
Abstract
In this paper, a multifunctional Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite catalyst with highly stabilized reactivity and magnetic recyclability was synthesized by a self-assembled method. The magnetic Fe3O4 nanoparticles were coated with a thin layer of the SiO2 to obtain a negatively charged surface. Then positively charged poly(ethyleneimine) polymer (PEI) was self-assembled onto the Fe3O4@SiO2 by electrostatic interaction. Next, negatively charged glutathione capped gold nanoparticles (GSH-AuNPs) were electrostatically self-assembled onto the Fe3O4@SiO2@PEI. After that, silver was grown on the surface of the nanocomposite due to the reduction of the dopamine in the alkaline solution. An about 5 nm thick layer of polydopamine (PDA) was observed to form the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite. The Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite was carefully characterized by the SEM, TEM, FT-IR, XRD and so on. The Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite shows a high saturation magnetization (Ms) of 48.9 emu/g, which allows it to be attracted rapidly to a magnet. The Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite was used to catalyze the reduction of p-nitrophenol (4-NP) to p-aminophenol (4-AP) as a model system. The reaction kinetic constant k was measured to be about 0.56 min−1 (R2 = 0.974). Furthermore, the as-prepared catalyst can be easily recovered and reused for 8 times, which didn’t show much decrease of the catalytic capability.
Similar content being viewed by others

Introduction
Novel metal nanoparticles and nanocomposites have drawn a lot of interest not only due to their unique physical, chemical and biological properties, but also their catalytic activities in many chemical reactions1,2,3,4. Recently, more and more attentions are focused on the gold nanoparticles (AuNPs), silver nanoparticles (AgNPs) and their alloys, because of their high catalytic activity, easy fabrication and recyclability4,5,6,7. In order to enhance their stability and catalytic activity, a number of materials have been used as the solid supports for the construction of the metal-organic/inorganic materials as hybrid catalysts. For example, graphene8, metal-organic framework (MOF)5,9, mesoporous silica (mSiO2)3,10,11, carbon4,12 and polypyrrole13,14 have been immobilized the surface of AuNPs. However, tedious centrifugation process is required during the synthesis of the metal-organic/inorganic catalysts. Besides, the recycling of the catalysts is difficult to achieve. Herein, integrating magnetic nanoparticles (especially Fe3O4 nanoparticles) into the metal–organic/inorganic materials as a magnetically recoverable catalyst provides a promising solution to these problems.
Magnetic particles have many applications in medicine15,16,17, biology18,19 and other fields20,21. Their excellent magnetic operability22,23 greatly favors separation and enrichment of targets15 by the external magnetic fields. Some of the magnetic metal-organic/inorganic catalysts were carefully fabricated6,8,9,24,25. Zheng et al. developed an in situ growth of the AuNPs on Fe3O4@SiO2 nanocomposite by the Sn2+ linkage and reduction6. The Sn2+ was first absorbed on the surface of the Fe3O4@SiO2 and then used to reduce Au3+ by adding sodium formate solution to induce the AuNPs growth. A novel strategy for in situ and controlled synthesis of Fe2O3 on PDA templates through a high temperature thermal decomposition approach was demonstrated by Huang and co-authors26. This magnetic nanoparticle was first encapsulated by surfactant-assisted silica followed by template removal by calcination, then functionalized with gold seeds and served as an ideal scaffold for hollow/permeable nanoreactors. This work is impressive, however, it needs high temperature reaction (290 °C) and calcination. Meanwhile, the existing efforts primarily focused on in situ synthesis of AuNPs on the supports, in which one typically lacked the degree of control over AuNPs growth as achieved in solution-based synthesis8. With the hypothesis that our self-assembly method to construct the functional nanocatalysts might solve these problems, we developed a facile, simple and low-cost method to obtain a novel magnetic–metal nanoparticle hybrid catalyst with enhanced catalytic activity, stability and reusability.
Herein, we proposed to fabricate multifunctional Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite catalyst in a simple self-assembled synthesis method (Fig. 1). The magnetic Fe3O4 nanoparticles were coated with a thin layer of SiO2 to obtain a negatively charged surface. Then positively charged PEI polymer was self-assembled onto the Fe3O4@SiO2 surface by the electrostatic interaction. The electrostatic interaction was also adopted to self-assemble the negatively charged AuNPs on the surfaces of PEI coated Fe3O4@SiO2. After that, the dopamine was used to reduce the Ag+ to Ag0 coated on the AuNPs or the AgNPs on the nanocomposites under an alkaline condition (Tris buffer, pH 8.5). The dopamine is a good reductant, which can be used to synthesize AgNPs via direct reduction of a silver salt solution27,28,29. Meanwhile, the dopamine can autopolymerize in the weak alkaline solution to generate a polydopamine layer, which could coat on almost all the surfaces30,31,32,33. Therefore, by the reduction, self-polymerization and the attractive adhesion, the dopamine could be directly used to generate the AgNPs and a layer of nanometer-thick PDA to encapsulate the Fe3O4@SiO2@PEI–Au/Ag nanocomposite. The advantage of this Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite prepared by this method is simple, moderate reaction condition and the nanocomposite could be easily separated from the reaction system by using a magnet. The Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite shows a high catalytic activity of reducing p-nitrophenol (4-NP) to the p-aminophenol (4-AP). Furthermore, thanks to the high magnetization and stability, the as-prepared catalyst can be easily recovered by a magnet and reused several times. This high efficient, stable and reusable catalyst might have various applications in catalysis, industrial wastewater treatment, pharmaceutical manufacture, petrochemical production and food processing.

Scheme of synthesis of the Fe3O4@SiO2@PEI–Au/Ag@PDA nanocomposite.
Results
Synthesis of the Fe3O4@SiO2–Au/Ag@PDA nanocomposite by the simple self-assembled method
Figure 1 shows the construction strategy of the Fe3O4@SiO2@PEI–Au/Ag@PDA nanocomposite. The electrostatic interactions between the positively charged PEI and negatively charged GSH-AuNPs were used to self-assemble the AuNPs to the Fe3O4@SiO2. The dopamine induced the reduction of the Ag+ to Ag0 where the Ag was grown either on the AuNPs or on the surfaces of the nanocomposite due to the weak reducing of the dopamine. The Fe3O4@SiO2@PEI–Au/Ag nanocomposite was also encapsulated by the self-polymerization of the dopamine to polydopamine under the alkaline condition. By using this process with moderate conditions, the stable magnetic catalyst could be obtained with a high yield. Our synthesis strategy has many advantages compared to other methods. First, the synthesis approach is simple and easy to repeat. Second, the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite is synthesized under moderate conditions which doesn’t require any high temperature reaction. Third, the dopamine reduced the Ag+ to Ag0 and coated the Au/AgNPs in a single step under the alkaline condition (Tris buffer, pH 8.5) because at this condition the dopamine can also be oxidized and generate the polydopamine. The coating of a nanometer thick layer of the polydopamine on the Fe3O4@SiO2@PEI-Au/Ag made our catalyst have a good hydrophilic property and excellent stability. During the synthesis process, we find that there is a much stronger adsorption of the Fe3O4@SiO2@AuNPs nanocomposite to the microcentrifuge tube than that of the Fe3O4@SiO2@Au/Ag@PDA (Supporting Information, Fig. S2), which demonstrates that the latter has a better recyclability than the Fe3O4@SiO2@AuNPs nanocomposite.
Characterization of Fe3O4@SiO2-Au/Ag@PDA nanocomposite
The magnetic Fe3O4 NPs were prepared via a simple hydrothermal reaction based on the high temperature reduction of Fe3+ salts with ethylene glycol (EG). As revealed by the SEM in Fig. 2a,b, the magnetic Fe3O4 NPs have an average diameter of ~300 nm. After coating a thin layer of the SiO2 by the sol-gel method in ethanol and NH3·H2O solution, the Fe3O4@SiO2 didn’t show much difference from the Fe3O4 NPs in the SEM images (Fig. 2c). The as-prepared Fe3O4@SiO2 showed an excellent monodispersion capability presumably because of its negatively charged surfaces. The zeta potential of the Fe3O4@SiO2 was measured to be about −39.0 mV in water (Fig. 3(B)). The negatively charged Fe3O4@SiO2 NPs could be used for self-assembly with positively charged polyelectrolytes, such as PEI or poly(diallyldimethylammonium chloride), by taking advantage of the electrostatic interaction. In this work, the PEI was adopted and used for coating the Fe3O4@SiO2 NPs. After the PEI coating, the Fe3O4@SiO2@PEI nanocomposite showed less charge in the SEM image because of the polymer on the surface (Fig. 2d), which suggested that PEI was successfully coated on the surfaces of the Fe3O4@SiO2 NPs. The zeta potential of the Fe3O4@SiO2@PEI nanocomposite was measured to be ~ +39.9 mV (Fig. 3(C)) in water, which further confirmed that the positively charged PEI polymer was successfully grafted on the surface of Fe3O4@SiO2 NPs. Because the GSH-capped AuNPs showed a strong negative charge (−35.1 mV, Fig. 3(A)), the AuNPs could be self-assembled on the positively charged Fe3O4@SiO2@PEI NPs through electrostatic interaction. The self-assembly of the GSH-AuNPs on the Fe3O4@SiO2@PEI NPs made the surface potential down to about +15.1 mV (Fig. 3(D)) and the color of the solution changed from gray to black (Data not shown). This is because a lot of the GSH-AuNPs self-assembled onto the Fe3O4@SiO2@PEI nanocomposite. In order to get a good dispersion of the Fe3O4@SiO2@PEI-AuNPs, mPEG-SH was used as a ligand exchange reagent and the PEG modified nanocomposite suspend better in hydrophilic environment. The zeta potential of the Fe3O4@SiO2@PEI-Au-SH-PEG only increased slightly to +18.2 mV (Fig. 3(E)) after the SH-PEG modification, but the hydrophilic solubility significantly improved. After the AgNPs growth by the reduction of Ag+ to Ag0 and generation a layer of the PDA shell, the zeta potential of the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite changed from +18.2 mV to ~−17.9 mV mainly due to the creation of the PDA shell (Fig. 3(F)). The PDA coating made the nanocomposite disperse better and adsorb less to the microcentrifuge tube than that of the Fe3O4@SiO2@PEI-Au nanocomposite (Supporting Information S3, Fig. S2). Figure 2e,f present the low and high magnification SEM images of the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite. Compared to Fig. 2d, there were many small Au/Ag nanoparticles on the surfaces of the Fe3O4@SiO2@PEI, which demonstrated the successful synthesis of the multifunctional nanocomposite catalyst.

SEM images of the Fe3O4 NPs (a,b) low and high magnification of the Fe3O4 NPs; Fe3O4@SiO2 (c) Fe3O4@SiO2 @PEI (d) Fe3O4@SiO2@PEI-Au/Ag@PDA (e,f) low and high magnification of the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite.

Zeta potentials of the GSH-AuNPs (A), Fe3O4@SiO2 (B), Fe3O4@SiO2@PEI (C), Fe3O4@SiO2@AuNPs (D), Fe3O4@SiO2@Au-SH-PEG (E) and the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite (F) respectively.
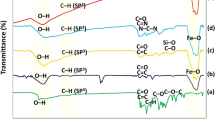
FT-IR spectra were used to study the transformation of the composition of composite of Fe3O4@SiO2, Fe3O4@SiO2 and Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite. As shown in Fig. 4, the strong absorption peak at about 585 cm−1 in the FT-IR of Fe3O4 NPs was assigned to the stretching vibration of Fe-O from the Fe3O4. For the Fe3O4@SiO2 NPs, new and broad absorption bands were observed in the range of 1000–1200 cm−1, which could be attributed to the Si-O stretching vibration of the SiO2 shell. After generation of the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite, besides the absorption peaks of the Fe3O4 core and the SiO2 shell, strong absorption peaks and some weak peaks were also observed in the range of 1420–1750 cm−1 and 2800–3000 cm−1 in the FT-IR spectrum. Compared to the Fe3O4@SiO2 NPs these peaks could be attributed to the stretching vibration of the aromatic rings and the C-O stretching of phenol compounds in the PDA shell.

FT-IR characterization of the Fe3O4, Fe3O4@SiO2 and Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite.
The HAADF STEM and TEM images of the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite were shown in Fig. 5. From the Fig. 5a,b, we can clearly observe the small AuNPs or AgNPs on the surface of the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite. Meanwhile, an about 5 nm PDA layer can be noticed directly in the TEM image (Fig. 5c,d). The thin layer of the PDA made the nanocomposite very stable.

HAADF STEM images of the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite.
(a,b) low and high magnification, respectively; TEM images of the Fe3O4@SiO2@PEI-Au/Ag@PDA (c,d) low and high magnification of the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite, respectively.
The crystal structures of the Fe3O4 NPs and the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite were recorded by the X-ray diffraction (XRD), as shown in Fig. 6. The XRD pattern of the Fe3O4 shows characteristic peaks at (220), (311), (400), (442), (511) and (440) in agreement to the JCPDS card No. 19-0629. XRD pattern contains no impurity peak indicating the high purity of the Fe3O4 sample and perfect phase transformation. Obviously, the XRD pattern of the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite has similar diffraction peaks as those of the Fe3O4 NPs. New diffraction peaks (labeled as *) at 38, 44 and 65 degree were observed, which could be attributed to the diffraction peaks of the AuNPs and AgNPs. However, it is hard to distinguish the diffraction peaks by this method because Au and Ag have very similar diffraction peaks in the XRD pattern.

XRD patterns of the Fe3O4 NPs (black) and the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite (red) (The new diffraction peaks on the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite were labeled with stars (*)).
The high angle annular dark field (HAADF) scanning transmission electron microscopy (STEM) was used to map the elements directly on the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite. As shown in the Fig. 7a, we can observe some small particles on the surfaces of the Fe3O4 in the HAADF STEM images directly, which confirmed that the gold nanoparticles assembled on the Fe3O4@SiO2@PEI nanospheres. The element mapping of the Fe, O, Au and Ag of Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite were shown in the Fig. 7a. After merge of the images of the Fe, Au and Ag, they suggest that there was a layer of the Au/Ag around the Fe3O4 core. From the merge image, we found the positions of Au and Ag elements were highly overlapped. However, it is hard to identify the formation of the Au@Ag core@shell structure by this HAADF mapping. The EDX spectrum of the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite (Fig. 7b) reveals the elements (Fe, O, C, N, Si, Au and Ag) of the composite, confirming the existence of iron oxide, gold, silver and the PDA organic layer on the nanocomposite. The separated characteristic peak of gold (around 2.1 keV and 9.8 keV) and silver (around 3.1 keV) proved that the AuNPs self-assembled on the nanocomposite and the silver grew onto the nanocomposite.

(a) HAADF SEM mapping of the elements of the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite; (b) The EDX spectrum of the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite. The Cu element comes from the substrate of the Cu grid.
The magnetic property of nanocomposite is beneficial for its convenient and fast separation, removing the requirements for repeated centrifugation in the practical applications. As shown in Fig. 8, the hysteresis loops of the Fe3O4, Fe3O4@SiO2 and the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite were measured by a superconducting quantum interface device (SQUID) magnetometer. From the results, the Fe3O4, Fe3O4@SiO2 and the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite all show strong magnetism characterization at room temperature. The Ms values of the Fe3O4 and Fe3O4@SiO2 and Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite are ~72.6 emu/g, 59.9 emu/g and 48.9 emu/g, respectively. As the introduction of nonmagnetic species, the Ms values of the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite are smaller than that of the Fe3O4@SiO2 and Fe3O4 nanoparticles. Even after the finally coating with the PDA, the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite still showed a rather high Ms value of 48.9 emu/g, which indicated that the catalyst could be rapidly separated by a common magnet. The Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite can be easily dispersed in aqueous solution and they can be rapidly collected from the mixture within 20s by a permanent magnet. These results showed that the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite can simplify the separation process in practical applications, because of its excellent magnetic response.

Magnetic hysteresis loops of the Fe3O4 (black), Fe3O4@SiO2 (red) and the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite (blue) at 300 K.
Inset: the photographs showing magnetic Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite separated by a magnet.
Catalytic performance of Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite to convert 4-NP to 4-AP
As a proof-of-concept, the catalytic property of the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite was studied by catalytic reduction of 4-NP to 4-AP with NaBH4 (Fig. 9)28. After adding NaBH4 into the 4-NP, the color of the solution changed from light yellow to bright yellow due to the deprotonation of the 4-NP that made the UV-vis characteristic peak shifted from 317 to 400 nm (Fig. 9A). Without addition of the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite as the catalyst, no obvious change of the UV-vis absorption spectra was observed over time, which demonstrated that the reduction reaction did not proceed without the catalyst. When the catalyst was added, the UV-vis absorption at 400 nm gradually decreased (Fig. 9B), which indicated that the reduction of 4-NP was in progress. Meanwhile, an absorption characteristic peak at 300 nm appeared due to the generation of the 4-AP (Fig. 9B). The conversion of 4-NP to 4-AP completed within 10 min, as the color of the solution transformed from bright yellow into colorless. The catalytic reaction from the 4-NP to 4-AP has been well studied and reported as a first-order reaction6,26,34. The plot of ln (Ct/C0) versus reaction time is linear (Fig. 9C), where C0 and Ct are the concentrations of 4-NP at time 0 and t measured from the relative absorbance At and A0, respectively. This linear relationship of the reduction reaction matched well with first-order kinetics and the rate constant k of the reaction with the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite was calculated to be ~0.56 min−1 (R2 = 0.974). The catalytic performance of the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite to convert the 4-NP to 4-AP is better than that of the self-assembling with only AuNPs or AgNPs into the nanocomposites (Supporting Information S4, Fig. S5). The reaction kinetic constants of Fe3O4@SiO2@PEI-Au@PDA and Fe3O4@SiO2@PEI-Ag@PDA nanocomposites to convert 4-NP to 4-AP are 0.26 min−1 and 0.19 min−1, respectively (Fig. S3, Fig. S4). Meanwhile, the catalytic activity of our Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite was higher than that of most reported catalysts for the same catalytic conversion based on Au or Ag nanoparticles as the catalytic elements (Supporting Information S5, Table S1).

Catalytic performance of Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite to convert 4-NP to 4-AP.
(a) UV–vis spectra of 4-NP (a) before and (b) after the addition of NaBH4; (b) Time-dependent UV–vis spectra of the reaction solution in the presence of the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite catalyst; (c) Plot of ln (Ct/C0) against the reaction time; (d) The recyclability of the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite as the catalyst for the reduction of 4-NP with NaBH4.
Furthermore, the reusable property of the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite catalyst was investigated by repeated use of the catalyst for several times (Fig. 9D). One significant advantage of this Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite is that the recovery process could be easily achieved by simple magnetic separation. The conversion percentage of 4-NP showed little decrease after 8 times cycles, indicating that the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite has an excellent reusability. This can be contributed to the very limited loss of the catalyst during the repeated magnetic separation process, an indication and demonstration of the excellent magnetic property of the Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite.
Conclusions
In summary, we demonstrated a self-assembly approach to fabricate a multifunctional Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite catalyst with highly stabilized reactivity and magnetic recyclability. The Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite exhibited an excellent catalytic capability. The catalytic reduction of 4-NP to 4-AP obeys a fast first-order kinetics with the reaction constant of 0.56 min−1. The Fe3O4@SiO2@PEI-Au/Ag@PDA nanocomposite shows a high saturation magnetization (Ms) of 48.9 emu/g, even after the self-assembly of Au/AgNPs on its surface and an about 5 nm layer of polydopamine coating which is much higher than some previously reported8,13,26. The Fe3O4@SiO2@PEI-Au/Ag@PDA NPs can be attracted to a magnet within about 20 s. Therefore, the Fe3O4@SiO2@PEI-Au/Ag@PDA NPs can be easy recyclable and reusable for the next catalytic reaction with a simple magnetic separation and washing process. The high saturation magnetization minimized the loss of the catalyst during the recovery process, which guaranteed a high efficient catalytic performance even after eight times. This simple self-assembled strategy could be used for construction of many magnetic nanocomposite catalysts and the high efficient, stable and reusable catalyst might find many applications in the catalytic field.
Methods
Chemicals
Hydrogen tetrachloroaurate (III) hydrate (HAuCl4), L-glutathione (GSH), silver nitrate (AgNO3) and sodium tetrahydridoborate (NaBH4) were purchased from Alfa Aesar. Ferric chloride hexahydrate (FeCl3·6H2O), ethylene glycol (EG) and sodium acetate (NaAc) were purchased from Alfa Aesar. Tetraethoxysilane (TEOS), poly(ethyleneimine) solution (PEI, 750 KD), dopamine hydrochloride and polyvinylpyrrolidone (PVP, 40 KD) were obtained from Sigma-Aldrich. Methoxy-poly(ethylene glycol)-thiol (mPEG-SH, 2 KD) was purchased from Laysan Bio, Inc. 4-nitrophenol (4-NP) was obtained form EMD Millipore (Billerica, MA, USA). All other chemical reagents used in this work were of analytical grade, obtained from VWR (Radnor, PA, USA) and used without further purification unless otherwise noted.
Synthesis of Magnetic Fe3O4, Fe3O4@SiO2 NPs and GSH-Capped AuNPs
Fe3O4 NPs were synthesized as a reported solvothermal approach without much modification35. Then the as-prepared Fe3O4 NPs were coated with a layer of the SiO2 according to the method previously reported with slight modification36. The detail information of the synthesis of the Fe3O4 NPs was shown in Supporting Information S1. The size of the Fe3O4 NPs was about 300 nm. The GSH-capped AuNPs (negative charge) were prepared as our previous report16 and the size of the GSH-capped AuNPs is characterized to be about 4 ~ 6 nm from the TEM images (Supporting Information S2, Fig. S1). The process of the synthesis of the AuNPs was described in the Supporting Information S2.
Electrostatic self-assembly of AuNPs onto the Fe3O4@SiO2 NPs
2.5 mg of Fe3O4@SiO2 nanocomposite was dissolved in 1.8 mL of 0.2 mg/mL PEI solution. After shaking continuously for 0.5 h at room temperature, the as-prepared Fe3O4@SiO2@PEI product was separated using a magnet, rinsed with pure water for three times and dispersed into 0.5 mL water. Then the negatively charged AuNPs were added into the above solutions under the ultrasonication for 5 min. The solution was kept shaking for 30 min at room temperature. After magnetic separation, the Fe3O4@SiO2@PEI-Au was collected and washed three times with pure water. In order to make sure the maximum electrostatic self-assembly of the AuNPs on the positively charged Fe3O4@SiO2@PEI, the process of adding AuNPs solution, magnetic separation and washing by water was repeated for several times until there were surplus of the AuNPs in the supernatant solution. Finally, the mPEG-SH was added into the as-prepared Fe3O4@SiO2@PEI-Au solution and the system was incubated for 24 h to obtain a good dispersion of the nanocomposite.
Synthesis of Fe3O4@SiO2@PEI–Au/Ag@PDA Nanocomposite
The Fe3O4@SiO2@PEI–Au/Ag@PDA nanocomposite was prepared by the mussel-inspired coating method. The PEGylated Fe3O4@SiO2@PEI–Au nanocomposite was dispersed in 2 mg/ml of dopamine solution (0.8 mL in 10 mM Tris-HNO3 buffer, pH 8.5), followed by slow addition of AgNO3 solution (2.5 mM, 0.8 mL). The reaction was allowed to react under shaking for 4 h at room temperature. The obtained Fe3O4@SiO2@PEI–Au/Ag@PDA nanocomposite was collected by a magnet and washed with ultrapure water for 5 times and finally dried in an oven at 60 °C for 12 h.
Catalytic Reaction of the 4-NP with Fe3O4@SiO2–Au/Ag@PDA nanocomposite
The reduction of 4-NP by NaBH4 was chosen as a model reaction for investigating the catalytic performance of the multifunctional Fe3O4@SiO2–Au/Ag@PDA nanocomposite. In a typical procedure, 400 μL of 0.2 M freshly prepared NaBH4 was added into the solution containing 12.5 μL of 5 mM 4-NP and 600 μL of H2O. Subsequently, 50 μL of 2 mg/mL catalyst was added into the above solution and the reaction started immediately. The color change of the solution from bright yellow to colorless was observed during the reaction process. The UV-vis absorption spectra of the solution were recorded to monitor the progress of the reaction by using a microplate reader with the scanning range from 250 nm to 550 nm. The kinetic rate constants of the reduction process were determined by measuring the change in absorbance at 400 nm as a function of time. In order to study the recyclability of the catalyst, the used Fe3O4@SiO2@PEI–Au/Ag@PDA nanocomposite was harvested from the reaction mixture by a magnet at the end of each run, washed with pure water by three times, then re-dispersed in 50 μL water and added in a fresh reaction solution. After reaction for 10 min, the Fe3O4@SiO2–Au/Ag@PDA nanocomposite was removed and the supernatant solution was measured to evaluate the recyclability of the catalysts. In total, this procedure was repeated 8 times.
Characterization
Scanning electron microscopy (SEM) images were obtained on a field-emission scanning electron microscope (NanoSEM 630, NOVA). Transmission electron microscopy (TEM) images were conducted with a Philips-420 microscope at an acceleration voltage of 120 kV. The high angle annular dark field (HAADF) images was obtained by a scanning transmission electron microscopy TITAN (FEI Titan3 G2). The zeta potential was measured by the Malvern Zetasizer ZS. Fourier transform infrared spectra were obtained by a Bruker Vertex V70 FT-IR spectrometer scanned from 400 to 4000 cm−1. The powder X-ray diffraction (PXRD) patterns were performed by a PANalytical Empyrean X-ray powder diffractometer with Cu target (45 kV, 40 mA) from 20 to 70 degrees. The magnetization measurement was collected by a superconducting quantum interface device (SQUID) magnetometer at 300 K.
Additional Information
How to cite this article: Yu, X. et al. Synthesis of Self-Assembled Multifunctional Nanocomposite Catalysts with Highly Stabilized Reactivity and Magnetic Recyclability. Sci. Rep. 6, 25459; doi: 10.1038/srep25459 (2016).
References
Wang, D. S. & Li, Y. D. One-pot protocol for Au-based hybrid magnetic nanostructures via a noble-metal-induced reduction process. J Am Chem Soc 132, 6280–6281 (2010).
Xu, Z., Hou, Y. & Sun, S. Magnetic core/shell Fe3O4/Au and Fe3O4/Au/Ag nanoparticles with tunable plasmonic properties. J Am Chem Soc 129, 8698–8699 (2007).
Deng, Y. et al. Multifunctional mesoporous composite microspheres with well-designed nanostructure: a highly integrated catalyst system. J Am Chem Soc 132, 8466–8473 (2010).
Liu, R. et al. Dopamine as a carbon source: the controlled synthesis of hollow carbon spheres and yolk-structured carbon nanocomposites. Angew Chem Int Ed 50, 6799–6802 (2011).
Jiang, H. L., Akita, T., Ishida, T., Haruta, M. & Xu, Q. Synergistic catalysis of Au@Ag core-shell nanoparticles stabilized on metal-organic framework. J Am Chem Soc 133, 1304–1306 (2011).
Zheng, J. et al. In situ loading of gold nanoparticles on Fe3O4@SiO2 magnetic nanocomposites and their high catalytic activity. Nanoscale 5, 4894–4901 (2013).
Zhou, J. et al. Interfacial assembly of mussel-inspired Au@Ag@polydopamine core-shell nanoparticles for recyclable nanocatalysts. Adv Mater 26, 701–705 (2014).
Zeng, T., Zhang, X. L., Ma, Y. R., Niu, H. Y. & Cai, Y. Q. A novel Fe3O4-graphene-Au multifunctional nanocomposite: green synthesis and catalytic application. J Mater Chem 22, 18658–18663 (2012).
Ke, F., Wang, L. & Zhu, J. Multifunctional Au-Fe3O4@MOF core-shell nanocomposite catalysts with controllable reactivity and magnetic recyclability. Nanoscale 7, 1201–1208 (2015).
Chen, J., Xue, Z., Feng, S., Tu, B. & Zhao, D. Synthesis of mesoporous silica hollow nanospheres with multiple gold cores and catalytic activity. J. Colloid Interface Sci. 429, 62–67 (2014).
Ge, J., Zhang, Q., Zhang, T. & Yin, Y. Core-satellite nanocomposite catalysts protected by a porous silica shell: controllable reactivity, high stability and magnetic recyclability. Angew Chem Int Ed 47, 8924–8928 (2008).
Liu, R., Qu, F., Guo, Y., Yao, N. & Priestley, R. D. Au@carbon yolk-shell nanostructures via one-step core-shell-shell template. Chem Commun 50, 478–480 (2014).
Zhang, X. et al. Polypyrrole-enveloped Pd and Fe3O4 nanoparticle binary hollow and bowl-like superstructures as recyclable catalysts for industrial wastewater treatment. ACS Appl. Mat. Interfaces 6, 450–458 (2014).
Yao, T. et al. A simple way to prepare Au@polypyrrole/Fe3O4 hollow capsules with high stability and their application in catalytic reduction of methylene blue dye. Nanoscale 6, 7666–7674 (2014).
Yu, X. et al. On-chip dual detection of cancer biomarkers directly in serum based on self-assembled magnetic bead patterns and quantum dots. Biosens Bioelectron 41, 129–136 (2013).
Yu, X., Zhang, Z. L. & Zheng, S. Y. Highly sensitive DNA detection using cascade amplification strategy based on hybridization chain reaction and enzyme-induced metallization. Biosens Bioelectron 66, 520–526 (2015).
Mizuno, M. et al. Magnetophoresis-integrated hydrodynamic filtration system for size- and surface marker-based two-dimensional cell sorting. Anal Chem 85, 7666–7673 (2013).
He, S. et al. Fast magnetic isolation of simple sequence repeat markers in microfluidic channels. Lab Chip 14, 1410–1414 (2014).
Kwon, D., Joo, J., Lee, J., Park, K. H. & Jeon, S. Magnetophoretic chromatography for the detection of pathogenic bacteria with the naked eye. Anal Chem 85, 7594–7598 (2013).
Lu, G. et al. Imparting functionality to a metal-organic framework material by controlled nanoparticle encapsulation. Nat Chem 4, 310–316 (2012).
Lattuada, M. & Hatton, T. A. Functionalization of monodisperse magnetic nanoparticles. Langmuir 23, 2158–2168 (2007).
Yu, X., Feng, X., Hu, J., Zhang, Z. L. & Pang, D. W. Controlling the magnetic field distribution on the micrometer scale and generation of magnetic bead patterns for microfluidic applications. Langmuir 27, 5147–5156 (2011).
Yu, X., Wen, C. Y., Zhang, Z. L. & Pang, D. W. Control of magnetic field distribution by using nickel powder@PDMS pillars in microchannels. RSC Adv 4, 17660–17666 (2014).
Park, H. H., Woo, K. & Ahn, J. P. Core-shell bimetallic nanoparticles robustly fixed on the outermost surface of magnetic silica microspheres. Sci Rep 3, 1497 (2013).
Lin, F. H. & Doong, R. A. Bifunctional Au-Fe3O4 Heterostructures for Magnetically Recyclable Catalysis of Nitrophenol Reduction. J Phys Chem C 115, 6591–6598 (2011).
Huang, L. et al. Iron oxide nanoparticle layer templated by polydopamine spheres: a novel scaffold toward hollow-mesoporous magnetic nanoreactors. Nanoscale 7, 806–813 (2015).
Cong, Y. et al. Mussel-inspired polydopamine coating as a versatile platform for synthesizing polystyrene/Ag nanocomposite particles with enhanced antibacterial activities. J Mater Chem B 2, 3450–3461 (2014).
Yu, X., Cheng, G., Zhou, M. D. & Zheng, S. Y. On-demand one-step synthesis of monodisperse functional polymeric microspheres with droplet microfluidics. Langmuir 31, 3982–3992 (2015).
Sileika, T. S., Kim, H. D., Maniak, P. & Messersmith, P. B. Antibacterial performance of polydopamine-modified polymer surfaces containing passive and active components. ACS Appl. Mat. Interfaces 3, 4602–4610 (2011).
Park, J. et al. Polydopamine-based simple and versatile surface modification of polymeric nano drug carriers. ACS Nano 8, 3347–3356 (2014).
Lee, H., Dellatore, S. M., Miller, W. M., Messersmith, P. B. & Messersmith P. B. Mussel-inspired surface chemistry for multifunctional coatings. Science 318, 426–430 (2007).
Lin, L. S. et al. Multifunctional Fe3O4@polydopamine core-shell nanocomposites for intracellular mRNA detection and imaging-guided photothermal therapy. ACS Nano 8, 3876–3883 (2014).
Liu, Y., Ai, K. & Lu, L. Polydopamine and Its Derivative Materials: Synthesis and Promising Applications in Energy, Environmental and Biomedical Fields. Chem Rev 114, 5057–5115 (2014).
Yang, H., Li, S., Zhang, X., Wang, X. & Ma, J. Imidazolium ionic liquid-modified fibrous silica microspheres loaded with gold nanoparticles and their enhanced catalytic activity and reusability for the reduction of 4-nitrophenol. J Mater Chem A 2, 12060–12067 (2014).
Deng, Y., Qi, D., Deng, C., Zhang, X. & Zhao, D. Superparamagnetic high-magnetization microspheres with an Fe3O4@SiO2 core and perpendicularly aligned mesoporous SiO2 shell for removal of microcystins. J Am Chem Soc 130, 28–29 (2008).
Graf, C., Vossen, D. L. J., Imhof, A. & van Blaaderen, A. A general method to coat colloidal particles with silica. Langmuir 19, 6693–6700 (2003).
Acknowledgements
We thank Ms Gray Jennifer for her kind help with the HAADF image process. We also thank the Penn State Materials Research Institute and Huck Institute of Life Sciences for their support. This work is partially supported by the Pennsylvania State University start-up fund to S. Y. Zheng and the National Cancer Institute of the National Institutes of Health under Award Number DP2CA174508.
Author information
Authors and Affiliations
Contributions
X.Y. designed and performed the experiments. S.-Y.Z. and G.C. conceived the idea. X.Y., G.C and S.-Y.Z. discussed and wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Yu, X., Cheng, G. & Zheng, SY. Synthesis of Self-Assembled Multifunctional Nanocomposite Catalysts with Highly Stabilized Reactivity and Magnetic Recyclability. Sci Rep 6, 25459 (2016). https://doi.org/10.1038/srep25459
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep25459
This article is cited by
-
Effective gene delivery based on facilely synthesized “core–shell” Ag@PDA@PEI nanoparticles
Journal of Nanoparticle Research (2022)
-
Glutathione-magnetite nanoparticles: synthesis and physical characterization for application as MRI contrast agent
SN Applied Sciences (2020)
-
Magnetic domain interactions of Fe3O4 nanoparticles embedded in a SiO2 matrix
Scientific Reports (2018)
-
Photo-reduced Cu/CuO nanoclusters on TiO2 nanotube arrays as highly efficient and reusable catalyst
Scientific Reports (2017)
-
Magnetically-Separable and Thermally-Stable Au Nanoparticles Encapsulated in Mesoporous Silica for Catalytic Applications
Topics in Catalysis (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
