
Abstract
Biofilms are complex communities of microbes that attach to biotic or abiotic surfaces causing chronic infectious diseases. Within a biofilm, microbes are embedded in a self-produced soft extracellular matrix (ECM), which protects them from the host immune system and antibiotics. The nanoscale visualisation of delicate biofilms in liquid is challenging. Here, we develop atmospheric scanning electron microscopy (ASEM) to visualise Gram-positive and -negative bacterial biofilms immersed in aqueous solution. Biofilms cultured on electron-transparent film were directly imaged from below using the inverted SEM, allowing the formation of the region near the substrate to be studied at high resolution. We visualised intercellular nanostructures and the exocytosis of membrane vesicles, and linked the latter to the trafficking of cargos, including cytoplasmic proteins and the toxins hemolysin and coagulase. A thick dendritic nanotube network was observed between microbes, suggesting multicellular communication in biofilms. A universal immuno-labelling system was developed for biofilms and tested on various examples, including S. aureus biofilms. In the ECM, fine DNA and protein networks were visualised and the precise distribution of protein complexes was determined (e.g., straight curli, flagella, and excreted cytoplasmic molecular chaperones). Our observations provide structural insights into bacteria-substratum interactions, biofilm development and the internal microbe community.
Similar content being viewed by others


Introduction
Biofilms are highly organized microbial communities on surfaces, such as the surfaces of medical implants and host organisms. The microbes in them, are embedded in a self-produced extracellular matrix (ECM)1 consisting of proteins2, polysaccharides3 and/or extracellular DNA (eDNA)4. The ECM has diverse functions to maintain the structural integrity of the biofilm and adapt to surrounding environments5. The resistance it confers to antimicrobial agents and host immune systems6 is a deleterious property, causative of various chronic human infectious diseases, including periodontal disease, bacteremia, pneumonia and meningitis. Biofilm-embedded bacteria cooperate and communicate with each other5. Thus, biofilms act as multi-cellular organisms, allowing microbes to survive in various environments and attack their host organisms1,6. Biofilms also cause familiar problems, such as those encountered in the maintenance of drinking water distribution systems.
Direct observation of the structure of biofilms is essential to understand their development and functions. Generally, optical microscopy (OM) is employed to observe bacterial cells or macro-organelles, and confocal laser scanning microscopy (CLSM) has made a large contribution to biofilm research7. Using fluorescence probes, CLSM can visualise the entire expanse of a biofilm, describe its overall structure and localize ECM constituents. However, the resolution of diffraction-limited OM is restricted to approximately 200 nm. Super-resolution OM overcomes this limitation8,9,10,11, and is providing exciting information about biofilms12. Nevertheless, this method images special fluorescence probes, and the visualisation of the surrounding structures is still awaited for the comprehensive understanding of biofilms.
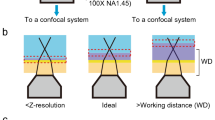
Conventional electron microscopy (EM) has been widely used to image biofilms at sub-nanometre resolution13. However, the sample is under vacuum, making pretreatments that might affect the soft and hydrophilic structure of biofilms (e.g., dehydration) basically unavoidable. Recently, atmospheric scanning electron microscopy (ASEM) was developed to observe biological samples at atmospheric pressure14. ASEM allows an inverted SEM to observe a wet sample from below while an OM simultaneously observes it from above15. Cells can be cultured, fixed and imaged in the specialized sample dish (ASEM dish) at atmospheric pressure16,17. The dish has an electron transparent, 100 nm-thick silicon nitride (SiNx is inevitably oxidized by the fabrication process, yielding SiNxOy18) film window in its base to allow SEM14,15, and holds a few millilitres of solution, enabling direct cell culture under stable conditions (Fig. 1). Importantly, at 30 kV acceleration voltage ASEM observes the 2–3 μm-thick specimen regions directly above the film, allowing structures near the film to be visualised at 8 nm resolution. In addition, the time-consuming sample preparation generally required for immuno-EM is not required for immuno-ASEM16,19 and the effort involved is comparable to that for immuno-OM.

(a) Bacteria were grown in an appropriate medium in the removable, 35-mm ASEM dish with SiNxOy film window at the centre of its base. After the indicated incubation time at 37 °C, biofilms were fixed with 1% glutaraldehyde (GA) and 4% paraformaldehyde (PFA) for heavy metal-labelling and charged Nanogold-labelling or only with 4% PFA for immuno-labelling. After the indicated labelling procedures, the solution in the ASEM dish was replaced by a radical scavenger solution (10 mg/ml ascorbic acid or 10 mg/ml glucose). (b) Diagram of the ASEM showing the inverted SEM, the detector and the specimen dish, which separates the atmosphere (above) and the column vacuum (below). The electron beam passes through the window and is projected up onto the biofilms immersed in solution, penetrating them to a depth of 2–3 μm. Backscattered electrons (BSE) are captured by a BSE imaging (BEI) detector.
In this study, we observed biofilms formed by a clinically important Gram-positive pathogen, Staphylococcus aureus, and a model Gram-negative bacterium, Escherichia coli. In combination with several labelling methods, ASEM could visualise the delicate structure of these biofilms and the fine structure of ECM components at the interface between the bacteria and the solid surface, in liquid (Fig. 1). Our results provide important information that will help to visualise biofilms immersed in solution by EM.
Results
Heavy metal staining of biofilms for ASEM
Here we report staining protocols developed to allow the detailed visualisation of biofilms by ASEM (Fig. 1) and the information acquired by their use. Heavy metal stains with the ability to readily permeate fixed cells and stain specific biological molecules, such as proteins, lipids and nucleic acids, have been widely developed for EM. Therefore, we expected that these stains should be also applicable to thick biofilms. We tested the ability of osmic acid (OA), uranyl acetate (UA) and lead citrate (LC) to stain 24-h biofilms of a clinically isolated strain of methicillin-resistant S. aureus (MRSA), MR23, which produces robust proteinaceous biofilms (Supplementary Table 1)20,21,22. ASEM imaging at 30 kV revealed that sequential staining with OA, UA, and LC drastically improved image contrast (Fig. 2a). Using this method, biofilm development was visualised over time (Fig. 2b). The number of surface-attached cells increased with the incubation period, and the proliferation pattern correlated well with quantification data obtained by a conventional crystal violet (CV)-staining method (Supplementary Fig. 1), indicating that the heavy metal stain permeated the biofilms. ASEM images of 24-h biofilms in an aqueous environment revealed that bacterial cells do not align in close proximity to each other at the bottom of biofilms (Fig. 2a) and that there are cell-free regions, which are probably so-called water channels (Supplementary Fig. 2a,c,e). Importantly, air-drying biofilms induced abnormal cell aggregation on the solid surface (Supplementary Fig. 2b,d,f), indicating that ASEM of wet samples is required to visualise native biofilm structure.

(a) S. aureus MR23 biofilm formed on an ASEM dish and, after 24 h, sequentially stained with heavy metals in the order: osmic acid (OA), uranyl acetate (UA), and lead citrate (LC). The ASEM images were recorded from the same area after each staining step. (b) Time course of biofilm formation on ASEM dishes. At the indicated time points, biofilms were stained with OA/UA/LC and observed by ASEM. (c) Higher magnification ASEM images of an MR23 biofilm at the indicated culture times. Arrows and arrowheads indicate filamentous and spherical structures, respectively. Scale bars, 1 μm in (a,c), and 10 μm in (b).
ASEM visualisation of the development of fibrillar nanostructures and vesicles in biofilms
Higher magnification ASEM images revealed that presence of nanostructures in 4- to 24-h biofilms of MR23 (Figs 2c and 3a,b). These included small spheres and fibrils. Careful observation also provided clear pictures of spheres associated with bacterial cells, and seem to be snapshots of the budding of membrane vesicles (MVs) (Fig. 3b). In agreement, when imaged by conventional SEM the cells were seen to have a rough-surface (Supplementary Fig. 3a–d). As demonstrated by time course images, the diameter of the spheres increased during biofilm development, and spheres with diameters ranging from 100 to 200 nm became major populations (Fig. 3c,d). Broken bacterial cells were also observed in 10- to 24-h biofilms (Fig. 3c). Serial thin-sectioning transmission electron microscopy (TEM) also captured snapshots of budding MVs, and indicated the presence of cytoplasmic substances within them (Fig. 4a, Supplementary Movies S1–S6). Next we isolated MVs from supernatants of planktonic and biofilm cultures. TEM images revealed that more MVs were produced under biofilm conditions than under planktonic conditions (Fig. 4b,c). The observed spherical structures were stained by the fluorescence-membrane probe FM4-64 and disappeared after detergent treatment, confirming their assigned identity (Supplementary Fig. 4). Sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS) and Western blotting indicated that cytoplasmic proteins, such as the molecular chaperone ClpB, existed in the MV fractions (Fig. 4d). The presence of these cytoplasmic cargos was confirmed by fluorescence microscopy using the ClpB::GFPuv translational fusion (Fig. 4e) and GFPuv (Supplementary Fig. 5) derived from the S. aureus genome and plasmid, respectively. Collectively, these results suggest that cytoplasmic macromolecules, including proteins, are excreted via an MV-dependent pathway, as well as by cell lysis and uncharacterized transporters (Supplementary Fig. 6). MVs might also mediate the delivery of toxins, such as hemolysin (Supplementary Fig. 7a) and coagulase (Supplementary Fig. 7b), from the microbes within biofilms, as previously reported for various bacteria23,24,25.

(a) ASEM image of an 8-h old S. aureus MR23 biofilm stained with OA/UA/LC. (b) Higher magnification image of the white rectangle in (a) revealing the presence of spherical nanostructures (arrowheads). (c) Production of spherical nanostructures (arrowheads) at the indicated time points during biofilm development. Broken bacterial cells (arrows) were also sometimes observed. (d) Histograms showing the number of spherical nanostructures with diameters in the indicated ranges at the indicated time points during biofilm development; as membrane vesicles (MVs) could only be observed by ASEM, the cultures were fixed at various time points, and arbitrary areas were imaged for manual statistical analysis. Scale bars, 1 μm.

(a) Production of membrane vesicles (MVs) within 4-h biofilms of MR23 observed by TEM. Epon812 embedded biofilm was thin-sectioned serially and imaged. Higher magnification images of the black rectangles are also shown. (b,c) Isolated MVs from planktonic (b) and biofilm cultures (c) observed by negative stain TEM. A higher magnification image is shown in the inset of (c). (d) Immunoblots of isolated membrane vesicles. Input, supernatant, and pelleted fractions obtained by ultracentrifugation, were separated by SDS-PAGE and the separated proteins were blotted onto a PVDF membrane. The cytoplasmic molecular chaperones ClpB and DnaK were detected using anti-ClpB and anti-DnaK antibodies, respectively. While DnaK was found in the supernatant, excreted ClpB mainly present the membrane pellet. (e) Images of the ClpB::GFPuv translational fusion produced from the genome of MR23. Membranes were stained with FM4-64 for the fluorescence microscopy. Phase contrast, green fluorescence (GFPuv), and red fluorescence (FM4-64) images are displayed in gray scales. The merged fluorescence image is also shown. Green: GFPuv; magenta: FM4-64. Lower panels: Higher magnification images of the white rectangles, documenting the secretion of ClpB in vesicles. Arrowheads indicate ClpB::GFPuv/FM4-64-positive spherical nanostructures. Scale bars, 100 nm in (a–c), 10 μm in (e).
Charged Nanogold labelling of fibrils and flagella in biofilms
Positively charged Nanogold (PCG)-labelling is an efficient and rapid method used to visualise the surfaces of biological samples, such as bacteria and cultured animal cells, exploiting their net negative surface charges26,27. Here, we compared the ability of PCG and recently available negatively charged Nanogold (NCG) to label biofilms produced by S. aureus SH1000, which harbour ECM mainly comprised of extracellular polysaccharides21 (Supplementary Fig. 3e–h). PCG visualised biofilm cell surfaces clearly (Fig. 5a), indicating that the surface of S. aureus, which is composed of peptidoglycans, teichoic acids and proteins, has a net negative charge. In contrast, NCG formed high contrast dots on the cell surfaces but did not stain them entirely. The alignment patterns of the bright dots resemble those observed by lectin-labelling using a colloidal gold-conjugated wheat germ agglutinin (WGA-gold; Fig. 5a), suggesting that some of them correspond to polysaccharides and/or glycoproteins, both of which contain N-acetylglucosamine residues28,29 and, thus, interact with WGA. This is the first observation of bacterial samples labelled with NCG. Sequential labelling with NCG and then PCG resulted in combined labelling, while labelling with PCG and then NCG resulted in similar or lower contrast images than those obtained with PCG alone. Simultaneous labelling with PCG and NCG was inefficient, probably because the net charge of one masked that of the other. Collectively, these results indicate that PCG-labelling is the simplest and most efficient of the five Nanogold methods tested, resulting in high contrast images of S. aureus biofilms.

(a) Two-hour biofilms of S. aureus SH1000 were labelled with positively charged Nanogold (PCG), negatively charged Nanogold (NCG), or colloidal gold-conjugated wheat germ agglutinin (WGA-gold). PCG and NCG were used sequentially or, if required, simultaneously, as indicated. After gold enhancement, specimens were observed by ASEM. (b) Four-hour biofilms of S. aureus MR10 labelled with PCG, gold-enhanced, and observed by ASEM. A higher magnification image of the white rectangle is shown on the right. The arrow indicates a PCG-positive fibril. (c) S. aureus MR10 biofilms were grown in BHIG medium supplemented with, or without, the indicated enzymes for 4 h at 37 °C, labelled with PCG, and observed by ASEM. Arrows mark PCG-positive fibrillar structures. (d) Four-hour biofilms of MR10 were labelled with anti-dsDNA mouse IgG primary antibody and colloidal gold-conjugated anti-mouse IgG secondary antibody (anti-dsDNA). The biofilms were subsequently counter-stained with PCG (anti-dsDNA/PCG). A higher magnification image of the white rectangle is shown on the right. Arrowheads and arrows mark linearly aligned colloidal gold particles and PCG-positive fibrillar structures, respectively. Scale bars, 1 μm.
PCG-labelling was further applied to visualise biofilms formed by another S. aureus strain, MR10, which also produces robust biofilms harbouring ECM mainly comprised of extracellular polysaccharides21. Fibrils connecting MR10 cells were apparent (Fig. 5b). Similar structures were observed in other extracellular polysaccharide-rich biofilms of Staphylococcus epidermidis SE422 by PCG-labelling and ASEM (Supplementary Fig. 8). Investigating MR10 biofilms with the fluorescent probe Alexa 488-conjugated WGA (WGA-Alexa 488) revealed that fibrils connected bacterial cells and were degraded by polysaccharide-hydrolysing enzyme dispersin B22 (Supplementary Fig. 9). However, we could not conclude that PCG-positive fibrils visualised by ASEM were in fact polysaccharide networks as the ECM of MR10 biofilms also harbours DNA22, a ubiquitous and pivotal component in diverse bacterial biofilms4. To assess this precisely, MR10 biofilms were formed in the presence or absence of ECM-degrading enzymes, including DNase I, proteinase K, and dispersin B and imaged from below by ASEM. PCG-positive S. aureus fibrils became invisible in the presence of DNase I, but were still observed in biofilms formed in the presence of dispersin B and proteinase K (Fig. 5c). Immuno-labelling with anti-double strand DNA (dsDNA) primary antibody and colloidal gold-conjugated secondary antibody resulted in linearly aligned gold particles, and counter-labelling with PCG enhanced the contrast of the regions between them revealing fibrils (Fig. 5d). In contrast, when primary antibody-labelling was omitted, such gold particles was not observed (Supplementary Fig. 10). These results indicate that the S. aureus fibrils observed by PCG-labelling and ASEM were DNA fibrils present at the bottom of the biofilms, whereas polysaccharides, main ECM components, might cover the top surface of the biofilms as observed by conventional SEM (Supplementary Fig. 3e–h).
Flagella are thought to be involved in surface attachment of bacteria at the initial stage of biofilm development30,31. PCG-labelling is potentially useful for visualising flagella as reported previously26. Here, we visualised flagella more carefully for various strains of E. coli, i.e., the K-12 wild type32 and the isogenic mutants: ΔfliC, ΔcsgA, ΔcsgD, ΔcsgG, and ΔfimA. The fliC gene is involved in the biosynthesis of flagella33; the csgA, csgD, and csgG genes are required for the production of curli, the extracellular functional amyloid that is crucial for biofilm formation34; the fimA gene encodes the type I pili subunit35. Delicate spiral fibrillar structures were observed in all of the strains examined, except the ΔfliC mutant (Fig. 6a, Supplementary Fig. 11). Interestingly, the number of flagella per cell for ΔcsgA, ΔcsgD, and ΔcsgG was more than for the wild type and ΔfimA (Fig. 6a, Supplementary Fig. 11). This observation was highly correlated with the motility measured on a soft agar plate (Fig. 6b,c), suggesting that curli production negatively regulates flagellum production. Cross-regulation between flagella and curli biosynthesis is mediated by the flagellar master regulator FlhDC, the stationary phase sigma factor σS (RpoS) and the key biofilm regulator CsgD. All have previously been shown to act as major hubs regulated by small RNAs36,37. According to the finding that the fliE and fliFGHIJK operons for flagellum formation are directly repressed by CsgD36, increased production of flagella in the ΔcsgD strain is understandable. On the other hand, the mechanism(s) causing increased flagellum production in the ΔcsgA and ΔcsgG strains is unclear. Curli themselves might induce cell envelope alterations, leading to the negative regulation of FlhDC by the RscCDB His-Asp phosphorelay system38.

(a) The indicated E. coli strains were labelled with PCG and observed in liquid by ASEM. Flagella were prominent. (b) Swimming activity of the E. coli strains on 0.3% soft agar plates. (c) Estimated diameters of the E. coli colonies in (b). The means and standard deviations of results from at least three independent experiments are shown. **P < 0.01; n.s., not significant. Scale bars, 1 μm.
Immuno-labelling of proteins in Staphylococcus aureus biofilms
The ease with which immuno-labelling can be achieved is a great advantage of ASEM over conventional EM16,27, however, the penetration of antibodies is critical for observing biofilms. Moreover, the presence of several IgG-binding proteins, including staphylococcal protein A (Spa) and the second IgG-binding protein (Sbi) in S. aureus biofilms (Supplementary Fig. 12a–c), prevents the immuno-detection of some proteins of interest22. We, therefore, constructed S. aureus mutants lacking the spa and/or sbi gene(s). Wild type and single deletion mutants displayed non-specific IgG-binding, but this was abolished in the double knockouts (Supplementary Fig. 12d). Of note, all mutants formed biofilms at the level of the wild type (Supplementary Fig. 12e). Using anti-Eap primary antibody and Nanogold-conjugated goat anti-rabbit Fab’, the extracellular adherence protein (Eap) was labelled in 2-h thin biofilms formed by the Δspa Δsbi strain, and cells were subsequently, counter stained by a modified National Center for Microscopy and Imaging Research (NCMIR) method (Supplementary Fig. 13)39. ASEM revealed gold particles localized on the surface of Δspa Δsbi cells and around them (Fig. 7a, upper panels), as previously demonstrated by fluorescence microscopy21. However, only few signals from gold particles were observed for the control Δspa Δsbi Δeap strain (Fig. 7a, lower panels). Fab fragments of primary antibodies and colloidal gold-conjugated Spa were also useful for immuno-labelling cell surface proteins, such as Spa and the excreted cytoplasmic molecular chaperones ClpB and DnaK, both of which were identified in biofilm matrices of S. aureus21 (Supplementary Figs 14 and 15). In both experiment series, counter staining by the modified NCMIR method enhanced the contrast of bacterial cells (Fig. 7a) more than counter staining with OA and UA (Supplementary Figs 14 and 15).

(a) Immuno-labelled biofilm cells of the MR23 Δspa Δsbi mutant (top), which was used to prevent nonspecific binding of IgGs to Spa and Sbi, and the similarly treated MR23 Δspa Δsbi Δeap mutant (bottom), which was used as a negative control. The same areas are shown in the left (immuno-labelling) and the right (counter staining) panels. (b,c) Curli production in the wild type and the ΔcsgA strains for 7 days at 25 °C observed by negative stain TEM after air-drying (b) and in solution by immuno-labelled ASEM (c). Curli were labelled with anti-curli rabbit IgG primary antibody and Nanogold-conjugated secondary antibody for the ASEM. Two typical images of these strains are shown. Scale bars, 1 μm.
Straight curli in wet biofilms imaged by immuno-labelling ASEM
We next visualised curli produced in E. coli colony biofilms using antibodies raised against purified curli22. Curli, but neither type I pili nor flagella, were crucial for biofilm formation in E. coli under the conditions tested (Supplementary Fig. 16). The importance of flagella for biofilm development depends on the culture media and exact conditions used30,40 and is still being debated41. We did not notice any differences in the number of surface-attached cells for the six strains under the conditions tested (Supplementary Fig. 11). Interestingly, TEM images of negatively stained air-dried curli revealed winding fibrils (Fig. 7b), as reported previously34, whereas, immuno-ASEM revealed linearly aligned gold-particles (Fig. 7c), indicating that curli present as straight filaments in an aqueous environment. Thus, air-drying for conventional EM might affect their morphology. Together the results presented show that immuno-ASEM in aqueous solution can be used to visualise the localization and structure of matrix components in biofilms.
Discussion
Here, we report the first observation of bacterial biofilms immersed in an aqueous solution by ASEM (Fig. 1). In combination with several labelling methods, including heavy metal-labelling (Figs 2 and 3), charged Nanogold-labelling (Figs 5 and 6), and immuno-labelling (Fig. 7), ASEM was able to visualise delicate hydrophilic nano-structures of the biofilm-surface interface (Supplementary Fig. 2). This was the case for various types of biofilms produced by Gram-positive cocci (Figs 2,3,5 and 7a) and Gram-negative bacilli (Figs 6 and 7c), suggesting the applicability of this cutting-edge technique to diverse bacterial biofilms. As ASEM can visualise various eukaryotic cells14,15,16,17,26,27,39,42, these should include biofilms formed by fungi.
Environmental cell (EC)-EM43 and environmental EM (EEM)44 were also developed to observe biological samples in aqueous solution. For EC-TEM, a capsule with two electron transparent film, now known as an environmental cell (EC) was developed and improved45,46,47, and has diverged to EC-SEM48. ECs can hold aqueous and gaseous samples, and are placed inside the microscope. However, their small volume (<20 μl) and dimensions probably making immuno-labelling experiments relatively difficult. EEM requires pressure-limiting apertures to allow the observation of samples in a low pressure gas by TEM and SEM. The hydration of the specimens is maintained for a limited period due to evaporation. Compared to EC-EM43,45,46,47,48,49,50 and EEM44, ASEM is easy to use and high throughput.
Atomic force microscopy (AFM) is also a powerful tool for the high-resolution imaging and analysis of biofilms, due to its capacity for imaging with a higher resolution than OM in both air and liquid51. Since it can visualise samples without fixing, labelling and staining, non-invasive imaging can be performed on surfaces in their native states. The force measurement capacity of AFM can provide unique insights into bacteria-bacteria and bacteria-surface interactions52. On the other hand, there are limitations of AFM for the study of biofilms. Imaging of rough surface biofilms with a large difference in height often become difficult. Similar to conventional SEM, AFM obtains top surface images of biofilms but is not able to visualise the interface structure between bacteria and surfaces51. Combining AFM with other techniques including ASEM is a way to study biofilm structures and physiology.
Among the labelling methods used for ASEM in our study, PCG-labelling was the most efficient, simplest and fastest, sample preparation taking less than half an hour and allowing both bacterial cells and extracellular structures to be visualised. PCG-labelling was also useful to counter-stain the immunogold-labelled samples (Fig. 5d). However, the permeation of PCG was insufficient for thick biofilms, probably due to the presence of a large amount of negatively charged molecules in biofilms, which would trap PCG particles. In this case, sequential heavy metal-labelling (OA/UA/LC) provided high contrast images and proved to be a powerful method (Figs 2 and 3), even though it requires a longer labelling/washing time. Immuno-labelling was also easy for E. coli biofilms (Fig. 7c). However, specific labelling in S. aureus biofilms was difficult due to the presence of the IgG-binding proteins, Spa and Sbi. As we demonstrate, this problem can be overcome in three ways: (i) by the use of a Δspa Δsbi strain (Fig. 7a); (ii) by labelling with Fab fragments of primary antibodies (Supplementary Fig. 14); (iii) by using colloidal gold-conjugated Spa instead of the gold-conjugated secondary antibody (Supplementary Fig. 15). These methods were effective for thin biofilms formed in a few hours (2–4 h), but not for thick biofilms due to the inability of antibodies to permeate them, like PCG. The modified NCMIR method proved to be an excellent way to counter stain immuno-labelled biofilms (Fig. 7a), and was superior to other staining methods (Supplementary Figs 14 and 15).
The ASEM system has an inverted SEM below the sample and an OM above it, providing a large potential for correlative OM/EM imaging16,17. CLSM can visualise structure of biofilms at higher resolution than conventional fluorescent microscopy12, and a recent advance including multiphoton confocal microscopy, is providing exciting results in biofilm biology7. These advanced OM may compensate the weak point of the present ASEM that can visualise only 2–3 μm thick specimen regions directly above the SiNxOy film. The development of an ASEM/CLSM hybrid machine is feasible, and would allow the 3D structure of biofilms and their ultrastructure at the bacteria-surface interface to be visualised. Our results provide a new methodology for observing biofilms, and include several findings of biological significance, as mentioned above. ASEM should shed light on unresolved questions, and may also lead to a new research direction in biology.
Methods
Bacterial strains and plasmids
The bacterial strains and plasmids used in this study are listed in Supplementary Table 1. Detailed procedures for the construction of S. aureus mutant strains and culture conditions are described in the Supplementary Methods.
Biofilm formation
Staphylococcal biofilms were formed in brain heart infusion (BHI) medium (Becton Dickinson) supplemented with 1% glucose (BHIG) in ASEM dishes (JEOL, Tokyo, Japan) (Fig. 1), 96-well plates (Corning, Corning, NY, USA), 35-mm plastic dishes (Nunc, Tokyo, Japan), and glass bottom dishes (Matsunami glass, Tokyo, Japan) over the indicated time periods at 37 °C. Biofilms formed in ASEM dishes were fixed, stained, labelled, and imaged by ASEM. Biofilms formed in 96-well plates were stained with 0.2% crystal violet (CV) and quantified by measuring the absorbance at 595 nm with a micro titre plate reader (Infinite F200 Pro, Tecan, Männedorf, Switzerland). Biofilms formed in 35 mm dishes were stained as previously reported20. Biofilms produced in glass bottom dishes were used for indirect immunofluorescence microscopy and lectin-labelling.
E. coli biofilms were formed in YESCA medium in 96-well plates for 7 days at 25 °C and were quantified as previously reported32. E. coli colony biofilms were formed on YESCA plates for 7 days at 25 °C22, and were imaged by ASEM and TEM32.
Fixation
Biofilms were fixed with 4% paraformaldehyde (PFA) and 1% glutaraldehyde (GA) for 10 min at room temperature for heavy metal staining and charged Nanogold labelling. Biofilms were fixed with 4% PFA for 10 min at room temperature for immuno-labelling. After immuno-labelling, biofilms were further fixed with 1% GA for 10 min at room temperature, to stabilize the antibody interactions prior to gold enhancement and/or counter staining with heavy metals.
Heavy metal staining
Biofilms on the ASEM dish were treated twice with 2% OA for 20 min, then with 2% UA for 25 min, and subsequently washed with double distilled water (DDW). If required, the biofilms were further stained with 0.4% LC in 0.4% sodium hydroxide for 5 min.
Modified NCMIR staining method
Immuno-labelled biofilms on the ASEM dish were stained using a slight modification of the NCMIR method developed by the Ellisman’s group53 as recently reported39.
Charged Nanogold labelling
For charged Nanogold-labelling, bacteria on the ASEM dish were incubated with 6 μM positively and/or negatively charged 1.4 nm Nanogold solution (Nanoprobes, Stony Brook, NY, USA) for 20 min at room temperature as previously reported26,42. After washing with DDW, the size of the gold particles was increased by gold enhancement using GoldEnhance-EM (Nanoprobes) for 10 min at room temperature, followed by washing with DDW. The bacteria were imaged by ASEM.
Immuno-labelling
PFA-fixed staphylococcal biofilms were incubated with 5% skimmed milk and 1% goat serum (Sigma, St. Louis, MO, USA) in buffer A [40 mM HEPES (pH 7.4) and 150 mM NaCl] for 1 h at room temperature. PFA-fixed E. coli cells were incubated with 5% skimmed milk in buffer A for 1 h at room temperature. Detailed information of the immuno-labelling is given in the Supplementary Methods.
Gold-conjugated lectin-labelling
For lectin labelling, one-hour biofilms of S. aureus SH1000 were fixed with 4% PFA and 1% GA for 10 min at room temperature, washed with PBS, and incubated with 30-nm colloidal gold-conjugated wheat germ agglutinin solution (WGA-Gold, EY Laboratories, 1/5 dilution in PBS) for 30 min at 37 °C. After washing with PBS, WGA-Gold was fixed with 1% GA for 10 min at room temperature, followed by washing with DDW. The biofilms were imaged by ASEM.
ASEM imaging
ASEM images were recorded using the ClairScope ASEM system (JASM-6200, JEOL, Ltd, Tokyo, Japan) (Fig. 1)14 as described in the Supplementary Methods.
TEM imaging
Thin sections of S. aureus MR23 biofilm cells, the MV fractions isolated from S. aureus biofilm and planktonic cultures, and E. coli colony biofilms32 were observed by TEM as described in the Supplementary Methods.
Fluorescence microscopy
S. aureus MR23 cells, expressing the ClpB::GFPuv translational fusion (Supplementary Table 1) or GFPuv from the plasmid pP1GFPuv21, were cultured in BHIG medium for 24 h at 37 °C. Aliquots (5 μl) of the cultures were spotted on a glass slide under a cover slip. Specimens were observed using a fluorescence microscope (Nikon, Tokyo, Japan) equipped with B2 (excitation, 450–490 nm; barrier, 520 nm), UV-1A (excitation, 360–370 nm; barrier, 420 nm) and G2A (excitation, 510–560 nm; barrier, 590 nm) filters.
Membranes and DNA were stained with FM4-64 (Life Technologies) and DAPI (Dojindo Laboratory, Kumamoto, Japan), respectively, as recently reported54. Extracellular polysaccharides of S. aureus were labelled with Alexa Fluor 488-conjugated WGA (WGA-Alexa 488, Life Technologies) and observed by fluorescence microscopy as previously reported22.
Congo red (CR)-binding assay
Curli produced by E. coli strains were analysed on YESCA plate containing 2% agar, 10 μg/ml Congo red, and 10 μg/ml Coomassie brilliant blue G-250 as recently reported32.
Isolation of MVs
MVs were isolated from the supernatants of biofilm cultures and planktonic cultures of S. aureus MR23 grown in BHIG medium for 6 h at 37 °C. The 50-ml cultures were centrifuged at 10,000 × g for 25 min at 4 °C. The supernatants were passed through a 0.2 μm filter, and the resulting solutions were concentrated to less than 1 ml using an Amicon 100 K filter (Millipore). The solutions were ultracentrifuged at 150,000 × g for 3 h at 4 °C. The supernatants were collected. The pellets were re-suspended in 1 ml of 10 mM HEPES (pH 7.0) and 100 mM NaCl, and imaged by TEM. The supernatant and pellet (MVs) fractions were used for further experiments.
SDS-PAGE and Western blotting
The concentrated supernatant of the biofilm culture and the supernatant and pellet fractions obtained by ultracentrifugation (see above) were analysed by SDS-PAGE, followed by Western blotting as described in the Supplementary Methods.
Swimming assay
Bacterial swimming activity was examined on a soft agar plate. In parallel, flagella were visualised by ASEM. Detailed information is provided in the Supplementary Methods.
Statistical analysis
The Student’s t test was used to assess biofilm formation and the swimming assays. A value of <0.05 was considered to indicate statistical significance. The means and standard deviations of the results from at least three independent experiments were also calculated.
Additional Information
How to cite this article: Sugimoto, S. et al. Imaging of bacterial multicellular behaviour in biofilms in liquid by atmospheric scanning electron microscopy. Sci. Rep. 6, 25889; doi: 10.1038/srep25889 (2016).
References
Costerton, J. W., Stewart, P. S. & Greenberg, E. P. Bacterial biofilms: a common cause of persistent infections. Science 284, 1318–1322 (1999).
Olsen, A., Jonsson, A. & Normark, S. Fibronectin binding mediated by a novel class of surface organelles on Escherichia coli . Nature 338, 652–655 (1989).
Eighmy, T. T., Maratea, D. & Bishop, P. L. Electron microscopic examination of wastewater biofilm formation and structural components. Appl. Environ. Microbiol. 45, 1921–1931 (1983).
Whitchurch, C. B., Tolker-Nielsen, T., Ragas, P. C. & Mattick, J. S. Extracellular DNA required for bacterial biofilm formation. Science 295, 1487 (2002).
Flemming, H. C. & Wingender, J. The biofilm matrix. Nature Rev. Microbiol. 8, 623–633 (2010).
Davies, D. Understanding biofilm resistance to antibacterial agents. Nat. Rev. Drug Discov. 2, 114–122 (2003).
Neu, T. R. & Lawrence, J. R. Innovative techniques, sensors, and approaches for imaging biofilms at different scales. Trends Microbiol. 23, 233–242 (2015).
Hell, S. W. & Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt. Let. 19, 780–782 (1994).
Gustafsson, M. G. Nonlinear structured-illumination microscopy: wide-field fluorescence imaging with theoretically unlimited resolution. Proc. Natl. Acad. Sci. USA 102, 13081–13086 (2005).
Betzig, E. et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science 313, 1642–1645 (2006).
Rust, M. J., Bates, M. & Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy. Nat. Methods 3, 793–795 (2006).
Berk, V. et al. Molecular architecture and assembly principles of Vibrio cholerae biofilms. Science 337, 236–239 (2012).
Marrie, T. J., Nelligan, J. & Costerton, J. W. A scanning and transmission electron microscopic study of an infected endocardial pacemaker lead. Circulation 66, 1339–1341 (1982).
Nishiyama, H. et al. Atmospheric scanning electron microscope observes cells and tissues in open medium through silicon nitride film. J. Struct. Biol. 169, 438–449 (2010).
Nishiyama, H. et al. Atmospheric scanning electron microscope system with an open sample chamber: configuration and applications. Ultramicroscopy 147, 86–97 (2014).
Maruyama, Y., Ebihara, T., Nishiyama, H., Suga, M. & Sato, C. Immuno EM-OM correlative microscopy in solution by atmospheric scanning electron microscopy (ASEM). J. Struct. Biol. 180, 259–270 (2012).
Hirano, K. et al. Electron microscopy of primary cell cultures in solution and correlative optical microscopy using ASEM. Ultramicroscopy 143, 52–66 (2014).
Zaluzec, N. J. When is Si3N4 not Si3N4? When it is a low stress SiNx membrane window. Microsc. Microanal. 21 (Suppl 3), 959–960 (2015).
Sato, C. et al. Rapid imaging of mycoplasma in solution using Atmospheric Scanning Electron Microscopy (ASEM). Biochem. Biophys. Res. Commun. 417, 1213–1218 (2012).
Sugimoto, S. et al. Cloning, expression and purification of extracellular serine protease Esp, a biofilm-degrading enzyme, from Staphylococcus epidermidis . J. Appl. Microbiol. 111, 1406–1415 (2011).
Sugimoto, S. et al. Staphylococcus epidermidis Esp degrades specific proteins associated with Staphylococcus aureus biofilm formation and host-pathogen interaction. J. Bacteriol. 195, 1645–1655 (2013).
Chiba, A., Sugimoto, S., Sato, F., Hori, S. & Mizunoe, Y. A refined technique for extraction of extracellular matrices from bacterial biofilms and its applicability. Microb. Biotechnol. 8, 392–403 (2015).
Wai, S. N. et al. Vesicle-mediated export and assembly of pore-forming oligomers of the enterobacterial ClyA cytotoxin. Cell 115, 25–35 (2003).
Vanhove, A. S. et al. Outer membrane vesicles are vehicles for the delivery of Vibrio tasmaniensis virulence factors to oyster immune cells. Environ. Microbiol. 17, 1152–1165 (2015).
Thay, B., Wai, S. N. & Oscarsson, J. Staphylococcus aureus alpha-toxin-dependent induction of host cell death by membrane-derived vesicles. Plos One 8, e54661 (2013).
Nishiyama, H., Teramoto, K., Suga, M. & Sato, C. Positively charged Nanogold label allows the observation of fine cell filopodia and flagella in solution by atmospheric scanning electron microscopy. Microsc. Res. Tech. 77, 153–160 (2014).
Kinoshita, T. et al. Immuno-electron microscopy of primary cell cultures from genetically modified animals in liquid by atmospheric scanning electron microscopy. Microsc. Microanal. 20, 469–483 (2014).
Mack, D. et al. The intercellular adhesin involved in biofilm accumulation of Staphylococcus epidermidis is a linear beta-1,6-linked glucosaminoglycan: purification and structural analysis. J. Bacteriol. 178, 175–183 (1996).
Hazenbos, W. L. et al. Novel staphylococcal glycosyltransferases SdgA and SdgB mediate immunogenicity and protection of virulence-associated cell wall proteins. PLoS Pathog. 9, e1003653 (2013).
O’Toole, G. A. & Kolter, R. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol. Microbiol. 30, 295–304 (1998).
Pratt, L. A. & Kolter, R. Genetic analysis of Escherichia coli biofilm formation: roles of flagella, motility, chemotaxis and type I pili. Mol. Microbiol. 30, 285–293 (1998).
Arita-Morioka, K., Yamanaka, K., Mizunoe, Y., Ogura, T. & Sugimoto, S. Novel strategy for biofilm inhibition by using small molecules targeting molecular chaperone DnaK. Antimicrob. Agents Chemother. 59, 633–641 (2015).
Kuwajima, G., Asaka, J., Fujiwara, T., Fujiwara, T., Node, K. & Kondo, E. Nucleotide sequence of the hag gene encoding flagellin of Escherichia coli . J. Bacteriol. 168, 1479–1483 (1996).
Chapman, M. R. et al. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 295, 851–855 (2002).
Klemm, P. The fimA gene encoding the type-1 fimbrial subunit of Escherichia coli. Nucleotide sequence and primary structure of the protein. Eur. J. Biochem. 143, 395–399 (1984).
Ogasawara, H., Yamamoto, K. & Ishihama, A. Role of the biofilm master regulator CsgD in cross-regulation between biofilm formation and flagellar synthesis. J. Bacteriol. 193, 2587–2597 (2011).
Mika, F. & Hengge, R. Small Regulatory RNAs in the control of motility and biofilm formation in E. coli and Salmonella . Int. J. Mol. Sci. 14, 4560–4579 (2013).
Francez-Charlot, A. et al. RcsCDB His-Asp phosphorelay system negatively regulates the flhDC operon in Escherichia coli . Mol. Microbiol. 49, 823–832 (2003).
Memtily, N. et al. Observation of tissues in open aqueous solution by atmospheric scanning electron microscopy: applicability to intraoperative cancer diagnosis. Int. J. Oncol. 46, 1872–1882 (2015).
Prigent-Combaret, C. et al. Developmental pathway for biofilm formation in curli-producing Escherichia coli strains: role of flagella, curli and colanic acid. Environ. Microbiol. 2, 450–464 (2000).
Hengge, R. Reply to “precedence for the structural role of flagella in biofilms”. mBio 4, e00245–00213 (2013).
Murai, T., Sato, M., Nishiyama, H., Suga, M. & Sato, C. Ultrastructural analysis of Nanogold-labeled cell surface microvilli in liquid by atmospheric scanning electron microscopy and their relevance in cell adhesion. Int. J. Mol. Sci. 14, 20809–20819 (2013).
Abrams, I. M. & McBain, J. W. A closed cell for electron microscopy. Science 100, 273–274 (1944).
Danilatos, G. D. Introduction to the ESEM instrument. Microsc. Res. Tech. 25, 354–361 (1993).
Evans, J. E. et al. Visualizing macromolecular complexes with in situ liquid scanning transmission electron microscopy. Micron 43, 1085–1090 (2012).
Williamson, M. J., Tromp, R. M., Vereecken, P. M., Hull, R. & Ross, F. M. Dynamic microscopy of nanoscale cluster growth at the solid-liquid interface. Nat. Materials 2, 532–536 (2003).
de Jonge, N. & Ross, F. M. Electron microscopy of specimens in liquid. Nat. Nanotechnol. 6, 695–704 (2011).
Thiberge, S. et al. Scanning electron microscopy of cells and tissues under fully hydrated conditions. Proc. Natl. Acad. Sci. USA 101, 3346–3351 (2004).
Fukushima, K., Katoh, M. & Fukami, A. Quantitative measurements of radiation damage to hydrated specimens observed in a wet gas environment. J. Electron Microsc. 31, 119–126 (1982).
de Jonge, N., Peckys, D. B., Kremers, G. J. & Piston, D. W. Electron microscopy of whole cells in liquid with nanometer resolution. Proc. Natl. Acad. Sci. USA 106, 2159–2164 (2009).
Wright, C. J., Shah, M. K., Powell, L. C. & Armstrong, I. Application of AFM from microbial cell to biofilm. Scanning 32, 134–149 (2010).
Beech, I., Smith, J. R., Steele, A. A., Penegar, I. & Campbell, S. A. The use of atomic force microscopy for studying interactions of bacterial biofilms with surfaces. Colloids Surf. B Biointerfaces 23, 231–247 (2002).
Deerinck, T. J., Bushong, E. A., Thor, A. & Ellisman, M. H. NCMIR methods for 3D EM: a new protocol for preparation of biological specimens for serial block face scanning electron microscopy. Microscopy 6–8 (2010). Available at http://gatan.actonservice.com/acton/attachment/11413/f-017e/1/-/-/-/-/sbfsem%20sample%20prep%20protocol.pdf?modal=1 (2010) (Accessed:19th April 2016).
Sugimoto, S., Arita-Morioka, K., Mizunoe, Y., Yamanaka, K. & Ogura, T. Thioflavin T as a fluorescence probe for monitoring RNA metabolism at molecular and cellular levels. Nucleic Acids Res. 43, e92 (2015).
Acknowledgements
We acknowledge Mrs. R. Shigemori and K. Sekiguchi for the construction of S. aureus mutants, all members of the Department of Bacteriology at Jikei University School of Medicine for valuable discussions and suggestions, Dr. T. Tachibana and all members of the Core Research Facilities at Jikei University School of Medicine for setting-up the ASEM instrument. This work was supported by a Grant-in-Aid for Young Scientists (A) to S.S. from JSPS (15H05619), by a Grant-in-Aid for Scientific Research (B) to Y.M. from JSPS (26293100), by a grant to Y.M. from MEXT-Supported Program for the Strategic Research Foundation at Private Universities, 2012–2016, by a grant from CREST JST to C.S., by a grant for Sparse Modeling from MEXT to C.S., by a grant from CANON to C.S., by a Grant-in-Aid for Scientific Research to C.S. from JSPS (15K14499), and by the Joint Usage/Research Center for Developmental Medicine, IMEG, Kumamoto University.
Author information
Authors and Affiliations
Contributions
S.S., K.O., Y.M. and C.S. planned the project. S.S. and C.S. designed the experiments, developed the assay, performed the experiments together with K.O., R.M., M.S., K.A.-M. and A.C. and analysed the data. S.S., K.O., K.Y., T.O., Y.M. and C.S. wrote the paper with input from all co-authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Sugimoto, S., Okuda, Ki., Miyakawa, R. et al. Imaging of bacterial multicellular behaviour in biofilms in liquid by atmospheric scanning electron microscopy. Sci Rep 6, 25889 (2016). https://doi.org/10.1038/srep25889
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep25889
This article is cited by
-
Instantaneous Clearing of Biofilm (iCBiofilm): an optical approach to revisit bacterial and fungal biofilm imaging
Communications Biology (2023)
-
Liquid-phase ASEM imaging of cellular and structural details in cartilage and bone formed during endochondral ossification: Keap1-deficient osteomalacia
Scientific Reports (2021)
-
Colony spreading of the gliding bacterium Flavobacterium johnsoniae in the absence of the motility adhesin SprB
Scientific Reports (2021)
-
Biofilm formation of Staphylococcus epidermidis imaged using atmospheric scanning electron microscopy
Analytical and Bioanalytical Chemistry (2021)
-
Session 2SHP report—decoding intracellular architecture using visualizing device development and mathematical modeling
Biophysical Reviews (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
