
Abstract
GABAA receptors are the main inhibitory neurotransmitter receptors in the brain and are targets for numerous clinically important drugs such as benzodiazepines, anxiolytics and anesthetics. We previously identified novel ligands of the classical benzodiazepine binding pocket in α1β2γ2 GABAA receptors using an experiment-guided virtual screening (EGVS) method. This screen also identified novel ligands for intramembrane low affinity diazepam site(s). In the current study we have further characterized compounds 31 and 132 identified with EGVS as well as 4-O-methylhonokiol. We investigated the site of action of these compounds in α1β2γ2 GABAA receptors expressed in Xenopus laevis oocytes using voltage-clamp electrophysiology combined with a benzodiazepine site antagonist and transmembrane domain mutations. All three compounds act mainly through the two β+/α− subunit transmembrane interfaces of the GABAA receptors. We then used concatenated receptors to dissect the involvement of individual β+/α− interfaces. We further demonstrated that these compounds have anesthetic activity in a small aquatic animal model, Xenopus laevis tadpoles. The newly identified compounds may serve as scaffolds for the development of novel anesthetics.
Similar content being viewed by others


Introduction
The search for novel anesthetics has been triggered by the rising age of patients and increasing use of anesthesia outside the operating room1,2. A key site of action of the potent anesthetics propofol and etomidate is the major inhibitory receptor in the mammalian central nervous system, the γ-aminobutyric acid type A (GABAA) receptor. These receptors are composed of five homologous subunits organized around a central Cl− selective channel3. Each subunit contains a large N-terminal extracellular domain (ECD), a transmembrane domain (TMD) with four alpha-helices (TM1 to TM4), and a variable-length intracellular domain (ICD) between TM3 and TM4. 19 subunits of the GABAA receptor have been cloned (for review see: 4, 5, 6), denoting that numerous types of receptor isoforms exist5. The most abundant GABAA receptor in the brain comprises α1, β2 and γ2 subunits3,4,5,7. The receptor possesses a 2α:2β:1γ subunit stoichiometry8,9,10,11, with a subunit arrangement of γβαβα anti-clockwise as seen from the synaptic cleft10,11,12,13. The receptor composition and arrangement influence its pharmacological properties14,15.
Benzodiazepines modulate α1β2γ2 GABAA receptor function by binding to a high affinity site located at the α+/γ− ECD interface, homologous to the agonist binding sites at β+/α− ECD interfaces16,17. In addition to the high affinity binding site for benzodiazepines (site 1), there are other low affinity sites. One of these, site 2, is located at the ECD α+/β− interface18,19. Others, together designated as site 3, are located in the TMD, based on abolition of benzodiazepine effects by combined isoleucine substitutions at the homologous residues α1S269, β2N265, and γ2S28020.
GABAA receptors are also targets for potent intravenous anesthetics, including barbiturates, propofol and etomidate21,22,23,24,25. Interestingly, receptor sensitivity to intravenous anesthetics is affected by benzodiazepine site 3 mutations26,27,28,29,30. Diverse anesthetics not only potentiate GABA-induced Cl− currents, but additionally at high concentrations directly activate GABAA receptors31,32. Photo-affinity labeling has located allosteric sites for the intravenous anesthetics etomidate and propofol to the TM1 of α and TM2, TM3 of β subunits23,24,33.
Previously, we reported a new method to identify ligands of the high affinity benzodiazepine pocket, experimental-guided virtual screening (EGVS), integrating experimental data with homology modeling of the GABAA receptor34. EGVS identified some ligands that only recognized site 1, others that recognized both site 1 and site 335, and another set that only recognize site 3.
Here we describe the actions of two compouds identified by EGVS, 31 and 13234 and 4-O-methylhonokiol36. Using mutations and concatenated receptors we determined that the three compounds act mainly through the TMD β+/α− interfaces (site 3), and particularly the γβ+/α−β site. The anesthetic action of these drugs was explored in vivo, revealing potencies similar to propofol.
Results
In an attempt to find novel ligands for the high affinity site for benzodiazepines on GABAA receptors we screened 198 compounds for displacement of the high affinity benzodiazepine site (called site 1 previously) antagonist [3H]-Ro 15-1788 at receptors expressed in HEK-cells. Many high affinity ligands were identified34. One compound, SJM3 acted as antagonist with high affinity at site 1, but allosterically potentiated receptor activation through sites in the membrane (called sites 3 previously)35. Compounds 31 and 132 either did not or weakly displaced [3H]-Ro 15-1788 from site 1 but potently enhanced GABAA receptor activation. 4-O-methylhonokiol shares these characteristics, potentiating α1β2γ2 receptors with an EC50 of 5.4 ± 1.8 μM, independent of the high affinity site for benzodiazepines36. Here, we report mechanistic and animal studies of these three compounds.
Compounds 31 and 132 are allosteric modulators of α1β2γ2 GABAA receptors
First, we investigated if compounds 31 and 132 were able to act as agonists. Figure 1 shows their chemical structures compounds. Both compounds at 3 and 30 μM elicited only very small currents by themselves in α1β2γ2 GABAA receptors expressed in Xenopus oocytes. These compounds elicited at the concentration of 3 μM and 30 μM currents amounting to 0.1 ± 0.06% (mean ± SD, n = 3) and 0.3 ± 0.13% (mean ± SD, n = 3), respectively of the maximal current amplitude elicited by GABA in the same oocytes. Thus neither of the compounds tested acts as an appreciable agonist on α1β2γ2 receptors.

Chemical structure of compounds 31, 132, 4-O-methylhonokiol, and the high-affinity benzodiazepine antagonist Ro 15-1788.
Both compounds strongly enhanced currents elicited by GABA. We established the concentration response curves of this positive allosteric modulation. After two applications of GABA at a concentration eliciting 0.5–1.5% of the maximal current amplitude, the same concentration of GABA was co-applied with increasing concentrations of the tested compounds. Figure 2a,b show current traces demonstrating positive modulation by different concentrations of compounds 31 and 132, respectively. Figure 2c summarizes the results of three of such experiments. Both compounds potentiate GABA elicited currents in oocytes expressing α1β2γ2 receptors. For compound 31, at high concentrations apparent desensitization was observed, that could be partly due to open channel blocker effect. For both compounds no saturation at the highest concentration was obtained, because of poor solubility we could not test higher concentrations.

α1β2γ2 receptors were expressed in Xenopus oocytes and electrophysiological experiments were performed. Original current traces of an experiment with compound 31 (a) and with compound 132 (b). Numbers indicate applied concentrations of the respective compounds. (c) Concentration dependence of the positive allosteric modulation by compound 31 (closed circles) and compound 132 (closed squares) in oocytes expressing α1β2γ2 receptors. Mean data ± SD for both compounds is shown, n = 3.
Potentiation by compounds 31 and 132 is not affected by Ro 15-1788
From the binding data we did not expect that the two compounds are acting through the high affinity benzodiazepine binding site 1 in α1β2γ2 receptors. Nevertheless, we tested whether 1 μM of the site 1 antagonist Ro 15-1788 inhibits potentiation of GABA currents by compounds 31 or 132. Either compound (3 μM) strongly potentiated currents elicited by GABA. Potentiation by either drug was not inhibited by 1 μM Ro 15-1788 (Fig. 3). Relative current amplitudes in the presence vs. absence of the antagonist were 112 ± 8% (mean ± SD, n = 4, p > 0.05, t test) for compound 31, and 115 ± 14% (mean ± SD, n = 4, p > 0.05, t test) for compound 132. This confirms that potentiation by compounds 31 and 132 does not result from action at the benzodiazepine site 1.

GABA at a concentration eliciting 0.5% of the maximal current amplitude (EC0.5, single bars) was applied until a stable response was obtained. Subsequently, the same concentration of GABA was co-applied with 3 μM of compounds 31 (a) or 132 (b), which resulted both in a large increase of current amplitude. Co-application of Ro 15-1788 with compound and GABA did not reduce the degree of modulation in both cases. Experiments were repeated 4 times, with three different batches of oocytes, with a similar outcome.
Compounds 31 and 132 and 4-O-methylhonokiol act at the low affinity benzodiazepine site 3 in α1β2γ2 receptors
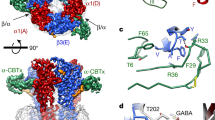
We next investigated whether benzodiazepine site 3 TMD mutations affect potentiation by compounds 31 and 132. Combining three homologous site 3 mutations in α1β2γ2 receptors, α1S269I, β2N265I and γ2S280I, eliminated the potentiation by high concentrations of diazepam20. We investigated the effects of these mutations individually and combined, abbreviating them as α1M, β2M and γ2M. Recently, we described the potency of GABA to activate all of these receptor subtypes expressed in Xenopus oocytes37. In order to exclude general gating effects caused by these mutations we showed that potentiation by low concentrations of diazepam and by THDOC are not affected37. For illustration, the localization of the mutations is shown in the crystalized homomeric β3 receptor38 where some of the β3 subunits were renamed α1, β2 and γ2 (Fig. 4a).

(a) Model structure of the GABAA receptor transmembrane domain. The major isoform of the GABAA receptor is composed of two α1, two β2, and one γ2 subunits. The model structure depicts the crystalized homomeric β3 GABAA receptor (PDB structure 4COF)38. In this figure, some of the β3 subunits were renamed α1 (yellow), β2 (blue) and γ2 (red); structures are shown in ribbon representation. The mutated residues α1S269, β2N265, and γ2S280 are located at the interfaces between subunits. (b) Potentiation of the GABA response by compound 31 (3 μM), compound 132 (3 μM), and 4-O-methylhonokiol (1 μM, abbreviated Mh) in wild-type α1β2γ2, single mutant (α1M, β2M, γ2M), and triple mutant receptors expressed in Xenopus oocytes. The bars indicate mean ± SD, n = 3.
Wild-type α1β2γ2, α1Mβ2γ2, α1β2Mγ2, α1β2γ2M and α1Mβ2Mγ2M receptors were expressed in Xenopus oocytes. Using electrophysiological techniques we determined the effect of these point mutations on the potentiation by compounds 31, 132 and 4-O-methylhonokiol, normalizing to the potentiation in wild type α1β2γ2 receptors.
Figure 4b summarizes the results obtained. The single mutations in the α1 and in the γ2 subunits did not significantly alter the degree of potentiation by 3 μM of either compound 31 or 132. Relative to wild-type α1β2γ2 receptors, modulation in mutated α1Mβ2γ2 receptors by compound 31 amounted to 77 ± 24% (mean ± SD, n = 3, p > 0.05, Tukey posthoc test), and by compound 132 to 118 ± 44% (mean ± SD, n = 3, p > 0.05, Tukey posthoc test). Modulation in α1β2γ2M receptors by compound 31 was 87 ± 52% (mean ± SD, n = 3, p > 0.05, Tukey posthoc test), and by compound 132 was 91 ± 21% (mean ± SD, n = 3, p > 0.05, Tukey posthoc test). In contrast, modulation by both compounds was strongly impaired in α1β2Mγ2 and triply mutated α1Mβ2Mγ2M receptors. Potentiation by 3 μM compound 31 in α1β2Mγ2 was 10.4 ± 8.5% (mean ± SD, n = 3, p = 0.007, Tukey posthoc test), and in α1Mβ2Mγ2M receptors was 2.3 ± 0.8% (mean ± SD, n = 3, p = 0.0043, Tukey posthoc test). Potentiation by compound 132 relative to that in α1β2γ2 was also dramatically reduced in α1β2Mγ2 7.3 ± 6.8% (mean ± SD, n = 3, p = 0.003, Tukey posthoc test) and α1Mβ2Mγ2M −0.4 ± 5.1% (mean ± SD, n = 3, p = 0.0021, Tukey posthoc test), respectively.
Potentiation by 1 μM 4-O-methylhonokiol was also significantly reduced only in α1β2Mγ2 and α1Mβ2Mγ2M receptors, similar to compounds 31 and 132. Relative to wild-type α1β2γ2 receptors, modulation in α1Mβ2γ2 mutated amounted to 63 ± 6% (mean ± SD, n = 3, p > 0.05, Tukey posthoc test), and in α1β2γ2M receptors to 79 ± 16% (mean ± SD, n = 3, p > 0.05, Tukey posthoc test). Relative to α1β2γ2, residual potentiation in α1β2Mγ2 receptors amounted to 15.6 ± 1.5% (mean ± SD, n = 3, p = 0.0022, Tukey posthoc test), and to 2.2 ± 0.2% (mean ± SD, n = 3, p = 0.0018, Tukey posthoc test) in α1Mβ2Mγ2M.
The above data suggests that the modulatory site(s) for the three compounds we studied is located in one or both of the β+/α− TMD subunit interfaces on α1β2γ2 GABAA receptors.
Role of the individual β+/α− subunit interfaces in channel modulation by compounds 31, 132 and 4-O-methylhonokiol
Each GABAA receptor contains two β+/α− subunit interfaces. Combined mutation at these interfaces greatly reduces the modulatory effects of compounds 31, 132 and 4-O-methylhonokiol. Using α1-β2-α1 and γ2-β2 subunit concatemers, we studied the effects of individual mutated interfaces. We designated receptors containing the mutant in the γ2-β2 construct interface 1 M, and the mutation in the α1-β2-α1 construct interface 2 M (Fig. 5a). The α1-β2M-α1 and γ2-β2M constructs were built, and were co-expressed with non-mutated dual or triple subunit constructs forming α1-β2-α1/γ2-β2M and α1-β2M-α1/γ2-β2 receptors. Both constructs were expressed together to form the double mutant receptor α1-β2M-α1/γ2-β2M.

(a) Scheme showing the four concatenated wild-type and mutant receptors. 1 and 2 refer to the two different β+/α− subunit interfaces, interface 1 and interface 2. The location of the β2N265I mutations is indicated in red color. Concatenated receptors were prepared containing no mutation (α1-β2-α1/γ2-β2, non M), a mutation at interface 1 (α1-β2-α1/γ2-β2M, interface 1 M), a mutation at interface 2 (α1-β2M-α1/γ2-β2, interface 2 M), or mutations in both sites (α1-β2M-α1/γ2-β2M, double M). Interface 2 harbors a binding site for GABA with higher apparent affinity for channel gating than the one positioned at the interface 154. (b) Potentiation by compound 31 (3 μM), compound 132 (3 μM), and 4-O-methylhonokiol (1 μM), using an EC0.5–1.5 concentration of GABA for each concatenated receptor subtype. Bars indicate mean ± SD, n = 3.
Wild type concatenated receptors α1-β2-α1/γ2-β2 were also expressed. This receptor has an EC50 for GABA of approximately 120 μM12. Results are standardized to the potentiation observed in α1-β2-α1/γ2-β2 concatenated receptors. As shown in Fig. 5b, in the double mutant concatemeric receptors (α1-β2M-α1/γ2-β2M) potentiation for all three compounds was abolished. Relative to α1-β2-α1/γ2-β2 concatenated receptors, in the double mutant α1-β2M-α1/γ2-β2M receptor modulation by compound 31 was reduced to 1 ± 5% (mean ± SD, n = 3, p < 0.0005, Tukey posthoc test), by compound 132 to −2 ± 0.4% (mean ± SD, n = 3, p = 0.0001, Tukey posthoc test), and by 4-O-methylhonokilol to 1 ± 0.6% (mean ± SD, n = 3, p = 0.0001, Tukey posthoc test). In receptors containing only one mutation, either interface 1 or interface 2, modulation by compound 31 was reduced significantly compared to concatenated wild type receptors α1-β2-α1/γ2-β2. With interface 1 M, residual relative potentiation was 28 ± 6% (mean ± SD, n = 3, p = 0.0005, Tukey posthoc test) and with interface 2 M it was 51 ± 3% (mean ± SD, n = 3, p = 0.0059, Tukey posthoc test) of wild-type. Potentiation of interface 1 M and interface 2 M receptors differed significantly (p = 0.0035, t test). Therefore, both TMD β+/α− sites seem to contribute differently to modulation by compound 31, interface 1 being more efficacious.
Likewise, modulation by compound 132 was sensitive to the mutations at both sites. In interface 1 M receptors relative potentiation was reduced to 8 ± 4% (mean ± SD, n = 3, p = 0.0002, Tukey posthoc test), and in interface 2 M receptors to 39 ± 19% (mean ± SD, n = 3, p = 0.0035, Tukey posthoc test). Again, interface 1 M produced a larger impact than 2 M, although the difference was at the statistical limit (p = 0.0510, t test). Modulation by 4-O-methylhonokiol in interface 1 M and 2 M receptors was reduced to 11 ± 6% (mean ± SD, n = 3, p < 0.0001, Tukey posthoc test) and 28 ± 8% (mean ± SD, n = 3, p = 0.0004, Tukey posthoc test), respectively. The two mutations produced significantly different effects (p = 0.043, t test), with the interface 1 M effect again larger.
Therefore, potentiation by all three compounds displayed similar sensitivity patterns with homologous mutations in distinct β+/α− TMD sites of α1-β2-α1/γ2-β2 receptors. Both sites are necessary for full modulation, while interface 1 produces a larger impact than interface 2.
Effect of the β2N265S mutation
The β2N265 residue is important for allosteric modulation of GABAA receptors by many compounds acting through the TMD. This residue was initially described as a determinant for the modulatory action of loreclezole, where the β2N265S mutation created a receptor unresponsive to this compound39. Mutations in this residue also abolish potentiation by the anesthetics etomidate and propofol28,40. As shown in Fig. 6, the β2N265S mutation in α1β2γ2 receptors significantly reduced potentiation by 3 μM compound 31, from 485 ± 230% in wild-type receptors (mean ± SD, n = 11), to 125 ± 25% in the mutated receptor (mean ± SD, n = 4, p = 0.009, Tukey posthoc test). In contrast, this mutation did not significantly reduce potentiation by 3 μM compound 132: 415 ± 126% in wild-type receptors (mean ± SD n = 6) versus 280 ± 36% in the mutated receptor (mean ± SD, n = 4, p > 0.05, Tukey posthoc test). We have shown earlier that potentiation by 4-O-methylhonokiol in receptors carrying the β2N265S mutation was greatly reduced to about 40%36.

Wild-type α1β2γ2 and mutated α1β2N265Sγ2 receptors were expressed in Xenopus oocytes and studied. Potentiation of GABA currents was determined using 3 μM of compound 31 or 132. Bars indicate mean ± SD, n = 4–11.
Subunit specificity of compounds 31 and 132
From the above experiments we inferred that the β+/α− TMD subunit interfaces mediate potentiation of compounds 31 and 132. We also wanted to know if potentiation by these compounds depends on subunit isoforms. First we replaced the α1 subunit by different α subunit isoforms: α1β2γ2, α2β2γ2, α3β2γ2, α4β2γ2, α5β2γ2 and α6β2γ2 (Fig. 7). Compound 31 displayed a similar degree of potentiation in α1β2γ2 receptors 485 ± 230% (mean ± SD, n = 11), α2β2γ2 receptors, 407 ± 172% (mean ± SD, n = 4, p > 0.05, Tukey posthoc test), α3β2γ2 receptors, 735 ± 234%, (mean ± SD, n = 5, p > 0.05, Tukey posthoc test), and α4β2γ2 receptors, 412 ± 175% (mean ± SD, n = 4, p > 0.05, Tukey posthoc test). The α5β2γ2 receptor showed a significant decrease in potentiation compared to α1β2γ2 receptors to 253 ± 92% (mean ± SD, n = 8; p = 0.023, Tukey posthoc test), and the α6β2γ2 receptor an increase in potentiation, amounting to 780 ± 236% (mean ± SD, n = 4; p = 0.048, Tukey posthoc test). This discrepancy in modulation of α5β2γ2 and α6β2γ2 receptors maybe explained by the fact that 3 of the first 7 residues of M1 located at the minus side of the α subunit are different (Fig. 8a). Compound 132 produced similar potentiation in receptors with all α subunits tested. Amounting to 415 ± 126% (mean ± SD, n = 6) in α1β2γ2 receptors, 694 ± 293% (mean ± SD, n = 4, p > 0.05, Tukey posthoc test) in α2β2γ2 receptors, 652 ± 215% (mean ± SD, n = 4, p > 0.05, Tukey posthoc test) in α3β2γ2 receptors, 476 ± 152% (mean ± SD, n = 4, p > 0.05, Tukey posthoc test) in α4β2γ2 receptors, 399 ± 147% (mean ± SD, n = 4, p > 0.05, Tukey posthoc test) in α5β2γ2 receptors, and 542 ± 175% (mean ± SD, n = 4, p > 0.05, Tukey posthoc test) in α2β2γ2 receptor type. These results indicate that although the type of α subunit has differential effects on potentiation between compounds 31 and 132, these compounds modulate all receptor subtypes studied.

Different subunit combinations were expressed in Xenopus oocytes. Potentiation of GABA currents at a GABA concentration eliciting 0.5–1.5% of the maximal current amplitude was determined using 3 μM of compound 31 (a) or 132 (b). Bars indicate mean ± SD, n = 4–11.

(a) Sequences preceding M1 and the first part of M1 in α subunits are shown. (b) Sequences preceding M3 and the first part of M3 in β subunits are shown.
Next, we examined the role of the β subunit, replacing the β2 by β1 or β3. For compounds 31 and 132, α1β3γ2 receptors showed a similar potentiation as α1β2γ2 receptors. Amounting to 381 ± 115% for compound 31 and 555 ± 169% for compound 132 (mean ± SD, n = 4, p > 0.05, Tukey posthoc test). In the case of α1β1γ2 receptors, potentiation by both compounds was significantly reduced compared to that in α1β2γ2, 41 ± 13% for compound 31 and 37 ± 7% for compound 132 (n = 4; p = 0.0023, Tukey posthoc test for compound 31; n = 4; p = 0.0004, Tukey posthoc test, for compound 132). These results indicate that the type of β subunit is important for the potentiation by both compounds. It is interesting to note in this context that β1 and β2/β3 differ not only in the residue 265, but also in the fourth residue of M3 predicted to be close to the latter residue (Fig. 8b).
When the γ2 subunit was omitted, no statistical difference was observed for either compound tested. Compound 31 displayed a similar degree of potentiation between α1β2γ2 receptors and α1β2 receptors, 339 ± 73% (mean ± SD, n = 4, p > 0.05, Tukey posthoc test). Compound 132 also showed a similar potentiation between both receptors, potentiation in α1β2 receptors amounting to 625 ± 219% (mean ± SD, n = 4, p > 0.05, Tukey posthoc test). When the γ2 subunit was replaced by a δ subunit, in the case of α1β2δ receptors, potentiation was affected only in the case of compound 31, where a significant reduction was observed relative to α1β2γ2 receptors. Where potentiation amounted to 168 ± 48% (mean ± SD, n = 4, p = 0.0026, Tukey posthoc test) for compound 31, and 640 ± 275% (mean ± SD, n = 4, p > 0.05, Tukey posthoc test) for compound 132. In α4β3δ receptors, potentiation by both compounds was statistically reduced relative to α4β3γ2 receptors. For compound 31 potentiation amounted to 345 ± 77% (mean ± SD, n = 4) in α4β3γ2 receptors, and was reduced to 125 ± 22% (mean ± SD, n = 4, p = 0.0015, Tukey posthoc test) in α4β3δ receptors. Potentiation of compound 132 in α4β3γ2 receptors was 728 ± 203% (mean ± SD, n = 4), and was reduced to 214 ± 71% (mean ± SD, n = 4, p = 0.0031 Tukey posthoc test, for compound 132) in α4β3δ receptors.
Previous work36 showed that potentiation by 4-O-methylhonokiol was dependent on the α subunit in a similar fashion as compound 31, since modulation was reduced by receptors containing α5 and α6 subunits. The type of β subunit was also important, as the presence of the β1 subunit strongly reduced potentiation by this compound. On the contrary, the presence of a γ or a δ subunit did not affect potentiation.
Anesthetic activity in tadpoles
The anesthetic activity for compounds 31, 132 and 4-O-methylhonokiol was determined as loss of righting reflex (LoRR) in Xenopus tadpoles, Fig. 9 shows the concentration dependence curve for each. Compound 31 yielded an EC50 of 2.7 μM (95% confidence interval = 2.0 to 3.7 μM), while the EC50 compound 132 was 1.2 μM (95% confidence interval = 0.73 to 2.0 μM), and 4-O-methylhonokiol EC50 = 1.0 μM (95% confidence interval = 0.46 to 2.2 μM). For comparison, the EC50 for propofol-induced LoRR in tadpoles is 1.3 μM41. Anesthesia was fully reversible for compound 31; for animals tested with compound 132, recovery was minimal at concentrations above 3 μM. For 4-O-methylhonokiol, animals tested at a concentration of 10 μM did not recover, whereas recovery was complete at 3 μM and lower concentrations.

The percent of animals anesthetized is plotted against aqueous anesthetic concentration, overlaid with logistic fits. Each point represents data from ten animals. Data were fitted to a Hill equation.
Discussion
Here we functionally characterized compounds 31, 132 and further investigated the properties of 4-O-methylhonokiol. All three compounds are potent allosteric potentiators of α1β2γ2 GABAA receptors that do not act at site 1. We suspected that they instead act through the low affinity TMD site(s) for benzodiazepines (site 3). Indeed, potentiation by compounds 31, 132 and 4-O-methylhonokiol was abolished in the triple mutant receptor α1S269Iβ2N265Iγ2S280I as well as by the single mutation β2N265I, but unaffected by the homologous mutations α1S269I and γ2S280I. Assuming that these TM2 mutations alter drug actions through local steric effects in adjacent TMD interfacial sites, our results indicate that of the five such sites, only the two β+/α− interfaces 1 and 2 mediate the potentiating effects of these three compounds.
We further dissected the contribution of the individual β+/α- subunit interfaces using concatenated subunit assemblies. The γβ+/α−β interface (interface 1) and αβ+/α−γ interface (2) participated differently in modulation by the three compounds studied. For all compounds the contribution of the interface 1 to drug modulation is apparently greater than that of the interface 2.
We and others have shown that the intravenous anesthetics etomidate, propofol and pentobarbital also act via TMD interfacial sites26,27,28,29,37. In α1β2γ2 receptors, β2N265I reduced potentiation by all compounds, α1S269I reduced potentiation exclusively by pentobarbital, and the γ2S280I mutation increased potentiation by etomidate, while reducing potentiation by propofol and pentobarbital37. Different sets of residues located at subunit interfaces have been photo-labeled by etomidate, barbiturate, and propofol analogs, revealing that some anesthetics selectively bind within different TMD interfaces23,24,33,42,43. Additionally, mutations of residues at the β+/α− subunit interface affecting anesthetic action have been shown to affect modulation by valerenic acid. This suggests that the binding pocket for this compound is also at or near the anesthetics binding site44. Other subunit interfaces were not investigated.
Work by our group using receptor concatenation determined that both β+/α− subunit interfaces participated equally in modulation by propofol. In contrast, modulation by etomidate was found to be more affected by the γβ+/α−β interface site (interface 1) than the αβ+/α−γ site (interface 2)37. Interestingly, studies using another mutation (β2M286W) and different concatenated subunit assemblies suggest that etomidate interactions are equivalent in the two β+/α− sites of α1β2γ2 receptors45.
The homologs of β2N265 in β1 and β3 are serine and asparagine, respectively, and this single residue dramatically influences sensitivity to loreclezole39, etomidate and propofol26,28,29,39,40. Potentiation by compound 31 was strongly affected by this mutation while that by compound 132 was affected less. On the other hand, potentiation by 132 was severely reduced in α1β1γ2 receptors, compared to α1β2γ2 receptors. Potentiation by 4-O-methylhonokiol is also reduced by the β2N265S mutation or substitution of β1 for β236.
In subunit specificity studies, compounds 31 and 132 potentiated receptors containing the α4 and α6 subunits, contrasting with benzodiazepine site 1 agonists. Both compounds also potentiated receptors carrying the δ subunit, although to different degrees. Thus, these compounds not only acted at receptors shown to be located synaptically as α1β2–3γ2, α2β2γ2 and α3β2γ246, but also at α5β2γ2 receptors and receptors containing the δ subunit, which are all located extra-synaptically5,46,47. Similarly, SJM-3 modulates both synaptic and extrasynaptic receptors35.
The similarities between the three compounds we studied and the clinical anesthetics propofol, etomidate and pentobarbital suggested their possible use as sedative-hypnotics in animals. Indeed, all three compounds induced reversible loss of righting reflexes (LoRR) in Xenopus laevis tadpoles with EC50s comparable to the anesthetics propofol41, and etomidate48. However, LoRR was not reversible with high concentrations of compound 132 and 4-O-methylhonokiol.
In summary, the newly identified compounds 31 and 132 modulate both synaptic and extrasynaptic GABAA receptors at molecular sites different from the classical benzodiazepine pocket. These compounds, together with 4-O-methylhonokiol, act through β+/α− TMD interfaces, with strongest effects through the interface 1. These compounds potently produced LoRR in aquatic animals and thus may be useful lead compounds in the search for novel anesthetic, sedative-hypnotic or anxiolytic drugs.
Methods
Construction of mutated receptor subunits
The point mutations α1S269I, β2N265I, β2N265S and γ2S280I were prepared using the QuikChangeTM mutagenesis kit (Stratagene, Agilent Technologies, Basel, Switzerland).
Construction of concatenated subunits
Construction of tandem and triple subunit cDNAs. The tandem construct γ2-β2, and triple construct α1-β2-α1 has been described previously12. Site-directed mutagenesis of β2N265 to I was done in the tandem construct and the triple construct using the QuikChangeTM mutagenesis kit (Stratagene, Agilent Technologies, Basel, Switzerland).
Expression of GABAA receptors in Xenopus oocytes
Capped cRNAs were synthesized (Ambion, Austin, TX, USA) from the linearized plasmids with a cytomegalovirus promotor (pCMV vectors) containing the different subunits, respectively. A poly-A tail of about 400 residues was added to each transcript using yeast poly-A polymerase (United States Biologicals, Cleveland, OH, USA). The concentration of the cRNA was quantified on a formaldehyde gel using Radiant Red stain (Bio-Rad) for visualization of the RNA. Known concentrations of RNA ladder (Invitrogen) were loaded as standard on the same gel. cRNAs were precipitated in ethanol/isoamylalcohol 19:1, the dried pellet dissolved in water and stored at −80 °C. cRNA mixtures were prepared from these stock solutions and stored at −80 °C.
Animal experiments were carried out in strict accordance to the Swiss ethical guidelines, and have been approved by the local committee of the Canton Bern Kantonstierarzt, Kantonaler Veterinärdienst Bern (BE85/15). Surgery was done under anesthesia, and all efforts were made to diminish animal suffering. Xenopus laevis oocytes were prepared, injected and defolliculated as described previously49,50. Oocytes were injected with 50 nL of the cRNA solution containing wild type or mutated rat α1, β2 and γ2 subunits of the GABAA receptors at a concentration of 10 nM:10 nM:50 nM51. For concatenated tandem and triple constructs, cRNA combinations ratios of 25: 25 nM were used. Injected oocytes were incubated in modified Barth’s solution at 18 °C for at least 24 h before the measurements.
Functional characterization of GABAA receptors
Currents were measured using a modified two-electrode voltage clamp amplifier Oocyte clamp OC-725 (Warner Instruments) in combination with a XY-recorder (90% response time 0.1 s) or digitized at 100 Hz using a PowerLab 2/20 (AD Instruments) using the computer programs Chart (ADInstruments GmbH, Spechbach, Germany). Tests with a model oocyte were performed to ensure linearity in the larger current range. The response was linear up to 15 μA. The holding potential was −80 mV. The perfusion medium contained 90 mM NaCl, 1 mM KCl, 1 mM MgCl2, 1 mM CaCl2, and 5 mM Na-HEPES (pH 7.4). Concentration response curves for the compounds were fitted with the equation I(c) = Imax/[1 + (EC50/c)n], where c is the concentration of the compound, EC50 the concentration eliciting half-maximal current amplitude, Imax is the maximal current amplitude, I the current amplitude, and n is the Hill coefficient. Maximal current amplitudes (Imax) were obtained from the fits of the concentration-response curves. For all receptors studied, modulation was measured at a GABA concentration eliciting 0.5–1.5% of the maximal GABA current amplitude. GABA was applied twice alone for 20–60 s, and then in combination with the different compounds for 45 s or 1 min. The duration of washout periods was 4 min in between agonist or agonist/drug aplications to prevent receptor desentization. At the beginning of the experiments, GABA applications were repeated when the elicited current amplitude altered by >5%. Potentiation was calculated by the following equation: (IModulator + GABA/IGABA − 1) * 100%. The perfusion solution was applied through a glass capillary with an inner diameter of 1.35 mm, the mouth of which was placed about 0.4 mm from the surface of the oocyte. This allowed fast changes in agonist concentration around the oocyte. The rate of change was estimated 70% in less than 0.5 s14. The perfusion system was cleaned between drug applications by washing with DMSO to avoid contamination. All media contained a final concentration of 0.5% DMSO (v/v) to ensure drug solubility.
All data are from at least two different batches of oocytes. Data represent mean ± SD. An unpaired t test was used to compare two means. One-way analysis of variance (ANOVA) was used for multiple comparisons followed by a Tukey post hoc test. *p < 0.05; **p < 0.01; ***p < 0.001.
Loss of righting reflex assay in Xenopus tadpole
Animals were used and experiments were carried out with approval and according to the guidelines of the MGH Institutional Animal Care and Use Committee. General anesthetic potency was assessed in Xenopus laevis tadpoles as previously described41,52,53. In brief, groups of 10 tadpoles were placed in aqueous solutions containing compound 31, 132, or 4-O-methylhonokiol, and tested every five minutes for loss of righting reflexes (LoRR). Each animal was assigned a score of either awake or LoRR, and the percent of animals anesthetized was plotted against the concentration of the compound tested. Concentration-response data was fitted by non-linear least squares to logistic functions of the form Y = 1/(1 +10^((LogEC50 − Log[Drug])*HillSlope)) using Graphpad Prism 6. Results are reported as EC50s and 95% confidence intervals.
Additional Information
How to cite this article: Maldifassi, M. C. et al. Novel positive allosteric modulators of GABAA receptors with anesthetic activity. Sci. Rep. 6, 25943; doi: 10.1038/srep25943 (2016).
References
Forman, S. A. Molecular approaches to improving general anesthetics. Anesthesiol. Clin. 28, 761–771 (2010).
Chitilian, H. V., Eckenhoff, R. G. & Raines, D. E. Anesthetic drug development: novel drugs and new approaches. Surg. Neurol. Int. 4, S2–S10 (2013).
Macdonald, R. L. & Olsen, R. W. GABAA receptor channels. Annu. Rev. Neurosci. 17, 569–602 (1994).
Barnard, E. A. et al. International union of pharmacology. XV. Subtypes of gamma-aminobutyric acid A receptors: classification on the basis of subunit structure and receptor function. Pharmacol. Rev. 50, 291–313 (1998).
Olsen, R. W. & Sieghart, W. International union of pharmacology. LXX. Subtypes of γ-aminobutyric acid type A receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol. Rev. 60, 243–260 (2008).
Sigel, E. & Steinmann, M. E. Structure, function, and modulation of GABAA receptors. J. Biol. Chem. 287, 40224–40231 (2012).
Rabow L. E., Russek, S. J. & Farb, D. H. From ion currents to genomic analysis: recent advances in GABAA receptor research. Synapse 21, 189–274 (1995).
Chang, Y., Wang, R., Barot, S. & Weiss, D. S. Stoichiometry of a recombinant GABAA receptor. J. Neurosci. 16, 5415–5424 (1996).
Farrar, S. J., Whiting, P. J., Bonnert, T. P. & McKernan, R. M. Stoichiometry of a ligand-gated ion channel determined by fluorescence energy transfer. J. Biol. Chem. 274, 10100–10104 (1999).
Tretter, V., Ehya, N., Fuchs & Sieghart, K. W. Stoichiometry and assembly of a recombinant GABAA receptor subtype. J. Neurosci. 17, 2728–2737 (1997).
Baumann, S. W., Baur, R. & Sigel, E. Subunit arrangement of gamma aminobutyric acid type A receptors. J. Biol. Chem. 276, 36275–36280 (2001).
Baumann, S. W., Baur, R. & Sigel, E. Forced subunit assembly in α1β2γ2 GABAA receptors. Insight into the absolute arrangement. J. Biol. Chem. 277, 46020–46025 (2002).
Baur, R., Minier, F. & Sigel, E. A GABAA receptor of defined subunit composition and positioning: concatenation of five subunits. FEBS Lett. 580, 1616–1620 (2006).
Sigel, E., Baur, R., Trube, G., Mohler, H. & Malherbe, P. The effect of subunit combination of rat brain GABAA receptors on channel function. Neuron. 5, 703–711 (1990).
Minier, F. & Sigel, E. Positioning of the α-subunit isoforms confers a functional signature to γ-aminobutyric acid type A receptors. Proc. Natl. Acad. Sci. USA 101, 7769–7774 (2004).
Sigel, E. & Lüscher, B. P. A closer look at the high affinity benzodiazepine binding site on GABAA receptors. Curr. Top. Med. Chem. 11, 241–246 (2011).
Smith, G. B. & Olsen, R. W. Functional domains of GABAA receptors. Trends Pharmacol. Sci. 16, 162–168 (1995).
Baur, R. et al. Covalent modification of GABAA receptor isoforms by a diazepam analogue provides evidence for a novel benzodiazepine binding site that prevents modulation by these drugs. J. Neurochem. 106, 2353–2363 (2008).
Ramerstorfer, J. R. et al. The GABAA receptor α+/β– interface: a novel target for subtype selective drugs. J. Neurosci. 31, 870–877 (2011).
Walters, R. J., Hadley, S. H., Morris, K. D. & Amin, J. Benzodiazepines act on GABAA receptors via two distinct and separable mechanisms. Nat. Neurosci. 12, 1274–1281 (2000).
Nishikawa, K., Jenkins, A., Paraskevakis, I. & Harrison, N. L. Volatile anesthetic actions on the GABAA receptors: contrasting effects of α1(S270) and β2(N265) point mutations. Neuropharmacol. 42, 337–345 (2002).
Chang, C. S., Olcese, R. & Olsen, R. W. A single M1 residue in the β2 subunit alters channel gating of GABAA receptor in anesthetic modulation and direct activation. J. Biol. Chem. 278, 42821–42828 (2003).
Li, G. D. et al. Identification of a GABAA receptor anesthetic binding site at subunit interfaces by photolabeling with an etomidate analog. J. Neurosci. 26, 11599–11605 (2006).
Yip, G. M. et al. A propofol binding site on mammalian GABAA receptors identified by photolabeling. Nat. Chem. Biol. 9, 715–720 (2013).
Rudolph, U. & Antkowiak, B. Molecular and neuronal substrates for general anaesthetics. Nat. Rev. Neurosci. 5, 709–720 (2004).
Belelli, D., Lambert, J. J., Peters, J. A., Wafford, K. & Whiting, P. J. The interaction of the general anesthetic etomidate with the gamma-aminobutyric acid type A receptor is influenced by a single amino acid. Proc. Natl. Acad. Sci. USA 94, 11031–11036 (1997).
Krasowski, M. D. et al. Propofol and other intravenous anesthetics have sites of action on the gamma-aminobutyric acid type A receptor distinct from that for isoflurane. Mol. Pharmacol. 53, 530–538 (1998).
Siegwart, R., Krahenbuhl, K., Lambert, S. & Rudolph, U. Mutational analysis of molecular requirements for the actions of general anaesthetics at the γ-aminobutyric acid type A receptor subtype, α1β2γ2 . BMC Pharmacol. 3, 13 (2003).
Stewart, D. S., Pierce, D. W., Hotta, M., Stern, A. T. & Forman, S. A. Mutations at βN265 in γ-aminobutyric acid type A receptors alter both binding affinity and efficacy of potent anesthetics. PLoS One 9, e111470 (2014).
Jurd, R. et al. General anesthetic actions in vivo strongly attenuated by a point mutation in the GABA(A) receptor β3 subunit. FASEB J. 2, 250–252 (2002).
Hill-Venning, C., Belelli, D., Peters, J. A. & Lambert, J. J. Subunit-dependent interaction of the general anaesthetic etomidate with the gamma-aminobutyric acid type A receptor. Br. J. Pharmacol. 120, 749–756 (1997).
Franks, N. P. General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal. Nat. Rev. Neurosci. 5, 370–386 (2008).
Jayakar, S. S. et al. Multiple propofol-binding sites in a γ-aminobutyric acid type A receptor (GABAAR) identified using a photoreactive propofol analog. J. Biol. Chem. 289, 27456–27468 (2014).
Middendorp, S. J., Puthenkalam, R., Baur, R., Ernst, M. & Sigel, E. Accelerated discovery of novel benzodiazepine ligands by experiment-guided virtual screening. ACS Chem. Biol. 9, 1854–1859 (2014).
Middendorp, S. J., Maldifassi, M. C., Baur, R. & Sigel, E. Positive modulation of synaptic and extrasynaptic GABAA receptors by an antagonist of the high affinity benzodiazepine binding site. Neuropharmacol. 95, 459–467 (2015).
Baur, R., Schuehly, W. & Sigel, E. Moderate concentrations of 4-O-methylhonokiol potentiate GABAA receptor currents stronger than honokiol. Biochim. Biophys. Acta. 1840, 3017–3021 (2014).
Maldifassi, M. C., Baur, R. & Sigel, E. Functional sites involved in modulation of the GABAA receptor channel by the intravenous anesthetics propofol, etomidate and pentobarbital. Neuropharmacol. 105, 207–214 (2016).
Miller, P. S. & Aricescu, A. R. Crystal structure of a human GABAA receptor. Nature 512, 270–275 (2014).
Wingrove, P. B., Wafford, K. A., Bain, C. & Whiting, P. J. The modulatory action of loreclezole at the γ-aminobutyric acid type A receptors is determined by a single amino acid in the β2 and β3 subunit. Proc. Natl. Acad. Sci. USA 91, 4569–4573 (1994).
Fernandez, S. P. et al. Flavan-3-ol esters: new agents for exploring modulatory sites on GABAA receptors. Br. J. Pharmacol. 165, 965–977 (2012).
Stein, M. et al. Azo-propofols: photochromic potentiators of GABAA receptors. Angew. Chem. Int. Ed. Engl. 51, 10500–10504 (2012).
Chiara, D. C. et al. Mapping general anesthetic binding site(s) in human α1β3 γ-aminobutyric acid type A receptors with [3H]TDBzl-etomidate, a photoreactive etomidate analogue. Biochemistry 51, 836–847 (2012).
Chiara, D. C. et al. Specificity of intersubunit general anesthetic binding sites in the transmembrane domain of the human α1β3γ2 GABAA receptor. J. Biol. Chem. 288, 19343–19357 (2013).
Luger, D. et al. Identification of the putative binding pocket of valerenic acid on GABAA receptors using docking studies and site-directed mutagenesis. Br. J. Pharmacol. 22, 5403–5413 (2015).
Guitchounts, G., Stewart, D. S. & Forman, S. A. The two etomidate sites in α1β2γ2 gamma-aminobutyric acid type A receptors contribute equally and noncooperatively to modulation of channel gating. Anesthesiology. 116, 1235–1244 (2012).
Jacob, T. C., Moss, S. J. & Jurd, R. GABAA receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat. Rev. Neurosci. 9, 331–343 (2008).
Farrant, M. & Nusser, Z. Variations on an inhibitory theme: Phasic and tonic activation of GABAA receptors. Nature Rev. Neurosci. 6, 215–229 (2005).
Husain, S. S. et al. 2-(3-Methyl-3H-diaziren-3-yl)ethyl 1-(1-phenylethyl)-1H-imidazole-5-carboxylate: a derivative of the stereoselective general anesthetic etomidate for photolabeling ligand-gated ion channels. J. Med. Chem. 46, 1257–1265 (2003).
Sigel, E. Properties of single sodium channels translated by Xenopus oocytes after injection with messenger ribonucleic acid. J. Physiol. Lond. 386, 73–90 (1987).
Sigel, E. & Minier, F. The Xenopus oocyte: system for the study of functional expression and modulation of proteins. Mol. Nutr. Food Res. 49, 228–234 (2005).
Boileau, A. J., Baur, R., Sharkey, L. M., Sigel, E. & Czajkowski, C. The relative amount of cRNA coding for γ2 subunits affects stimulation by benzodiazepines in GABAA receptors expressed in Xenopus oocytes. Neuropharmacol. 43, 695–700 (2002).
Desai, R. et al. Contrasting actions of a convulsant barbiturate and its anticonvulsant enantiomer on the α1β3γ2 GABAA receptor account for their in vivo effects. J. Physiol. 593, 4943–4961 (2015).
Ge, R. L. et al. Pharmacological studies of methoxycarbonyl etomidate’s carboxylic acid metabolite. Anesth. Analg. 115, 305–308 (2012).
Baumann, S. W., Baur, R. & Sigel, E. Individual properties of the two functional agonist sites in GABAA receptors. J. Neurosci. 23, 11158–11166 (2003).
Acknowledgements
This work was supported by the Swiss National Science Foundation grant 315230_156929/1. M.C.M. is a recipient of a fellowship (Beca Chile Postdoctorado from CONICYT, Ministerio de Educacion, Chile). Andrea Szabo (Department of Anesthesia Critical Care & Pain Medicine at Massachusetts General Hospital, Boston, MA, USA) helped to perform tadpole loss of righting reflexes studies. 4-O-methylhonokiol was a kind gift by Dr. Wolfgang Schuehly (Karl-Franzens-University, Graz, Austria).
Author information
Authors and Affiliations
Contributions
M.C.M. and R.B. performed electrophysiological experiments. D.P. and A.N. performed LoRR experiments. M.C.M., S.A.F. and E.S. designed the experiments, analysed the data, and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Maldifassi, M., Baur, R., Pierce, D. et al. Novel positive allosteric modulators of GABAA receptors with anesthetic activity. Sci Rep 6, 25943 (2016). https://doi.org/10.1038/srep25943
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep25943
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
