
Abstract
Interstitial cystitis/bladder pain syndrome (IC) is associated with significant morbidity, yet underlying mechanisms and diagnostic biomarkers remain unknown. Pelvic organs exhibit neural crosstalk by convergence of visceral sensory pathways, and rodent studies demonstrate distinct bacterial pain phenotypes, suggesting that the microbiome modulates pelvic pain in IC. Stool samples were obtained from female IC patients and healthy controls, and symptom severity was determined by questionnaire. Operational taxonomic units (OTUs) were identified by16S rDNA sequence analysis. Machine learning by Extended Random Forest (ERF) identified OTUs associated with symptom scores. Quantitative PCR of stool DNA with species-specific primer pairs demonstrated significantly reduced levels of E. sinensis, C. aerofaciens, F. prausnitzii, O. splanchnicus, and L. longoviformis in microbiota of IC patients. These species, deficient in IC pelvic pain (DIPP), were further evaluated by Receiver-operator characteristic (ROC) analyses, and DIPP species emerged as potential IC biomarkers. Stool metabolomic studies identified glyceraldehyde as significantly elevated in IC. Metabolomic pathway analysis identified lipid pathways, consistent with predicted metagenome functionality. Together, these findings suggest that DIPP species and metabolites may serve as candidates for novel IC biomarkers in stool. Functional changes in the IC microbiome may also serve as therapeutic targets for treating chronic pelvic pain.
Similar content being viewed by others

Introduction
Patients with urologic chronic pelvic pain syndromes (UCPPS) suffer chronic pelvic pain and dramatically lower quality of life, yet diagnostic markers and effective therapies remain elusive for these costly syndromes1. Interstitial cystitis/bladder pain syndrome (IC/BPS or IC) is a debilitating UCPPS condition of pelvic pain and voiding dysfunction that afflicts as many as 8 million U.S. women where depression is a common co-morbidity2,3,4,5. IC etiology remains unknown, but urothelial lesions and lamina propria mast cells are associated with patient symptoms6,7,8. HPA axis dysfunction has been implicated in female and male patients and cats with feline IC9,10,11,12,13, and thus may be common among UCPPS, but mechanisms that integrate pelvic pain, voiding dysfunction, HPA activity, and depression are lacking. Because of these long-standing questions, NIDDK has launched its flagship urology study, the Multi-Disciplinary Approaches to Chronic Pelvic Pain (MAPP) Research Network with the most comprehensive studies to date of UCPPS including clinical characterization and epidemiologic and mechanistic studies14,15.
The microbiome is increasingly appreciated as driving diverse physiologic processes in both health and disease16,17. With the notable exception of C. difficile colitis, early studies implicated pathogenic dysbioses primarily at the phylum and genus levels, but more recent studies identify individual species associated with disease. Moreover, individual species within the microbiome have recently been associated with driving disease through altered innate metabolism or altered pharmaceutical metabolism18. Despite these advances, it remains unclear whether microbiota contribute to pain syndromes generally and to UCPPS in particular. Consistent with the recent finding of a normal female urinary microbiome19, the MAPP Network is also exploring UCPPS microbiota with a focus on the urinary microbiome. MAPP Network studies have identified an association between fungi and symptom flares, but a signature UCPPS urinary microbiome has not yet been found that is associated with baseline symptoms. As a result, there is currently no understanding of the UCPPS gut microbiome that may modulate urinary sensation.
We recently defined bacterial pain phenotypes in murine UTI, where transient bladder infection can elicit a spectrum of pain responses, including chronic pelvic pain20,21. Because pelvic pain is subject to modulation via organ crosstalk22,23,24,25,26, we hypothesized that GI and/or reproductive tract microbiota modulate IC pelvic pain. Here, we characterized microbiota of IC patients and healthy controls, including MAPP participants. We observed significant differences between microbiota of patients and controls, including specific species, thereby potentially identifying novel biomarkers and potential therapeutic targets for IC.
Results
Clinical samples obtained from IC patients and controls
Female healthy controls and patients were recruited from MAPP participants at Northwestern and through separate advertising to yield a cohort of average age of 35 years (see Table 1). Individuals receiving antibiotic therapy within the prior 3 months were excluded from the study. Study participants exhibited racial skewing such that all IC patients were Caucasian, and three IC patients were on disability due to IC. Patients had a mean duration of symptoms of 9 years and exhibited significantly higher combined GUPI scores and significantly higher GUPI subscores. Patients also reported higher incidence of conditions previously associated with IC including allergies/respiratory tract disorders and gastrointestinal disease as well as urologic/gynecologic surgeries that represent a potential risk factor for pelvic pain.
IC patients have altered stool microbiome
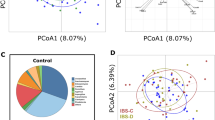
DNA was purified from stool and vaginal samples, and 16S sequences of the V3–V5 hypervariable regions were amplified and subjected to deep sequencing27. Sequencing reads were subsequently assigned to operational taxonomic units (OTUs) using QIIME with a threshold for homology of 97%. Preliminary characterization performed on a pilot cohort revealed no trend of segregation of vaginal samples, whereas similar analyses identified stool as a potential site where microbiota of IC patients segregated from controls (Fig. 1A,B). As a result, subsequent analyses focused on the stool microbiome (Fig. 2; n = 34). Several phyla and bacterial orders were differentially represented in controls and IC patients, however these differences were restricted to less abundant phyla (Fig. 2A). For example, the three orders of Chloroflexi (Herpetosiphonales, Ktedonobacterales, and Sphaerobacterales) were virtually absent from healthy controls, yet their presence in IC represented only 0.02% of reads (Fig. 2B and data not shown). Nonetheless, these differences suggest that the IC microbiome may be functionally distinct from the normal adult fecal microbiome. Moreover, these differences at the family level suggest differences at the genera level that are associated with symptom severity.

(A) Principle component analysis of pilot cohort suggested segregation of stool microbiome among IC patients and controls (P = 0.055). (B) Principle component analyses of vaginal samples from pilot cohort did not segregate by diagnoses (P = 0.326).

(A) Comparison of phyla abundance between IC patients and controls as determined by 16S reads. (B) Comparison of bacterial order abundance shows numerous orders that are differentially abundant in controls (red) and IC (green; n = 34). Phyla are indicated by colors in the outer ring. Data were compared to M5RNA using a maximum e-value of 1e-20, a minimum identity of 97%, and a minimum alignment length of 15 bp.
IC is a symptom-based diagnosis, so we sought to identify features of the IC microbiome specifically associated with patient symptoms. To identify specific taxa that discriminate IC stool microbiota from controls in association with symptom score, individual taxa were evaluated for significance as features using Extended Random Forest (ERF). Briefly, ERF uses the Random Forest (RF) algorithm followed by stability analysis and feature permutation to determine all relevant taxa. RF shows excellent performance with this type of complex data where the number of variables is orders of magnitude larger than the number of patient samples28. ERF analyses of stool operational taxonomic units (OTUs) revealed 26 features with a likelihood of relevance exceeding 70%. In ERF, the best performing shadow (or “noise”) feature is set at 50% and every OTU above is likely to contribute to discriminating IC microbiome from controls (Fig. 3). Significant OTUs were identified among diverse phyla including Actinobacteria, Firmicutes, and Proteobacteria. More importantly, these findings support the possibility that individual species reside within the identified taxa that are significantly altered in IC.

Box plot of likelihood of relevance for fecal OTUs by (vertical) and importance (x axis; n = 34).
Fecal species altered in IC
To determine whether individual species are differentially represented in IC patient stool samples relative to controls, quantitative PCR was performed with primers designed against the top taxa ranked by ERF. DNA was purified from all stool samples of patients and controls. Species-specific primers were designed against unique regions of 16S rDNA for the top three ranked OTUs in the pilot cohort (Odoribacter, Faecalibacterium, and Lachnospiriceae) and the top six ranked OTUs in the ranking for the combined cohort (Faecalibacterium, Colinsella, Eggerthella, Lachnospiriceae, Lactonifactor and Roseburia). Because several of these OTUs were classified only at the genus level, species-specific primers were designed against all available 16S sequences in Genbank within a genus. Ultimately, specific primers were designed for O. splanchnicus, O. denticanus, O. laneus, F. pruasnitzii, Lachnospiriceae pectinoschiz, C. aerofaciens, C. intestinalis, C. stercoris, E. lenta, E. sinensis, Lactonifactor longiviformis, and R. intestinalis. We omitted analyses of genera for which many species were reported (e.g., Clostridia). Each primer pair was then tested for amplicon specificity by melt curve analysis of amplification products using a small panel of stool DNAs purified from healthy controls. Using qPCR of stool DNA from IC patients and controls, five significant species were identified: O. splanchnicus, F. prausnitzii, C. aerofaciens, E. sinensis, and L. longoviformis (Fig. 4A, P < 0.05). Interestingly, these species were deficient among IC patients and thus were dubbed “deficient in interstitial cystitis pelvic pain” (DIPP). Connectivity analyses were also performed by Maximal Information Coefficient (MIC) analysis to identify co-varying OTUs29. C. aerofaciens, O. splanchnicus, E. sinensis, and F. prausnitzii were found to co-vary, therefore suggesting that DIPP species are members of a community that is altered in IC (Fig. 4B).

(A) qPCR was performed on healthy control (n = 10) and IC stool DNA (n = 16) using species-specific primers (±SEM). Species with significant, differential abundance are indicated (*P < 0.05). (B) Interactive network diagram integrating qPCR findings with 16S data by MIC analysis to detect relationships between features. (C) ROC curves for C. aerofaciens qPCR data.
DIPP species and stool metabolites as candidate IC biomarkers
To complement the microbiome analyses, we performed metabolomic studies of stool from healthy controls and IC patients by GC-MS to quantify distinct metabolites30. As most samples were initially stored directly in preservative, analysis was necessarily confined to a subset of samples from patients where a portion of unadulterated stool was retained (n = 15). Nonetheless, metabolomic data were subsequently analyzed for significant pathways and biomarker relevance using MetaboAnalyst 3.031,32,33. Pathway analysis identified multiple candidate pathways, including lipid metabolism pathways for linoleic acid, butanoic acid, and arachidonic acid (Fig. 5A). Arachidonic acid signaling was identified as significant (P < 0.05).

(A) ROC analysis (left panel) and box-and-whisker plot (right panel) identified glyceraldehyde as significantly elevated in IC and a candidate biomarker (AUM = 0.98, P<0.05). (B) Pathway analysis of stool metabolites in MetaboAnalyst 3.0 identified arachidonic acid signaling as significantly altered in IC (P<0.05). (C) PICRUSt metagenome analyses identified fatty acid biosynthesis, homologous recombination and nicotinate/nicotinamide metabolism as significantly different.
Metabolites and DIPP species were evaluated as potential biomarkers for IC. Receiver operator characteristics (ROC) were determined for each metabolite by classic univariate analysis in MetaboAnalyst, where a marker was considered significant with area under the curve (AUC) in excess of 0.70. The metabolite with the best predictive value was identified as glyceraldehyde, a metabolite elevated in IC stool (Fig. 5B and Table 2; HMDB01051, AUC 0.98). Among the bacterial DIPP species, ROC analyses revealed C. aerofaciens, E. sinensis, F. prausnitzii, and O. splanchnicus as exceeding AUC > 0.70 (Table 2). Together, these findings suggest that candidate stool-based biomarkers for IC may be identified among both DIPP species and metabolites.
Discussion
UCPPS remain diagnoses of exclusion due to a lack of biomarkers. Here, we used a combination of microbiome and metabolome analyses as a strategy to identify novel stool-based biomarkers for IC. Individual IC patients can be exquisitely sensitive to specific comestibles, where dietary constituents such as caffeine, acid, and alcohol often exacerbate pain and urinary symptoms34. As a result, a front-line approach to symptom management is dietary modification through an “elimination diet” to identify and then eliminate any sensitizing comestibles that vary with the individual patient35. We anticipated that such dietary modification among IC patients might complicate analysis of any UCPPS pathogenesis-associated shifts in stool microbiota. Thus, it was not surprising that the global level of analysis represented by PCA revealed only modest segregation of control and IC stool microbiota in the pilot cohort (Fig. 1), but these findings did not preclude identification of differences between IC and controls at the levels of phyla and order (Fig. 2), suggesting the presence of specific taxa as candidate biomarkers for IC. Indeed, more detailed analyses revealed multiple such genera.
ERF identified 26 taxa as highly significant features, including Actinobacteria, Firmicutes, and Proteobacteria (Fig. 3). An important asset of ERF as applied in these studies is that feature rankings were based on symptom scores, thus significant taxa were not identified merely on the basis of prevalence but instead reflected IC symptom severity. This is a particularly important consideration, given that UCPPS are symptom-based diagnoses that are made after excluding other conditions that might elicit similar symptoms (e.g., bladder cancer). We used the ERF findings to develop species-specific primers and validated O. splanchnicus, F. prausnitzii, C. aerofaciens, E. sinensis, and L. longiviformis as DIPP species with significantly lower abundance in IC stool (Fig. 4). Although all significant species were DIPP species, we speculate that the IC microbiome is not simply characterized by species deficiency but also harbors species that are increased in IC pelvic pain (IIPP). For example, albeit not significant, we note that E. lenta is somewhat elevated and thus is a candidate IIPP species that will be examined in future UCPPS studies (Fig. 4). Similarly, several bacterial orders were elevated in IC relative to controls (Fig. 2), suggesting that multiple genera may harbor IIPP species in the IC microbiome. As the DIPP species were initially identified on the basis of ERF association with symptom scores, it will also be interesting to determine if any or all of these species vary with individual symptom scores, such as during “flares” or periods of remission.
The IC fecal microbiome and metabolome suggest a GI contribution to UCPPS symptomatology. Little is known of O. splanchnicus metabolism, but L. longoviformis metabolizes phytoestrogens, and IC has a strong gender bias that is mimicked in a clinically relevant murine model36,37. E. lenta is associated with bacteremia but more recently has been implicated in digoxin metabolism, thereby shaping pharmacologic therapy18. Thus it is possible that E. sinensis also alters the IC metabolome and may therefore contribute to alterations in the gut-brain axis or pelvic organ crosstalk22,38,39. F. prausnitzii is an anaerobe that is highly abundant in the human GI tract. Interestingly, F. prausnitzii provides butyrate to the gut, and F. prausnitzii deficiency is associated with irritable bowel disease40,41,42,43. Short chain fatty acids are appreciated as beneficial mediators of the microbiome44,45, and we note that butyrate (a.k.a. butanoate) is one of the top-ranked pathways in the IC fecal metabolome (Fig. 5C). Therefore, F. prausnitzii deficiency may be a major mediator of the IC microbiome. Similarly, C. aerofaciens deficiency is significantly associated with symptoms in IBS patients46. Consistent with these features of the IC microbiome, PICRUSt functional prediction of the IC metagenome revealed that fatty acid biosynthesis was the most significant functionality (Fig. 5C, P = 0.03). These metagenome findings dovetail with the metabolome analyses that identified arachidonic acid, butyrate/butanoate, and linoleic acid (Fig. 5A). These findings, in turn, are supported by recent suggestions that the balance of omega-3 and omega-6 polyunsaturated fatty acids influence urologic inflammation (reviewed in47). Taken together, as the abundances of individual DIPP species co-vary as an apparent community (Fig. 4B), it is possible that the lipid-specific effects of any individual DIPP species on IC symptoms is compounded as the DIPP community declines and impacts multiple lipid classes that mediate urologic health and disease.
These studies have potential for important clinical implications for IC. First, IC diagnosis remains difficult for the clinician and patient alike, but stool-based biomarkers identified here offer important promise (Table 2). These candidates identified are likely not exhaustive, as these initial studies are marked by a limited cohort size, and as some genera were excluded in our analyses as a matter of convenience due to the many known species. Moreover, it is likely additional biomarker species exist beyond the DIPP species identified here because they reside within genera that did not reach the significance threshold ERF. For example, O. splanchnicus emerged as a candidate because ERF analyses performed on the pilot cohort initially identified Odoribacter as a significant OTU (this fluctuation is due to the small patient sample size of the pilot cohort). We nonetheless retained Odoribacter as a taxon of interest and thereby identified O. splanchnicus as a DIPP species. Despite these limitations, it is clear that stool is a convenient source of sensitive and specific IC biomarkers, and we identified here 6 candidate biomarkers for further study (Table 2) and others are likely to emerge. Second, and perhaps more importantly, the IC microbiome suggests probiotic/prebiotic therapeutic opportunities. For example augmenting DIPP species themselves or supplying other species with similar metabolic activities may complement metabolic defects that contribute IC directly, by the gut-brain axis, or by pelvic organ crosstalk39,48. Thus in summary, the IC microbiome and metabolome identify important new diagnostic and therapeutic opportunities for IC patients. Future studies already underway in the MAPP Network will determine if men suffering chronic prostatitis/chronic pelvic pain syndrome also exhibit an altered fecal microbiome, and it will be of great interest to determine the extent to which our IC microbiome findings can be generalized to other UCPPS conditions.
Methods
Study Design and Oversight
Studies were conducted under Protocol STU00055668. All experimental protocols within STU00055668 were approved by the Institutional Review Board of Northwestern University and carried out in accordance with the approved procedures, and the study was posted on ClinicalTrials.gov. Study design was unblinded, and all samples were de-identified. Participants were enrolled by a research coordinator, and all participants provided written, informed consent by consent form with valid IRB date stamp; signed consents obtained after 01 August 2014 were logged immediately into eNOTIS/NITRO. Clinical samples were collected in the Urology Clinic or by self-collection by study participants (see below). Analyses were performed at Northwestern University and under IRB exemption at University of Illinois at Urbana–Champaign.
Patients
Female participants were recruited into the Urology Clinic of Northwestern University Feinberg School of Medicine in two separate cohorts. A pilot cohort of 15 individuals (8 healthy controls, 7 IC) was recruited from individuals participating in the NIDDK MAPP Network study at Northwestern15, and stool and vaginal swabs were obtained from this group. A second cohort was recruited independent of MAPP Network participation for collection of stool samples only (i.e., no vaginal swabs were collected; 9 healthy controls, 10 IC). In all cases, demographic data, medical history, and symptom scores were captured using an abbreviated MAPP demographic and medical history, and a female-specific MAPP genitourinary pain index (GUPI) questionnaire at the time of enrollment49. Individuals attesting to antibiotic use within the prior three months were excluded.
Sample collection and preparation
Clinical samples of vaginal swabs and/or stool were collected from IC patients or controls at Northwestern according to protocols of the HMP Manual of Procedures. Briefly, participants were provided with a stool collection kit for at-home sample collection. Participants were instructed to freeze samples overnight and then ship to the laboratory within 48h on wet ice. Upon receipt in the laboratory, stool was aliquoted, placed into RNAlater (MoBio) and stored at −80°. For vaginal swabs, pH was determined with pH strips, and swabs were collected from the introitus, midpoint, and fornynx. Swabs were placed immediately into RNAlater and stored at −80°. Collected samples were shipped on dry ice to University of Illinois Urbana–Champaign for analyses.
Microbiome analyses
Phylotype profiles of the microbiome populations were generated using deep amplicon sequencing of the hypervariable V3–V5 region of the 16S ribosomal RNA (rRNA) gene, which has been validated by the HMP for use with human microbiomes27. Barcoding samples prior to MiSeq tag sequencing will yield approximately 50,000 reads/sample, ensuring detection of both dominant (core microbiome) and poorly represented taxa (variable microbiome). The hypervariable V3–V5 region was selectively amplified from total genomic DNA by 30 cycles of PCR using conserved primer sequences 357F (CCTACGGGAGGCAGCAG) and 926R (CCGTCAATTCMTTTRAGT) using protocols established by the HMP27. Amplicon pools were quantified using a Qubit fluorimeter, and the average fragment sizes were determined on an Agilent bioanalyzer High Sensitivity DNA LabChip (Agilent Technologies, Wilmington, DE) and diluted to 10nM. Amplicons were spiked with 20% of PhiX control library to provide for accurate calculation of matrix, phasing and prephasing. The mixture was sequenced on an Illumina MiSeq V2 sequencer for 250nt from each end of the amplified fragments.
Key OTUs mediating UCPPS were identified using an unbiased protocol that incorporated machine learning and decision tree algorithms (see also below). Data from 16S sequence reads were binned at 97% sequence identity using QIIME and Galaxy to define OTU abundance profiles and phylogenic relationships50,51. Metagenome analyses were performed using MG-RAST version 3.6 at the Argonne National Laboratory52.
Extended Random Forest
Extended Random Forest was applied to identify and rank OTUs predictive of pain phenotype. This process uses the Random Forest (RF) algorithm followed by stability analysis and feature permutation to determine all relevant OTUs. RF has been used on similar problems in other fields53. RF shows excellent performance with this type of complex data and is well suited for datasets where the number of variables is orders of magnitude larger than the number of patient samples28,54.
For this study of IC/BPS, we first pre-processed the data by dropping features with zero variance, and then we incorporated a parameter-space mapping algorithm to determine the model parameters for RF. Using these parameters (Ntree: 25000, Mtry: 200), RF constructed an ensemble of decision trees and provided VIM scores for each variable. VIM represents the benefit of having a feature included in a tree compared to random noise. For feature ranking, the advantage of the RF permutation-accuracy measure compared to univariate filtering measures is that it captures the impact of each feature individually, as well as conditional importance stemming from multivariate interactions between features55.
These steps were followed by stability analysis and feature permutation to identify a minimal optimal set of highly relevant OTUs. The original feature set was extended with a permuted set of features, called shadow features. For each original feature, a shadow feature was created by randomly permuting the values from all observations. When permuted features are used for prediction, the prediction accuracy decreases substantially. Thus a reasonable measure for feature relevance is the likelihood that a feature has a higher mean VIM than its shadow feature. This was estimated by the p-value of a one-sided t-test between each feature and the shadow with the highest mean VIM.
DIPP species quantitation
Relative levels of species in controls and IC patients were measured by quantitative real-time PCR. DNA was purified from stool samples were using the Qiagen Stool DNA purification kit according to the manufacturer’s instructions. Primers were designed against unique regions of 16S rRNA. The following species-specific primers were confirmed by melt-curve analyses: O. splanchnicus forward TCGAAGGCTTGACCTTACGC and reverse TTCATTCGTACGTCCGGTGG; F. prausnitzii forward GAGGTTGAACGGCCACATTG and reverse ACCTAGTAAACATCGGCC AC; E. sinensis forward GGGATCTCTAATCCGAGGGC and reverse TAATGCGTTAGCTGCGGGC; E. lenta forward GGAATGCGCAGATATCGGGA and reverse CCCCCACACCTAGTATCCATC; C. aerofaciens forward GTGGAACACCGATGGCGAA and reverse CCTGGTAAGGTTCTTCGCGT; L. pectinoschiz forward AGCAGTTGGAAACGGCTGAT and reverse TTACTGACCGGGCAAGGA G; L. longoviformis forward TGCATTGGAAACTGTGCAGC and reverse TTCTTGCGAACGTACTCCCC; and R. intestinalis forward CGGCTTAAATACGTGCCAGC and reverse AGCCTCAGCGTCAGTAATCG. Duplicate amplification reactions were carried out in a volume of 20 μL that included 10 μL of SYBR Green (BioRad), 0.5 nM stool DNA, and 0.5 nM each primer using the following cycling conditions: 95° for 5 minutes, 40 cycles of denaturing 20″ at 95°, annealing 30″ at 70°, and extension 30″ 72°. Differences in threshold cycles between the positive control and each sample were quantified by the ΔΔCT method and expressed as fold change. ROC estimates were constructed from 5-fold cross validation using a linear threshold model.
Metabolomics
Two 1ml fractions of the lavage samples were taken from each subject and dried. One fraction was derivatized according to Roessner and colleagues30 with minor modifications: 90 minutes at 500 °C with 80 μl of methoxyamine hydrochloride in pyridine (20 mg/ml) followed by a 60 minute treatment at 500 °C with 80 μl MSTFA. A 5 μl aliquot of an internal standard (C31 fatty acid) was added to each, in the derivatized samples this occurred prior to trimethylsilylation. Sample volumes of 1 mL were injected with a split ratio of 7:1 into a GC-MS system consisting of an Agilent 7890A (Agilent Inc, Palo Alto) gas chromatograph, an Agilent 5975C mass selective detector and Agilent 7683B autosampler. Gas chromatography was performed on 60 m HP-5MS columns with 0.25 mm inner diameter and 0.25 mm film thickness (Agilent Inc, Palo Alto) and an injection temperature of 2500 °C, the interface set to 2500 °C, and the ion source adjusted to 2300 °C. The helium carrier gas was set at a constant flow rate of 1.5 ml min−1. The temperature program of 5-min isothermal heating at 700 °C, followed by an oven temperature increase of 50 °C min−1 to 3100 °C and a final 20 min at 3100 °C. The mass spectrometer was operated in positive electron impact mode (EI) at 69.9 eV ionization energy in m/z 30–800 scan range. The spectra of all chromatogram peaks were then compared with electron impact mass spectrum libraries NIST08 (NIST, Gaithersburg), WILEY08 (Palisade Corporation), and to a custom library of the University of Illinois metabolomics center. To allow direct comparisons between samples all data was normalized to the internal standard in each chromatogram. The chromatograms and smass spectra were evaluated using the MSD ChemStation (Agilent, Palo Alto) and AMDIS (NIST, Gaithersburg). The retention time and mass spectra were implemented within the AMDIS method formats.
Metabolomic data were analyzed using Pathway Analysis and ROC Analysis modules in MetaboAnalyst 3.031,32,33. Pathway analysis utilized normalization to sample mass for analytes in the Human Metabolome Database ID. ROC analyses were performed by univariate analysis.
Predictive functional analysis
Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) was used to predict function from 16S sequence data, PICRUSt predicts function from phylogeny based on the shared gene content and does not consider the variations among species56. The PICRUSt compatible OTU table was further normalized and used to predict to KEGG Orthologs (KOs) using the online PICRUSt Galaxy version.
Additional Information
How to cite this article: Braundmeier-Fleming, A. et al. Stool-based biomarkers of interstitial cystitis/bladder pain syndrome. Sci. Rep. 6, 26083; doi: 10.1038/srep26083 (2016).
References
Clemens, J. Q., Markossian, T. & Calhoun, E. A. Comparison of economic impact of chronic prostatitis/chronic pelvic pain syndrome and interstitial cystitis/painful bladder syndrome. Urology 73, 743–746, doi: S0090-4295(08)01836-010.1016/j.urology.2008.11.007 (2009).
Berry, S. H. et al. Prevalence of symptoms of bladder pain syndrome/interstitial cystitis among adult females in the United States. J Urol 186, 540–544, doi: 10.1016/j.juro.2011.03.132 (2011).
Rabin, C., O’Leary, A., Neighbors, C. & Whitmore, K. Pain and depression experienced by women with interstitial cystitis. Women & health 31, 67–81 (2000).
Watkins, K. E. et al. Depressive disorders and panic attacks in women with bladder pain syndrome/interstitial cystitis: a population-based sample. General hospital psychiatry 33, 143–149, doi: 10.1016/j.genhosppsych.2011.01.004 (2011).
Rothrock, N. E., Lutgendorf, S. K., Hoffman, A. & Kreder, K. J. Depressive symptoms and quality of life in patients with interstitial cystitis. J Urol 167, 1763–1767 (2002).
Hanno, P. Campbell-Walsh Urology Vol. 1 (eds Wein, A. J. et al.) Ch. 14, 334–370 (Elsevier, 2016).
Leiby, B. E., Landis, J. R., Propert, K. J. & Tomaszewski, J. E. Discovery of morphological subgroups that correlate with severity of symptoms in interstitial cystitis: a proposed biopsy classification system. J Urol 177, 142–148 (2007).
Tomaszewski, J. E. et al. Biopsy features are associated with primary symptoms in interstitial cystitis: results from the interstitial cystitis database study. Urology 57, 67–81 (2001).
Yilmaz, U., Liu, Y. W., Berger, R. E. & Yang, C. C. Autonomic nervous system changes in men with chronic pelvic pain syndrome. J Urol 177, 2170–2174; discussion 2174, doi: 10.1016/j.juro.2007.01.144 (2007).
Dimitrakov, J. et al. Adrenocortical hormone abnormalities in men with chronic prostatitis/chronic pelvic pain syndrome. Urology 71, 261–266, doi: 10.1016/j.urology.2007.09.025 (2008).
Lutgendorf, S. K. et al. Diurnal cortisol variations and symptoms in patients with interstitial cystitis. J Urol 167, 1338–1343 (2002).
Anderson, R. U., Orenberg, E. K., Chan, C. A., Morey, A. & Flores, V. Psychometric profiles and hypothalamic-pituitary-adrenal axis function in men with chronic prostatitis/chronic pelvic pain syndrome. J Urol 179, 956–960, doi: 10.1016/j.juro.2007.10.084 (2008).
Buffington, C. A. Idiopathic cystitis in domestic cats–beyond the lower urinary tract. Journal of veterinary internal medicine/American College of Veterinary Internal Medicine 25, 784–796, doi: 10.1111/j.1939-1676.2011.0732.x (2011).
Clemens, J. Q. et al. The MAPP research network: a novel study of urologic chronic pelvic pain syndromes. BMC urology 14 (2014).
Landis, J. R. et al. The MAPP research network: design, patient characterization and operations. BMC urology 14, 58, doi: 10.1186/1471-2490-14-58 (2014).
Blaser, M., Bork, P., Fraser, C., Knight, R. & Wang, J. The microbiome explored: recent insights and future challenges. Nat Rev Microbiol 11, 213–217, doi: 10.1038/nrmicro2973 (2013).
Human Microbiome Project, C. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214, doi: 10.1038/nature11234 (2012).
Haiser, H. J., Seim, K. L., Balskus, E. P. & Turnbaugh, P. J. Mechanistic insight into digoxin inactivation by Eggerthella lenta augments our understanding of its pharmacokinetics. Gut Microbes 5, 233–238, doi: 10.4161/gmic.27915 (2014).
Hilt, E. et al. Urine is not sterile: use of enhanced urine culture techniques to detect resident bacterial flora in the adult female bladder. Journal of clinical microbiology, doi: 10.1128/JCM.02876-13 (2013).
Rudick, C. N. et al. Host-pathogen interactions mediating pain of urinary tract infection. The Journal of infectious diseases 201, 1240–1249, doi: 10.1086/651275 (2010).
Rudick, C. N. et al. O-antigen modulates infection-induced pain states. PLoS ONE 7, e41273, doi: 10.1371/journal.pone.0041273 (2012).
Malykhina, A. P. Neural mechanisms of pelvic organ cross-sensitization. Neuroscience 149, 660–672, doi: 10.1016/j.neuroscience.2007.07.053 (2007).
Malykhina, A. P. et al. Differential effects of intravesical resiniferatoxin on excitability of bladder spinal neurons upon colon-bladder cross-sensitization. Brain research 1491, 213–224, doi: 10.1016/j.brainres.2012.11.003 (2013).
Pezzone, M. A., Liang, R. & Fraser, M. O. A model of neural cross-talk and irritation in the pelvis: implications for the overlap of chronic pelvic pain disorders. Gastroenterology 128, 1953–1964 (2005).
Rudick, C. N. et al. Uropathogenic Escherichia coli induces chronic pelvic pain. Infect Immun 79, 628–635, doi: IAI.00910-10 10.1128/IAI.00910-10 (2011).
Rudick, C. N., Chen, M. C., Mongiu, A. K. & Klumpp, D. J. Organ Crosstalk Modulates Pelvic Pain. Am J Physiol Regul Integr Comp Physiol 293, R1191–1198 (2007).
Evaluation of 16S rDNA-based community profiling for human microbiome research. Plos One 7, e39315, doi: 10.1371/journal.pone.0039315 (2012).
Diaz-Uriate, R. S. & de Andres, S. Gene selection and classification of microarray data using random forest. BMC bioinformatics 7, 3 (2006).
Reshef, D. N. et al. Detecting novel associations in large data sets. Science 334, 1518–1524, doi: 10.1126/science.1205438 (2011).
Roessner, U., Wagner, C., Kopka, J., Trethewey, R. N. & Willmitzer, L. Technical advance: simultaneous analysis of metabolites in potato tuber by gas chromatography-mass spectrometry. Plant J 23, 131–142 (2000).
Xia, J., Mandal, R., Sinelnikov, I. V., Broadhurst, D. & Wishart, D. S. MetaboAnalyst 2.0–a comprehensive server for metabolomic data analysis. Nucleic Acids Res 40, W127–133, doi: 10.1093/nar/gks374 (2012).
Xia, J., Psychogios, N., Young, N. & Wishart, D. S. MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Res 37, W652–660, doi: 10.1093/nar/gkp356 (2009).
Xia, J., Sinelnikov, I. V., Han, B. & Wishart, D. S. MetaboAnalyst 3.0–making metabolomics more meaningful. Nucleic Acids Res 43, W251–257, doi: 10.1093/nar/gkv380 (2015).
Shorter, B., Lesser, M., Moldwin, R. M. & Kushner, L. Effect of comestibles on symptoms of interstitial cystitis. J Urol 178, 145–152, doi: 10.1016/j.juro.2007.03.020 (2007).
Friedlander, J. I., Shorter, B. & Moldwin, R. M. Diet and its role in interstitial cystitis/bladder pain syndrome (IC/BPS) and comorbid conditions. BJU international 109, 1584–1591, doi: 10.1111/j.1464-410X.2011.10860.x (2012).
Rudick, C. N., Pavlov, V. I., Chen, M. C. & Klumpp, D. J. Gender specific pelvic pain severity in neurogenic cystitis. J Urol 187, 715–724, doi: 10.1016/j.juro.2011.10.048 (2012).
Theoharides, T. C., Pang, X., Letourneau, R. & Sant, G. R. Interstitial cystitis: a neuroimmunoendocrine disorder. Annals of the New York Academy of Sciences 840, 619–634. (1998).
Cryan, J. F. & O’Mahony, S. M. The microbiome-gut-brain axis: from bowel to behavior. Neurogastroenterol Motil 23, 187–192, doi: 10.1111/j.1365-2982.2010.01664.x (2011).
Klumpp, D. J. & Rudick, C. N. Summation model of pelvic pain in interstitial cystitis. Nature clinical practice 5, 494–500, doi: 10.1038/ncpuro1203 (2008).
Khan, M. T. et al. The gut anaerobe Faecalibacterium prausnitzii uses an extracellular electron shuttle to grow at oxic-anoxic interphases. ISME J 6, 1578–1585, doi: 10.1038/ismej.2012.5 (2012).
Sokol, H. et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. PNAS 105, 16731–16736 (2008).
Sokol, H. et al. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflammatory bowel diseases 15, 1183–1189, doi: 10.1002/ibd.20903 (2009).
Willing, B. et al. Twin studies reveal specific imbalances in the mucosa-associated microbiota of patients with ileal Crohn’s disease. Inflammatory bowel diseases 15, 653–660, doi: 10.1002/ibd.20783 (2009).
De Vadder, F. et al. Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell 156, 84–96, doi: 10.1016/j.cell.2013.12.016 (2014).
Kelly, C. J. et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe 17, 662–671, doi: 10.1016/j.chom.2015.03.005 (2015).
Malinen, E. Association of symptoms with gastrointestinal microbiota in irritable bowel syndrome. World Journal of Gastroenterology 16, 4532, doi: 10.3748/wjg.v16.i36.4532 (2010).
Tamma, S. M., Shorter, B., Toh, K. L., Moldwin, R. & Gordon, B. Influence of polyunsaturated fatty acids on urologic inflammation. Int Urol Nephrol 47, 1753–1761, doi: 10.1007/s11255-015-1108-8 (2015).
Klumpp, D. & Rudick, C. N. Dietary sensitivity of interstitial cystitis: bane and opportunity. European Urological Review 4, 54–56 (2009).
Clemens, J. Q. et al. The MAPP research network: a novel study of urologic chronic pelvic pain syndromes. BMC urology 14, 57, doi: 10.1186/1471-2490-14-57 (2014).
Blankenberg, D. et al. Galaxy: a web-based genome analysis tool for experimentalists. Current protocols in molecular biology/edited by Frederick M. Ausubel. et al. Chapter 19, Unit 19 10 11–21, doi: 10.1002/0471142727.mb1910s89 (2010).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nature methods 7, 335–336, doi: 10.1038/nmeth.f.303 (2010).
Meyer, F. et al. The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC bioinformatics 9, 386, doi: 10.1186/1471-2105-9-386 (2008).
Breiman, L. Random forests. Machine Learning 45, 5–32 (2001).
Nilsson, R., Pena, J., Bjorkegren, J. & Tegner, J. Consistent feature selection for pattern recognition in polynomial time. Journal of Machine Learning Research 8, 612 (2007).
Friedman, J. Greedy function approximation: a gradient boosting machine. The Annals of Statistics 29, 1189–1232 (2001).
Langille, M. G. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31, 814–821, doi: 10.1038/nbt.2676 (2013).
Acknowledgements
We are grateful to all study subjects who gave of their time and effort to provide samples for this research. Studies were supported by NIDDK award R24 DK094575 (MPI award to D.J.K., A.J.S. and B.A.W.).
Author information
Authors and Affiliations
Contributions
All authors contributed to multiple aspects of this study. Experimental Design: A.B-F., M.Y.N., D.S.M., C.B.B., L.B., S.C.F., M.E.W., B.A.W., A.J.S. and D.J.K. Conduct of Experiments: A.B-F., N.T.R., M.Y.N., R.E.Y., M.B., L.B., S.C.F., D.S.M. and C.B.B. Data Analysis: A.B-F., N.T.R., M.Y.N., W.Y., M.B., C.B.B., M.E.W., B.A.W., A.J.S. and D.J.K. Manuscript Preparation: A.B-F., N.T.R., M.Y.N., W.Y., R.E.Y., C.B.B., M.E.W., B.A.W., A.J.S. and D.J.K.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Braundmeier-Fleming, A., Russell, N., Yang, W. et al. Stool-based biomarkers of interstitial cystitis/bladder pain syndrome. Sci Rep 6, 26083 (2016). https://doi.org/10.1038/srep26083
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep26083
This article is cited by
-
S2k-Leitlinie Diagnostik und Therapie der interstitiellen Zystitis (IC/BPS)
Der Urologe (2022)
-
S2k-Leitlinie Diagnostik und Therapie der interstitiellen Zystitis (IC/BPS)
Journal für Urologie und Urogynäkologie/Österreich (2022)
-
Biomarkers for Bladder Pain Syndrome/Interstitial Cystitis
Current Bladder Dysfunction Reports (2021)
-
Identification of early liver toxicity gene biomarkers using comparative supervised machine learning
Scientific Reports (2020)
-
Interstitielle Zystitis/Blasenschmerzsyndrom (IC/BPS)
Der Urologe (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
