
Abstract
The underlying mechanisms responsible for the general increase in species richness from temperate regions to the tropics remain equivocal. Many hypotheses have been proposed to explain this astonishing pattern but additional empirical studies are needed to shed light on the drivers at work. Here we reconstruct the evolutionary history of the cosmopolitan diving beetle subfamily Colymbetinae, the majority of which are found in the Northern hemisphere, hence exhibiting an inversed latitudinal diversity gradient. We reconstructed a dated phylogeny using 12 genes, to investigate the biogeographical history and diversification dynamics in the Colymbetinae. We aimed to identify the role that phylogenetic niche conservatism plays in the inversed diversification pattern seen in this group. Our results suggest that Colymbetinae originated in temperate climates, which supports the hypothesis that their distribution is the result of an ancestral adaptation to temperate environmental conditions rather than tropical origins, and that temperate niche conservatism can generate and/or maintain inverse latitudinal diversity gradients.
Similar content being viewed by others


Introduction
The increase in species diversity with decreasing latitude, or high tropical species richness, is an ecological pattern that has long intrigued naturalists1. Best known as the latitudinal diversity gradient (LDG), numerous empirical studies have investigated the relative contribution of macroevolutionary drivers such as diversification rate dynamics and biogeographical history2,3,4,5,6,7,8. It was suggested, for example, that LDGs resulted from a reduced number of lineages evolving adaptations to cold and dry climates with strong seasonal oscillations typical of non-tropical areas9. Tropical regions are also viewed as both cradles of diversity that continuously generate new species, and museums that harbour ancient species persisting throughout geological times10,11. Several hypotheses attempted to summarize the available evidence explaining the origin and evolution of the LDG11,12,13,14,15. The ‘out of the tropics’ (OTT) hypothesis suggests that tropical regions harbour a high number of species eventually expanding their distributional ranges towards the poles and/or high altitudes13,14. However, according to the tropical niche conservatism (TNC16,17) and the tropical conservation hypothesis (TCH - Zanne et al. 201418), dispersal out of the tropics towards temperate zones is limited by the tendency of lineages to retain their tropical niche-related traits throughout the speciation process. These hypotheses both focus on the historical biogeography and/or physiological niche conservatism limiting the expansion of clades out of the tropics.
In contrast to niche conservatism, other hypotheses are based on the assumption that tropical environments are geologically older, have occupied larger areas and have been more stable over time (as summarized in18). The time for speciation hypothesis (TFS) predicts that the time required for speciation to build up diversity in a region explains the high species diversity in the tropics in contrast to the geologically younger and less stable temperate environments19. The Centre of Origin (COO) hypothesis suggests that lineage diversification took place in older and historically larger tropical regions, resulting in higher species richness compared to the younger temperate regions20. Other hypotheses invoke higher rates of tropical speciation versus higher rates of temperate extinction21. Recent studies suggest that major environmental change (e.g. glaciations or volcanism) might have supported LDGs2,22. Instead of focussing on niche conservatism, these hypotheses aim at explaining LDGs as results of migration, speciation and extinction rates. While these hypotheses generally focus on explaining high tropical biodiversity, similar mechanisms might also serve as explanations as to why some taxa are more diverse in temperate regions18.
These hypotheses are not necessarily mutually exclusive, but rather provide a framework for a comprehensive explanation of complex and partly idiosyncratic processes15. For instance, Wiens et al.23 suggest that the higher tropical diversity of Old World Ranidae frogs can neither be accounted for by accelerated tropical speciation nor by higher temperate extinction, but rather by relatively recent colonization of the temperate regions. This highlights the importance of comprehensive biogeographical analyses in a phylogenetic context, which have been conducted in many case studies on LDGs.
However, few comprehensive studies have investigated the opposite case, namely the inverse Latitudinal Diversity Gradient (iLDG). Taxa presenting an iLDG exhibit relatively high species richness in temperate areas compared to the tropics. This pattern was first shown for a number of Holarctic bird families24, later for parasitic ichneumonid wasps25,26,27,28, marine benthic algae29, aphids30, Emydidae turtles19, shallow-water molluscs (as summarized in31), Holarctic tree frogs32, New World Lampropeltini snakes18, and cosmopolitan Poaceae grasses9. The macroevolutionary and ecological factors during lineage diversification that led to iLDGs remain little explored to date. Thus, there is a need for additional empirical studies based on comprehensive, large scale datasets to study the triggers of iLDG18,31. Molecular phylogenetics and recent methodological developments provide tools for a more accurate inference of diversification dynamics and biogeographical histories. These can then be used to empirically test the origins and causes of heterogeneous clade diversity distributions2,23.
Here, we study Colymbetinae diving beetles, which are comprised of 140 described species occurring in a wide variety of aquatic habitats on all continents but Antarctica. Colymbetinae show a marked iLDG, being most diverse in the Holarctic region with decreasing species diversity towards the Equator, but with a conspicuous equatorial “peak” in the Eastern Old World tropics (Figure 1). They are almost completely absent from tropical lowlands but occur in tropical montane and alpine habitats, and in the southern temperate regions33,34,35.

Species richness is declining towards the equator (red dotted line). The many species endemic to single mountain tops in the Eastern Old World cause an extratropical diversity peak. Species data was compiled from the world catalogue of Dytiscidae by36. Map (from Wikipedia) and species richness graphs were created using Microsoft Power Point 2010.
We aim to (i) infer the temporal and spatial origins of the group using fossil-based calibration and likelihood-based historical biogeography methods, (ii) test for possible diversification rate shifts as an explanation for the iLDG, (iii) calculate the extent of phylogenetic niche conservatism within the lineage and (iv) identify the putative mechanisms explaining the iLDG as well as species ecological preferences during lineage diversification.
Material and Methods
Taxon sampling
We used 87 Colymbetinae species (Table S1) mostly preserved in 96% ethanol, representing 62% of the ca. 140 described species and all extant genera and subgenera36. We also included 17 species of Agabinae, the sister-group of Colymbetinae37 as outgroups. We rooted the tree with Batrachomatus daemeli (Sharp 1882) (Matinae), a related subfamily clearly outside Colymbetinae plus Agabinae.
Molecular biology
Genomic DNA was extracted and purified using the Qiagen DNeasy tissue kit (Qiagen, Hilden, Germany). DNA samples were then used to amplify five mitochondrial gene fragments: 12S (352bp in the combined dataset), 16S (797bp), cytochrome b (326bp), cytochrome oxidase 1 (1,336bp) and cytochrome oxidase 2 (527bp)) in addition to seven nuclear gene fragments: 18S (1,951bp), arginine kinase (674bp), enolase (677bp), wingless (496bp), elongation factor 1 alpha (553bp), histone 4 (159bp) and carbamoyl-phosphate synthetase 2 (815bp) using standard procedures following Balke et al.33 and Tänzler et al.38. PCR products were purified and processed for sequencing, using BigDye v3.1 (ABI, Darmstadt, Germany). Assembling and editing of the sequences were performed using Sequencher 4.10.1 (Gene Codes, Ann Arbor, MI, USA). All protein coding genes were then imported into Mesquite v2.75 (Maddison and Maddison 201539) in order to translate to amino acids and screen for anomalies. Gene alignments were concatenated using Geneious R8 (Biomatters, http://www.geneious.com). All sequences are available online (BOLD Process IDs ZSMDB032-15 - ZSMDB138-16, Genbank Accession Numbers KJ638060 - 607997).
Phylogenetic inference
Phylogenetic inferences were conducted using maximum parsimony (MP), maximum likelihood (ML) and Bayesian inference (BI). The MP analyses were carried out using the TNT v1.1 program40 applying the Tree Ratchet, Tree Fusing and Tree Drifting Sectorial Searches and 1000 Jackknife replicates (P = 36) to assess the stability of nodes. ML analyses were conducted using RAxMLGUI v.0.9341 with the autoFC command for automatic determination of sufficient fast bootstrap repeats. A partitioning scheme with protein coding genes partitioned into 1st, 2nd, and 3rd codon positions, as well as corresponding models of substitutions were obtained using PartitionFinder v1.1.142 (Table S2). BI analyses of the combined dataset were performed on the workstations and the cluster of the entomology department of the Bavarian State Collection of Zoology with MrBayes v.3.243. The analyses started with a random tree and consisted of two runs each with four chains (Markov Chain Monte Carlo, one cold and three incrementally heated) running for 50 million generations and sampling every 1000 cycles. A conservative burn-in of 25% was applied after checking that convergence was reached through the split frequencies of each run in Tracer 1.544 (Table S3). The resulting trees were then combined to calculate a 50% majority rule consensus topology mapping the posterior probabilities (PP) of each node.
Estimation of divergence times
We estimated absolute divergence times using a combination of calibration sets in order to improve the robustness of the estimates. At first, we applied three substitution rates calculated for the 3’ end of COI gene in recent studies focussing on Coleoptera lineages (as in ref. 45). We used an interval comprising these three distinct rate values, instead of performing multiple independent analyses applying each rate individually (recent examples are38,45). We applied the mean substitution rate from a dated phylogeny of carabid beetles (r = 0.0145; r is the substitutions per site per million years per lineage, subs/s/Myr/l), based on multiple geological and fossil records. We then used the divergence rate calculated for Tenebrionidae beetles (r = 0.0177 subs/s/Myr/l). Finally, we used the rate calculated for the Rhantus suturalis clade (r = 0.0195 subs/s/Myr/l). We applied the introduced interval (0.0145–0.0195 subs/s/Myr/l) to specify a normal and a uniform distribution on the ucld.mean in BEAUTi v1.5.444. By incorporating the means of the three rates, we took into account the substitution rate heterogeneity across beetle lineages. For the very same analysis, three different fossil calibrations were implemented in BEAUTi. To constrain the root of the tree, we applied a uniform distribution (Lower = 0, Upper = 155), restricting the root not to be older than 155 million years ago (Ma). This is the approximate age of the oldest known dytiscid fossil †Palaeodytes guttata. We used a uniform distribution for two fossils, namely †Colymbetes aemulus Heer from the Miocene and †Agabus rathbuni Scudder from the Oligocene. The two fossils were respectively used to enforce a minimum constraint on the crown groups of the genus Colymbetes and the subfamily Agabinae. The youngest estimate of the geological strata they were embedded in was used as a minimum bound for each calibration (11.6 for †Colymbetes aemulus and 37.2 for †Agabus rathbuni), whereas the maximum bounds were set by the age of the defined root (155). The Tree Model was set to a birth-death model in an analysis consisting of 50 million generations sampled every 5000 generations. BEAST v.1.746 analyses were conducted on the workstations and the cluster of the entomology department of the Bavarian State Collection of Zoology. As a starting tree for the BEAST analysis, the BI topology was used in order to optimize the search of optimal ages by starting at high likelihood in topology space. The convergence of the runs was investigated using statistics in Tracer inclusive ESS values. A conservative burn-in of 25% was applied after checking the log-likelihood curves and a maximum credibility tree with median ages and their 95% highest posterior density (HPD) were subsequently generated using TreeAnnotator 1.7.446.
Hypothesis testing overview
In order to test the hypothesis of iLDGs being a result of temperate niche conservatism, we needed to test for congruence between the biogeographical origins and the historical climatic preferences for Colymbetinae. To do this, we gathered information on their modern distributions and performed historical biogeographical analyses to test for their ancestral ranges. To test for Phylogenetic Niche Conservatism (PNC), we performed ancestral state reconstructions for temperature and precipitation. We then used the biogeographical results in combination with the ancestral climate reconstructions to ultimately test for evidence of PNC triggering the iLDG.
Historical biogeography analyses
We used the R package BioGeoBEARS to test between different biogeographical model-based approaches47,48. As BioGeoBEARS requires an ultrametric tree, we used the BEAST chronogram from which all outgroup species (including Agabinae) were pruned using Mesquite v.2.75. Information on their current distributions was taken from the world catalogue of Dytiscidae36. Seven biogeographical regions were defined, namely Nearctic (A), Neotropics (B), Western Palaearctic (C), Afrotropics (D), Eastern Palaearctic (West of the Ural mountains including Asia and the Oriental region) (E), Australia (F) and the Pacific region (G). In an effort to reduce the large amount of computation time, the Eastern Palaearctic was merged with the South Asian region, as only a few species inhabit the latter area. We compared three main models: the Dispersal-Extinction-Cladogenesis model (DEC49,50), the DIVA-like model51 and the BAYAREA-like model52 to infer the ancestral ranges and colonization history of the Colymbetinae. They were implemented with and without the jump dispersal parameter (J), where ranges can change to include new areas during speciation48, for a total of six distinct models (DEC, DEC + J, DIVA-like, DIVA-like + J, BAYAREA-like, BAYAREA-like + J). We first ran a model in which dispersal between regions was not penalized (all rates are 1.0) and later designed and ran other models with varying dispersal probabilities reflecting past climatic and geological events53,54 . These included (i) adjacency matrix constraints, (ii) varying dispersal probabilities over time and (iii) a combination of both. After comparing models using AICc weights, we ran additional tests to explore what effect varying stringency on long-distance dispersal (LDD) had on model selection. In the second set of analyses, the LDD events were penalized differently in each model, in particular for LDD events between the Neotropical and Afrotropical or Pacific regions, as well as dispersal between the Palaearctic and the Nearctic regions (Table 1). We chose four time slices (0–5, 5–30, 30–45 and 45–70 MYA) for all models to account for major climatic and geological events throughout the entire evolution of the group. To reduce the set of possible regions, the maximum number of ancestral regions for each node was set to four.
Climate Niche Modelling
Distribution data of the species used was obtained from the entomological collection of the Bavarian State Collection of Zoology (ZSM), the Global Biodiversity Information Facility (GBIF – www.gbif.de) and the catalogue of Dytiscidae36. Georeferenced coordinates were obtained using www.gpso.de/maps. The number of occurrence points ranged from 10 to 127. For species with very restricted ranges (endemic to small islands or isolated mountain tops) 10 points were evenly scattered across the spatial extents of these geographic features (see examples of the same procedure in refs 55 or 56). Current climatic conditions data (~1950–2000) on 19 BioClim variables was downloaded at a resolution of 30 arc-seconds (ca. 1km) (http://www.worldclim.org). We used a subset of the BioClim variables to capture information about general attributes of the climate that are known to be relevant for dytiscids (temperature and precipitation) (Table 2)57,58.
We used Maxent v.3.3.3k59,60 to predict habitat suitability given our environmental variables and georeferenced records. Maxent has previously been shown to work well with small number of occurrence points (minimum 10)55,56,61. We used the default settings for model training in Maxent. We evaluated model performance using AUC (Area Under the Receiver Operator Curve). For AUC model evaluation we withheld 20% of the samples for testing. AUC values range from 0.5, which are no better than a random coin flip, to 1.0 for optimal predictive accuracy of presence versus absence. We used the R packages raster, maps, rgdal, maptools, sp and dismo for this process62. The environmental niche modelling showed that BIO1, BIO2 and BIO4 contributed the most to the model predictions among the different biogeographical regions (Figure S1). The results of the niche models for each species can be found in the supplementary information. The calculated AUC values ranged from 0.95 to 0.53, 90% of the species had values > 0.5 (Table S4). These niche models allowed us to obtain an estimate of suitable environmental conditions for each species.
Predicted Niche Occupancy (PNO) and Niche overlap
To identify whether clades can rapidly change their environmental preferences, we calculated the mean for each species from the range of values in our predicted niche occupancy (PNO) profiles. The PNO takes the likelihood surface from the Maxent output and relates it to the raster input layers to calculate a species probability of occurrence at a given environmental value. We cropped the BioClim layers and models produced by Maxent according to each species’ spatial extent in order to accommodate limited computational memory. PNO profiles were first constructed in the R package phyloclim63. One hundred random samples were then drawn from the PNO profile and then the mean was calculated as in Evans et al.56. The mean calculated from the PNOs was used in further ancestral state reconstructions. PNOs for each BioClim variable used were then merged and binned by 10 (1°C/10mm). Niche overlap was computed in phyloclim from the PNO profiles, using the summary statistics Schoener’s D and Hellinger Distances as in Warren et al.64. As we were interested in looking at the niche overlap between biogeographical regions, species were combined according to their geographic region and a mean PNO was calculated, which gave an estimate of the niche overlap between regions.
Testing for Phylogenetic Niche Conservatism
In order to test for PNC, we calculated the environmental mean for the given BioClim variables for each species in phyloclim. We tested for PNC using recently proposed methods65,66. In order to test for evidence of a phylogenetic signal in the bioclimatic variables used here, we first calculated Blomberg’s K values67. We then tested for PNC amongst 3 evolutionary models of trait evolution: Brownian Motion (BM; genetic drift), White Noise (WN; no phylogenetic signal) and Ornstein-Uhlenbeck (OU; stabilizing selection)68. Support of the WN model indicates that traits are evolving independently without phylogenetic signal, whereas the BM and OU models indicate that traits evolve with an underlying phylogenetic pattern. Selection of the OU over the BM model indicates more stabilizing selection over drift.
Diversification rate analyses
To investigate potential diversification rate shifts in the evolution of Colymbetinae while taking into account the missing taxon sampling in our phylogenetic reconstruction we used the function ‘bd.shifts.optim’ in the R package TreePar (as in ref. 45). We used the BEAST chronogram as input and fitted several birth-death models including 0 (null model, constant-rate model) to multiple diversification rate shifts during the evolution of the group. We then tested different models ranging from 0 to 5 rate shifts. All analyses were carried out with the following non-default settings: taxon sampling 87/140, start = 0, end = 56.0 and grid = 0.1 Myr for a fine-scale estimation of rate shifts. We finally calculated AICc scores and computed Likelihood Ratio Tests (LRT) to select the best-fit between the different models allowing incrementally more shifts during the evolution of the clade.
We also used Bayesian Analysis of Macroevolutionary Mixtures (BAMM71) and its R implementation BAMMtools72 to identify clades with higher or lower speciation rates in the Colymbetinae phylogeny. We performed multiple BAMM runs on the BEAST chronogram, with five million generations of Markov Chain Monte Carlo (MCMC) sampling per run and sampling evolutionary parameters every 1000 generations. We assessed the convergence of the different BAMM runs by computing effective sample sizes of log-likelihoods, number of processes and evolutionary rate parameters using the package CODA73. We reconstructed marginal distributions of net diversification rates for each branch in the BEAST chronogram using the posterior distribution of evolutionary parameters sampled by the reversible jump MCMC algorithm in BAMM.
In order to address the question that the observed iLDG pattern might be the result of an association between latitude and diversification rates, we compared their net-diversification rates using the geographical state speciation and extinction model (GoeSSE)6,74,75 in the R package diversitree76. We incorporated sampling fraction where 72% tropical, 65% temperate, and 100% of species occurring in both regions were included in our phylogeny. A model in which diversification was independent of geographic state using AIC model weights was rejected. Next we compared AIC model weights between the full 7 parameter model and one with 6 where between-region speciation was not estimated (sAB = 0). We conducted both a BI and ML estimation for the 6 free parameters (speciation in area A and B; extinction in area A and B; dispersal in area A and B). For the Bayesian inference we set the tuning parameter w to the distance between the 5% and 95% quantiles from the marginal distribution of a preliminary run, then ran 1e + 05 MCMC generations, sampling every 100th generation, to obtain an estimate of the parameters.
Results
Phylogenetic relationships
The aligned dataset comprised 8,663 bp. Protein coding genes in general showed no insertions or deletions (indels), but we found an amino acid deletion (3 indels) in the CAD alignment in Rhantus orbignyi. The CAD sequences were therefore realigned and translated into AA-sequences for quality control, but no obvious pseudogenes were recognized. The 18S and 16S rRNA genes showed several single or multi base indels between regions of high nucleotide conservation. No indels were found in the 12S rRNA gene. Within the Colymbetinae, most of the internal nodes were supported by bootstrap values > 80, or posterior probabilities > 0.95. With the exception of the clade containing Melanodytes pustulatus, the phylogenies inferred by the model based approaches were highly compatible with the MP analysis (Supplemental Material A, Figure S2). Colymbetinae was always recovered monophyletic with strong support. The cosmopolitan genus Rhantus was polyphyletic with strong support in all analyses.
Divergence time estimation
The results of the BEAST analysis and details for each node (HPD intervals) are shown in Figure S3, median ages of each node are indicated in Fig. 2. After checking convergence of the runs, the molecular dating approach of the model incorporating substitution rates calculated for different beetle lineages of the COI gene under the uniform prior distribution was selected (Table S5). However, both divergence time estimations using normal and uniform distributions were mostly compatible. The optimal model recovered an origin of the Colymbetinae at the Palaeocene-Eocene border, approximately 56 million years ago (Ma) (HPD 68.6–44.6 Ma). Our divergence time estimates of the “southern” species of the Rhantus suturalis clade (HPD 3.6–8.0 Ma) (R. bacchusi, R. ekari, R. dani, R. supranubicus, R. suturalis) were mostly congruent with the findings of Balke et al.33 and Toussaint et al.35 (2.7–4.3 versus 1.5–4.7 Ma respectively).

We applied the BI topology as a starting tree in the BEAST analysis. The BioGeoBEARS approach was used to calculate the most probable ancestral biogeographical region at each node. 95% HPD intervals are indicated as bars at each node. Paleo climatic conditions (adapted by77) are illustrated for the Lower Eocene, Oligocene and Miocene in the lower part of the figure. Maps (from Wikipedia and the work of 77) were adapted, redrawn and colorized using Microsoft Power Point 2010.
Historical biogeography and diversification rate analysis (LTT)
The results of the BioGeoBEARS analysis are shown in Fig. 2 and Table 3. The model receiving the strongest support from the different analyses performed in BioGeoBEARS was the time constrained model 3 DEC + J. The ancestral areas of extant Colymbetinae diving beetles were reconstructed to be the warm temperate to temperate Eastern Palaearctic and Australian regions. Our results suggest that extant Colymbetinae persisted in the Eastern Palaearctic for 20 to 25 million years (Myr), and that modern lineages are a result of continuous colonization events from these ancestral areas. The results of the diversification rate analyses are shown in Table S6 and Figure S4. Neither the TreePar nor BAMM analysis detected a significant shift in diversification rate, therefore supporting a constant rate of diversification throughout the evolution of the group.
The results of the GeoSSE show that the full model was favoured over the model where the geographic range was independent of diversification. The 6 parameter model was favoured over the full 7 parameter model where between-region speciation was estimated (Table S7).
The results of the Bayesian analyses indicate that the different parameters were distinct (not overlapping 95% credible intervals) except for extinction. Net-diversification between tropical and temperate species also overlapped between their 95% credible intervals (Figure S5).
Predicted Niche Occupancy (PNO) and Niche overlap
We used the results from the PNO profiles to calculate the niche overlap between biogeographical regions. The Pacific region stood out as having low niche overlap with the other regions, except for BIO3 and BIO4, which showed a different pattern. These two variables show low niche overlap between the Palaearctic and the Pacific, Neotropics, Afrotopics and Australian regions. The combined niche overlap showed a similar pattern with low overlap between the Pacific, Neotropics, Afrotropics and Australian regions with the Palaearctic regions (Figures S6 and S7).
Testing for Phylogenetic Niche Conservatism (PNC)
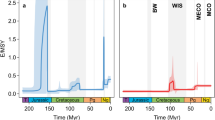
For all bioclimatic variables, Blomberg’s K-value showed significant phylogenetic signal (p < 0.05) (Table S8). Among models of trait evolution, the OU model was selected as the best fitting model given the dAICc statistic for all bioclimatic variables (Table S9). We used the OU model, instead of the BM model, to reconstruct ancestral climate preferences using a modified version of the phytools function ‘contMap’ (Fig. 3).

Discussion
While many animal and plant taxa show increasing species richness towards the Equator, Colymbetinae exhibit a different pattern–they are most diverse in temperate areas (Fig. 1). In this study, we investigate if this pattern was a recent switch or if the group historically originated and diversified in temperate zones. The results of the biogeographical reconstruction indicate an Eastern Palaearctic and Australian origin of Colymbetinae at the Palaeocene-Eocene boundary, approximately 56 million years ago (Fig. 2). Since at least the late Jurassic, the climate of these areas were mostly temperate or warm temperate (Fig. 2)77. Thus, the biogeographical reconstruction infers temperate or warm temperate climate niches for the ancestral taxa. This niche was also inferred by the climate ancestral state reconstructions (Fig. 3). However, the inferred ancestral state is only one piece of information towards understanding phylogenetic niche conservatism, as we also need to know how conserved these traits are. For instance, a rapidly shifting ancestral climate preference indicates that this trait is not conserved and may have little to do with limiting where a taxon can reside. In contrast, if the preference is conserved beyond that expected under descent with modification, the ancestral state’s lability may strongly influence the evolutionary history of a clade and its diversity and distribution patterns (e.g.65,66,78). Instead, the ancestral state may not be representative for later climate preferences in a clade in which these preferences shift rapidly. The reason ancestral niche conservatism is important is because of the exponential accumulation of lineages through time even under a constant speciation model79,80. So the under PNC older nodes have a propagative effect, disproportionally contributing to current niche preferences. In order to test our hypothesis that the iLDG pattern in Colymbetinae is the result of ancestral temperate origin in combination with niche conservatism, we tested for PNC using standard methods as in Wiens et al.66 and Pyron et al.65. This is supported by the results of our analyses, as a significant amount of phylogenetic signal with additional support of the OU model is generally interpreted as an indication of trait conservatism65,66,78. Thus, we infer the ancestral area with its associated climatic conditions in combination with PNC as the underlying factors largely responsible for the observed iLDG pattern in Colymbetinae.
The importance of niche conservatism in various aspects of ecology, evolution and diversification was recently reviewed by Wiens et al.66. Biologists have perhaps disproportionally focused on examples of adaptation and (rapid) change, while the tendency among species to retain similar traits over long periods of time may be a factor just as important to understand diversity-distribution patterns. Wiens et al.66 broadly defined niche conservatism as “the retention of niche-related ecological traits over time”, applying whenever phylogenetic signal is stronger than expected under a pure BM model. A narrower concept of phylogenetic niche conservatism proposes that closely related species are ecologically more similar than would be expected under BM inheritance of traits with genetic drift (Losos 200881). The preference of the OU model over the BM model for Colymbetinae serves as partial evidence for phylogenetic niche conservatism. However, observing a pattern of phylogenetic niche conservatism may have several explanations, such as evolutionary constraints from various factors (physiological, host choice, developmental and genetic), stabilizing selection or “phylogenetic inertia” (Cooper et al. 201082). Our biogeographical reconstruction highlights multiple colonization and long distance dispersal events into the Indomalayan region out of the Palaearctic (Rhantus pacificus and R. suturalis clade). These results also indicate that the Neotropical clade containing Rhantus calidus originated as a result of colonization out of the Australian region via LDD or transitions through Antarctica. Transitions throughout Antarctica and Australia into other temperate regions appear plausible for highly vagile organisms because climatic conditions were temperate to cold temperate until the onset of the Oligocene83. Colymbetinae qualify as a highly vagile taxa; for example, the Neotropical species Rhantus signatus has colonized the island of Tristan da Cunha over 3500 km into the Atlantic34, suggesting long distance dispersal seems plausible. According to our analyses, the Afrotropical region was colonized twice by species of the Rhantus bohlei clade from the Nearctic and Neotropical regions (Fig. 2). The results indicate that Rhantus capensis colonized the Afrotropical region via long distance dispersal out of the Neotropics, whereas the remaining Afrotropical members of the clade are a result of long distance dispersal out of the Nearctic region. When present in tropical regions, Colymbetinae are mostly restricted to high altitude habitats and subalpine biomes33,34. Forty species can be found in the well sampled Indomalayan-Australasian archipelago, including thirty endemics, resulting in a peak of diversity in this otherwise temperate group. Most of these species are restricted to single high valleys or mountain tops, which are the sole regions in tropical latitudes in which low temperatures predominate84. The niche overlap results demonstrate that these habitats are roughly similar to the temperate regions. Adaptations to temperate climates would facilitate the colonization of tropical cool highlands. Similar processes might also explain the origin of the tropical lineages within the Agabinae, the sister group of Colymbetinae. While being mostly restricted to temperate regions, the genus Agabus also inhabits Afrotropical mountain ranges including the Cape region of South Africa with Mediterranean climate and has a small radiation in the high altitude areas of Ethiopia. The genus Platynectes inhabits the montane regions of the Andes. In the latter case, as well as in lineages of the Oriental and Australian Platynectes, lowland lineages are also known, but the phylogenetic relationships and their ancestral origin is not currently known at present37.
Our results suggest that the physiological niche conservatism hypotheses for high species diversity in the tropics can be equally applicable to explaining high temperate species richness9,18. Explanations for high species diversity in temperate regions, for groups that originated in temperate zones could also be explained by the hypotheses we discussed attempting to explain tropical biodiversity gradients based on their geological age and physiological niche adaptation of species. The ancestral biogeographical and environmental preference reconstructions results indicate that niche conservatism was largely responsible for restricting the distribution of the Colymbetinae.
While the vast majority of Earth’s biodiversity exhibits a gradient of species richness declining with increasing latitude3, others manifest the opposite pattern of biodiversity. However, the appearance of this inverted pattern of species diversity is likely to be the consequence of multiple interleaved processes of evolution. For Colymbetinae diving beetles, the most important processes inferred here are captured by the general theories of the centre-of-origin20, the time-for-speciation-effect19 and niche conservatism (reviewed in ref. 66). The generality of these conclusions remains to be tested by forthcoming empirical studies on other groups of organisms showing a clear iLDG pattern. But as these processes and effects are of a very general nature we would not be surprised if they turn out to be mechanisms governing iLDG patterns also in a range of other groups.
Additional Information
How to cite this article: Morinière, J. et al. Phylogenetic niche conservatism explains an inverse latitudinal diversity gradient in freshwater arthropods. Sci. Rep. 6, 26340; doi: 10.1038/srep26340 (2016).
References
Bates, H. W. The Naturalist on the River Amazons: A Record of Adventures, Habits of Animals, Sketches of Brazilian and Indian Life, and Aspects of Nature Under the Equator, During Eleven Years of Travel. With a Memoir of the Author, by Edward Clodd. J. Murray (1892).
Condamine, F. L., Sperling, F. A., Wahlberg, N., Rasplus, J. Y. & Kergoat, G. J. What causes latitudinal gradients in species diversity?Evolutionary processes and ecological constraints on swallowtail biodiversity. Ecol. Let. 15(3), 267–277 (2012).
Hillebrand, H. On the generality of the latitudinal diversity gradient. Am. Nat. 163(2), 192–211 (2004).
Mittelbach, G. G. et al. Evolution and the latitudinal diversity gradient: speciation, extinction and biogeography. Ecol. Let. 10(4), 315–331 (2007).
Pianka, E. R. Latitudinal gradients in species diversity: a review of concepts. Am. Nat. 910(100) 33–46 (1966).
Rolland, J., Condamine, F. L., Jiguet, F. & Morlon, H. Faster speciation and reduced extinction in the tropics contribute to the mammalian latitudinal diversity gradient. PLoS Biol. 12(1), e1001775 (2014).
Weir, J. T. & Schluter, D. The latitudinal gradient in recent speciation and extinction rates of birds and mammals. Science. 315(5818), 1574–1576 (2007).
Willig, M. R., Kaufman, D. M. & Stevens, R. D. Latitudinal gradients of biodiversity: pattern, process, scale, and synthesis. Annu. Rev. Ecol. Evol. Syst. 34, 273–309 (2003).
Visser, V., Clayton, W. D., Simpson, D. A., Freckleton, R. P. & Osborne, C. P. Mechanisms driving an unusual latitudinal diversity gradient for grasses. Global Ecol. Biogeogr. 23(1), 61–75 (2014).
McKenna, D. D. & Farrell, B. D. Tropical forests are both evolutionary cradles and museums of leaf beetle diversity. PNAS. 103(29), 10947–10951 (2006).
Stebbins, G. L. Flowering plants: evolution above the species level. (E. Arnold, London 1974).
Chown, S. L. & Gaston, K. J. Areas, cradles and museums: the latitudinal gradient in species richness. Trends Ecol. Evol. 15(8), 311–315 (2000).
Fischer, A. G. Latitudinal variations in organic diversity. Evolution 14(1), 64–81 (1960).
Jablonski, D., Roy, K. & Valentine, J. W. Out of the tropics: evolutionary dynamics of the latitudinal diversity gradient. Science. 314(5796), 102–106 (2006).
Kerkhoff, A. J., Moriarty, P. E. & Weiser, M. D. The latitudinal species richness gradient in New World woody angiosperms is consistent with the tropical conservatism hypothesis. PNAS. 111(22), 8125–8130 (2014).
Crisp, M. D. & Cook, L. G. Phylogenetic niche conservatism: what are the underlying evolutionary and ecological causes? New Phytol 196(3), 681–694 (2012).
Wiens, J. J. & Donoghue, M. J. Historical biogeography, ecology and species richness. Trends Ecol, Evol. 19(12), 639–644 (2004).
Zanne, A. E. et al. Three keys to the radiation of angiosperms into freezing environments. Nature 506(7486), 89–92 (2014).
Stephens, P. R. & Wiens, J. J. Explaining species richness from continents to communities: the time‐for‐speciation effect in emydid turtles. Am. Nat. 161(1), 112–128 (2003).
Ricklefs, R. E. & Schluter, D. Species diversity in ecological communities: historical and geographical perspectives. (University of Chicago Press, 1993).
Rohde, K. Latitudinal gradients in species diversity: the search for the primary cause. 514–527 (Oikos, 1992).
Mannion, P. D., Upchurch, P., Benson, R. B. & Goswami, A. The latitudinal biodiversity gradient through deep time. Trends Ecol. Evol. 29(1), 42–50 (2014).
Wiens, J. J., Sukumaran, J., Pyron, R. A. & Brown, R. M. Evolutionary and biogeographic origins of high tropical diversity in Old World frogs (Ranidae). Evolution 63(5), 1217–1231 (2009).
Cook, R. E. Variation in species density of North American birds. Syst. Biol. 18(1), 63–84 (1969).
Gauld, I. D. Latitudinal gradients in ichneumonid species richness in Australia. Ecol. Entomol 11(2), 155–161 (1986).
Hawkins, B. A. Pattern and process in host-parasitoid interactions. (Cambridge University Press, 1994).
Janzen, D. H. The peak in North American ichneumonid species richness lies between 38 degrees and 42 degrees N. Ecology 532–537 (1981).
Owen, D. F. & Owen, J. Species diversity in temperate and tropical Ichneumonidae. Nature. 249(5457), 583–584 (1974).
Santelices, B. Phytogeographic characterization of the temperate coast of Pacific South America. Phycologia 19(1), 1–12 (1980).
Dixon, A. F. G., Kindlmann, P., Leps, J. & Holman, J. Why there are so few species of aphids, especially in the tropics. Am. Nat. 129, 580–592 (1987).
Kindlmann, P., Schödelbauerová, I. & Dixon, A. F. Inverse latitudinal gradients in species diversity. Scaling biodiversity. 246–257 (Cambridge University Press, 2007).
Smith, S. A., Stephens, P. R. & Wiens, J. J. Replicate patterns of species richness, historical biogeography, and phylogeny in Holarctic treefrogs. Evolution. 59(11), 2433–2450 (2005).
Balke, M. et al. New Guinea highland origin of a widespread arthropod supertramp. Proc. Soc. London,Ser. B. 276, 2359–2367 (2009).
Morinière, J. et al. Anisomeriini diving beetles–an Atlantic–Pacific Island disjunction on Tristan da Cunha and Robinson Crusoe Island, Juan Fernández? Cladistics 31(2), 166–176 (2015).
Toussaint, E. F., Sagata, K., Surbakti, S., Hendrich, L. & Balke, M. Australasian sky islands act as a diversity pump facilitating peripheral speciation and complex reversal from narrow endemic to widespread ecological supertramp. Ecol. Evol. 3(4), 1031–1049 (2013).
Nilsson, A. N. A world catalogue of the family Dytiscidae, or the diving beetles (Coleoptera, Adephaga). (Umeå, 2013).
Ribera, I., Nilsson, A. N. & Vogler, A. P. Phylogeny and historical biogeography of Agabinae diving beetles (Coleoptera) inferred from mitochondrial DNA sequences. Mol. Phylogenet Evol. 30, 545–562 (2004).
Tänzler, R., Toussaint, E. F., Suhardjono, Y. R., Balke, M. & Riedel, A. Multiple transgressions of Wallace’s Line explain diversity of flightless Trigonopterus weevils on Bali. Proc. Soc. London, Ser. B. 281(1782), 20132528 (2014).
Maddison, W. P. & Maddison, D. R. Mesquite: a modular system for evolutionary analysis. Version 3.04, http://mesquiteproject.org. (2015).
Goloboff, P. A., Farris, J. S. & Nixon, K. C. TNT, a free program for phylogenetic analysis. Cladistics. 24(5), 774–786 (2008).
Silvestro, D. & Michalak, I. raxmlGUI: a graphical front-end for RAxML. Org. Divers. Evol. 12(4), 335–337 (2012).
Lanfear, R., Calcott, B., Ho, S. Y. & Guindon, S. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 29(6), 1695–1701 (2012).
Ronquist, F. et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61(3), 539–542 (2012).
Drummond, A. J. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7(1), 214 (2007).
Toussaint, E. F. et al. The towering orogeny of New Guinea as a trigger for arthropod megadiversity. Nat. comm. doi: 10.1038/ncomms5001 (2014).
Drummond, A. J., Suchard, M. A., Xie, D. & Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29(8), 1969–1973 (2012).
Matzke, N. J. BioGeoBEARS: BioGeography with Bayesian (and likelihood) evolutionary analysis in R Scripts. R package ‘BioGeoBEARS’. URL:http://CRAN.R-project.org/package=BioGeoBEARS (2013).
Matzke, N. J. Model selection in historical biogeography reveals that founder-event speciation is a crucial process in island clades. Syst. Biol. 63(6), 951–970 (2014).
Ree, R. H. Detecting the historical signature of key innovations using stochastic models of character evolution and cladogenesis. Evolution 59(2), 257–265 (2005).
Ree, R. H. & Smith, S. A. Maximum likelihood inference of geographic range evolution by dispersal, local extinction, and cladogenesis. Syst. Biol. 57(1), 4–14 (2008).
Ronquist, F. Dispersal-vicariance analysis: a new approach to the quantification of historical biogeography. Syst. Biol. 46(1), 195–203 (1997).
Landis, M. J., Matzke, N. J., Moore, B. R. & Huelsenbeck, J. P. Bayesian analysis of biogeography when the number of areas is large. Syst. Biol. doi: 10.1093/sysbio/syt040 (2013).
Miller, K. G. et al. The Phanerozoic record of global sea-level change. Science. 310(5752), 1293–1298 (2005).
Seton, M., Müller, R. D., Zahirovic, S., Gaina, C., Torsvik, T. & Shephard, G. et al. Global continental and ocean basin reconstructions since 200Ma. Earth Sci. Rev. 113(3), 212–270 (2012).
Hernandez, P. A., Graham, C. H., Master, L. L. & Albert, D. L. The effect of sample size and species characteristics on performance of different species distribution modeling methods. Ecography 29(5), 773–785 (2006).
Evans, M. E., Smith, S. A., Flynn, R. S. & Donoghue, M. J. Climate, Niche Evolution, and Diversification of the “Bird‐Cage” Evening Primroses (Oenothera, Sections Anogra and Kleinia). Am. Nat. 173(2), 225–240 (2009).
Hawlitschek, O., Porch, N., Hendrich, L. & Balke, M. Ecological niche modelling and nDNA sequencing support a new, morphologically cryptic beetle species unveiled by DNA barcoding. PLoS One. 6(2), 1–14 (2011).
Hawlitschek, O. et al. Pleistocene climate change promoted rapid diversification of aquatic invertebrates in Southeast Australia. BMC Evol. Biol. 12(1), 142 (2012).
Elith, J. et al. A statistical explanation of Maxent for ecologists. Divers. Distrib. 17(1), 43–57 (2011).
Phillips, S. J. & Dudík, M. Modeling of species distributions with Maxent: new extensions and a comprehensive evaluation. Ecography. 31(2), 161–175 (2008).
Pearson, R. G., Raxworthy, C. J., Nakamura, M. & Townsend Peterson, A. Predicting species distributions from small numbers of occurrence records: a test case using cryptic geckos in Madagascar. J. Biogeogr. 34(1), 102–117 (2007).
Hijmans, R. J. & van Etten, J. raster: Geographic analysis and modeling with raster data. R package ‘raster’. URL:http://cran.r-project.org/web/packages/raster.pdf (2012).
Heibl, C., Calenge, C. & Heibl, M. C. R package ‘phyloclim’. URL: http://cran.r-project.org/web/packages/phyloclim/index.html (2013).
Warren, D. L., Glor, R. E. & Turelli, M. ENMTools: a toolbox for comparative studies of environmental niche models. Ecography 33(3), 607–611 (2010).
Pyron, R. A., Costa, G. C., Patten, M. A. & Burbrink, F. T. Phylogenetic niche conservatism and the evolutionary basis of ecological speciation. Biol.Rev. 90(4), 1248–1262 (2014).
Wiens, J. J. et al. Niche conservatism as an emerging principle in ecology and conservation biology. Ecol. Let. 13(10), 1310–1324 (2010).
Blomberg, S. P., Garland Jr, T. & Ives, A. R. Testing for phylogenetic signal in comparative data: behavioral traits are more labile. Evolution 57(4), 717–745 (2003).
Butler, M. A. & King, A. A. Phylogenetic comparative analysis: a modeling approach for adaptive evolution. Am. Nat. 164(6), 683–695 (2004).
Felsenstein, J. Maximum likelihood and minimum-steps methods for estimating evolutionary trees from data on discrete characters. Syst. Zool. 240–249 (1973).
Kozak, K. H. & Wiens, J. J. Accelerated rates of climatic‐niche evolution underlie rapid species diversification. Ecol . Let. 13(11), 1378–1389 (2010a).
Rabosky, D. L. Automatic detection of key innovations, rate shifts, and diversity-dependence on phylogenetic trees. PLoS One. 9(2), e89543 (2014a).
Rabosky, D. L. et al. BAMMtools: an R package for the analysis of evolutionary dynamics on phylogenetic trees. Methods Ecol. Evol. 5(7), 701–707 (2014b).
Plummer, M., Best, N., Cowles, K. & Vines, K. CODA: Convergence diagnosis and output analysis for MCMC. R news. 6(1), 7–11 (2006).
Goldberg, E. E., Lancaster, L. T. & Ree, R. H. Phylogenetic inference of reciprocal effects between geographic range evolution and diversification. Syst. Biol. 60(4), 451–465 (2011).
Rolland, J., Condamine, F. L., Beeravolu, C. R., Jiguet, F. & Morlon, H. Dispersal is a major driver of the latitudinal diversity gradient of Carnivora. Global Ecol. Biogeogr. 24(9), 1059–1071 (2015).
FitzJohn, R. G. Diversitree: comparative phylogenetic analyses of diversification in R. Methods Ecol . Evol. 3(6), 1084–1092 (2012).
Boucot, A. J., Chen, X. & Scotese, C. R. Phanerozoic paleoclimate: An atlas of lithologic indicators of climate. Society of Economic Paleontologists and Mineralogists (2013).
Kozak, K. H. & Wiens, J. J. Niche Conservatism Drives Elevational Diversity Patterns in Appalachian Salamanders. Am. Nat. 176(1), 40–55 (2010b)
Nee, S. Birth-death models in macroevolution. Annu. Rev. Ecol. Evol. Syst. 37, 1–17 (2006).
Ricklefs, R. E. Estimating diversification rates from phylogenetic information. Trends Ecol. Evol. 22(11), 601–610 (2007).
Losos, J. B. Phylogenetic niche conservatism, phylogenetic signal and the relationship between phylogenetic relatedness and ecological similarity among species. Ecology letters 11(10), 995–1003 (2008).
Cooper, N., Jetz, W. & Freckleton, R. P. Phylogenetic comparative approaches for studying niche conservatism. Journal of evolutionary biology. 23(12), 2529–2539 (2010).
Almeida, E. A., Pie, M. R., Brady, S. G. & Danforth, B. N. Biogeography and diversification of colletid bees (Hymenoptera: Colletidae): emerging patterns from the southern end of the world. J. Biogeogr. 39(3), 526–544 (2012).
Sarmiento, G. Ecological features of climate in high tropical mountains. High altitude tropical biogeography, 11–45 (Oxord University Press, 1986).
Author information
Authors and Affiliations
Contributions
M.B. conceived the study, J.M. and M.B. designed the study, J.M. and M.B. performed molecular labwork and analysed the data with E.F.A.T., M.H.V.D. and M.B., J.M. and E.F.A.T. wrote the first draft of the MS and all other co-authors (M.H.V.D., O.H., J.B., M.C.M., L.H. and I.R.) contributed substantially. Some specimen and/or DNA-extracts were provided by M.B., I.R., L.H. and M.C.M.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Morinière, J., Van Dam, M., Hawlitschek, O. et al. Phylogenetic niche conservatism explains an inverse latitudinal diversity gradient in freshwater arthropods. Sci Rep 6, 26340 (2016). https://doi.org/10.1038/srep26340
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep26340
This article is cited by
-
On the relative importance of landscape variables to plant diversity and phylogenetic community structure on uninhabited islands, South Korea
Landscape Ecology (2021)
-
Closely related species show species-specific environmental responses and different spatial conservation needs: Prionailurus cats in the Indian subcontinent
Scientific Reports (2020)
-
Spatial distribution pattern of taxonomic and phylogenetic diversity of woody flora in Andaman and Nicobar Islands, India
Forest Ecosystems (2019)
-
The Evolution of Climatic Niches and its Role in Shaping Diversity Patterns in Diprotodontid Marsupials
Journal of Mammalian Evolution (2019)
-
Role of way of life, latitude, elevation and climate on the richness and distribution of orchid species
Biodiversity and Conservation (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
