinics 50, 54–65, 10.1097/AIA.0b013e31826e3438 (2012).
Axelrod, D. A. Economic and financial outcomes in transplantation: whose dime is it anyway? Current opinion in organ transplantation 18, 222–228, 10.1097/MOT.0b013e32835f0757 (2013).
Balistreri, W. F. Growth and development of a new subspecialty: pediatric hepatology. Hepatology 58, 458–476, 10.1002/hep.26580 (2013).
Kelly, D. A. et al. Long-term medical management of the pediatric patient after liver transplantation: 2013 practice guideline by the American Association for the Study of Liver Diseases and the American Society of Transplantation. Liver transplantation: official publication of the American Association for the Study of Liver Diseases and the International Liver Transplantation Society 19, 798–825, 10.1002/lt.23697 (2013).
Juranka, P. F., Zastawny, R. L. & Ling, V. P-glycoprotein: multidrug-resistance and a superfamily of membrane-associated transport proteins. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 3, 2583–2592 (1989).
Kluth, M. et al. A mutation within the extended X loop abolished substrate-induced ATPase activity of the human liver ATP-binding cassette (ABC) transporter MDR3. The Journal of biological chemistry 290, 4896–4907, 10.1074/jbc.M114.588566 (2015).
Kwak, J. O. et al. Selective inhibition of MDR1 (ABCB1) by HM30181 increases oral bioavailability and therapeutic efficacy of paclitaxel. European journal of pharmacology 627, 92–98, 10.1016/j.ejphar.2009.11.008 (2010).
Kim, W. J. et al. A nonsynonymous variation in MRP2/ABCC2 is associated with neurological adverse drug reactions of carbamazepine in patients with epilepsy. Pharmacogenetics and genomics 20, 249–256, 10.1097/FPC.0b013e328338073a (2010).
Acknowledgements
This study was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2010-0027945) and the Ministry of Education (2010-0003262) to J.H.C. This study was also supported by a faculty research grant of Yonsei University College of Medicine (6-2015-0175) to H.Y.G.
Author information
Authors and Affiliations
Contributions
H.Y.G. and J.H.C. designed the study. H.J.P., T.H.K., S.W.K., S.H.N., K.J.C., C.C., E.Y.K. and Y.G.C. carried out the experiments and analyzed data. H.J.P., T.H.K., H.Y.G. and J.H.C. wrote the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Park, H., Kim, T., Kim, S. et al. Functional characterization of ABCB4 mutations found in progressive familial intrahepatic cholestasis type 3. Sci Rep 6, 26872 (2016). https://doi.org/10.1038/srep26872
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep26872
This article is cited by
-
Clinical and genetic characterization of pediatric patients with progressive familial intrahepatic cholestasis type 3 (PFIC3): identification of 14 novel ABCB4 variants and review of the literatures
Orphanet Journal of Rare Diseases (2022)
-
Case report: progressive familial intrahepatic cholestasis type 3 with compound heterozygous ABCB4 variants diagnosed 15 years after liver transplantation
BMC Medical Genetics (2020)
-
Structural analogues of roscovitine rescue the intracellular traffic and the function of ER-retained ABCB4 variants in cell models
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
