
Abstract
Phosphatidylinositolbinding clathrin assembly protein (PICALM) gene is one novel genetic player associated with late-onset Alzheimer’s disease (LOAD), based on recent genome wide association studies (GWAS). However, how it affects AD occurrence is still unknown. Brain reserve hypothesis highlights the tolerant capacities of brain as a passive means to fight against neurodegenerations. Here, we took the baseline volume and/or thickness of LOAD-associated brain regions as proxies of brain reserve capacities and investigated whether PICALM genetic variations can influence the baseline reserve capacities and the longitudinal atrophy rate of these specific regions using data from Alzheimer’s Disease Neuroimaging Initiative (ADNI) dataset. In mixed population, we found that brain region significantly affected by PICALM genetic variations was majorly restricted to posterior cingulate. In sub-population analysis, we found that one PICALM variation (C allele of rs642949) was associated with larger baseline thickness of posterior cingulate in health. We found seven variations in health and two variations (rs543293 and rs592297) in individuals with mild cognitive impairment were associated with slower atrophy rate of posterior cingulate. Our study provided preliminary evidences supporting that PICALM variations render protections by facilitating reserve capacities of posterior cingulate in non-demented elderly.
Similar content being viewed by others
Introduction
The global situation of dementia is not optimistic. The prevalence of dementia was estimated 5–7% in most global regions and 35.6 million people lived with dementia in 2010, with numbers predicted to almost double every 20 years, to 65.7 million in 2030 and 115.4 million in 20501,2, leading to an increasing burden on caregivers and society3. The recently released Alzheimer Report 2015 reflects a same trend but lousier prospect. (http://www.alzforum.org/news/research-news/world-alzheimer-report-2015-revised-estimates-hint-larger-epidemic) As the most common type (roughly 60%) of dementia, Alzheimer’s disease (AD) significantly inflicts both reduced life-span and lowered life quality on patients4,5,6.
In confrontation of this situation, scientific efforts to elucidate its etiology has never been stopped. It is now widely accepted that AD is a complex disease entity, with occurrence underpinned by both genetic and environmental components7,8. APOE4 was a widely validated genetic risk but merely accounted for a limited percentage of LOAD risk, several genome-wide association studies (GWAS) and meta-analyses had revealed a series of new risk loci associated with the late-onset type of AD (LOAD; >65 years of age)9,10,11,12, to some extent filling up the vacant area of its genetic etiology.
The gene encoding phosphatidylinositolbinding clathrin assembly protein (PICALM) was one of these new players. Its association with AD was revealed in large GWAS9,10,11,12 and further validated in a series of larger replication studies in both European13,14,15,16,17 and Asian population18, in spite of some conflicting results from those with smaller sample sizes19. However, the concrete pathways by which PICALM gene are involved in AD occurrence are still an enigma.
More than a decade ago, Stern20,21 proposed the concept of reserve to explain the disjunction between AD pathology degree and severity of clinical performances. The hypothesis proposed a passive protective model named “brain reserve”, positing that the quantity of available neural substrate (e.g., brain size, synaptic count, or dendritic branching) can be the basis of cerebral tolerance to abnormal insults (Fig. 1A)8,22. However, it seemed that previous understandings put more focus on the global situation of the whole brain than on some brain regions specifically associated with the disease, such as hippocampus (CA1 subregion), middle temporal area, entorhinal area, posterior cingulate, precuneus, and parahippocampal area. Based on these findings, we thus supposed that LOAD-associated genetic variations may be involved in AD occurrence by modulating the brain reserve capacities of these brain sub-regions which has been proved vulnerable in AD process (Fig. 1B).

(A) Depiction of how brain reserve operates to protect the brain. The x-axis represents time, over which AD pathology slowly accumulates. The y-axis represents cognitive function. We assume that AD pathology accumulates over time at the same rate in two individuals with high and low brain reserve (BR). The amount of pathology needed before cognitive function is affected is greater for individual with higher CR, leading to a later change point of time. It follows that greater pathology will be needed for the person with higher BR to meet clinical diagnostic criteria for AD, thus delaying the onset of the disease. Once cognitive decline arises, it is faster in the person with higher BR22. (B) We proposed a hypothesis that PICALM genetic variations were associated with brain reserve (baseline thickness/volume and atrophy rate) of specific regions associated with AD in non-dementia elderly. We hypothesized that individuals carrying specific PICALM variations might have higher baseline thickness/volume of specific brain areas and/or slower atrophy rate in confrontation with impacts of pathological impairments and/or normal aging for some unknown reasons. Based on model depicted in (A), these trends equivalently render two powerful “weapons” for the individuals to maintain normal cognition and stay away from AD over a period longer than others. Abbreviation: BR = brain reserve; AD = Alzheimer’s disease.
Herein, we took the baseline volume and/or thickness of AD-associated brain regions (as mentioned above) as proxies of brain reserve capacities and investigated whether PICALM genetic variations can influence the reserve capacities and longitudinal atrophy rate of these specific regions using data from Alzheimer’s Disease Neuroimaging Initiative (ADNI) dataset.
Results
Demographic, cognitive, and clinical characteristics
Demographic, cognitive, and clinical characteristics of the included subjects are shown in Table 1. In brief, a total of 281 NC (145 female, 74.51 ± 5.56 years), 483 MCI (201 female, 72.28 ± 7.45 years) and 48 AD patients (18 female, 75.51 ± 9.23 years) were enrolled in the present study. The frequency for the ε4 allele of APOE gene was AD > MCI > NC. For the cognitive function, AD patients displayed the worst cognitive function according to various neuropsychological scales, including CDRSB, MMSE, ADAS-cog, RAVLT, FAQ and MoCA. For the brain reserve capacity, AD patients showed the most severe atrophy in hippocampus, middle temporal and entorhinal cortex.
Brain structures and PICALM genotypes in the mixed population
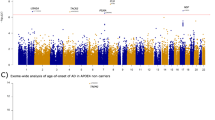
At baseline, no loci showed significant association with volume of either hippocampus or hippocampal CA1 region. A allele of rs3851179 showed trend of association with larger thickness of right entorhinal area. G allele of rs561655 showed trend of association with larger thickness of parahippocampal region. C allele of rs592297 showed trend of association with smaller volume of left middle temporal area and larger thickness of parahippocampal region. C allele of rs642949 showed trends of association with larger volume of left middle temporal area and right posterior cingulate, larger thickness of left precuneus and smaller thickness of left parahippocampal area. However, all these associations failed to survive the FDR correction (Fig. 2 and Supplementary Table 2).

In mixed population, we identified multiple brain regions showing trend of association with PICALM genetic variations (Deep green block). However, only posterior cingulate survived the FDR correction (Red block); We further tested this genetic predisposition in NC and MCI population. Results in NC individuals indicated that variations of seven loci were associated with baseline or one year or two years atrophy rate of posterior cingulate; Results in MCI individuals indicated that variations of two loci were associated with one year or two years atrophy rate of posterior cingulate. Abbreviations: NC = normal cognition; MCI = mild cognition impairment; AD = Alzheimer’s disease.
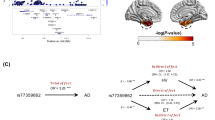
Analysis after one year follow-up indicated faster atrophy rate of right hippocampal CA1 for individuals carrying variations in rs543293 (A allele) and rs1237999 (G allele). C allele of rs64249 showed trends of associations with slower atrophy rate of left hippocampus and faster atrophy rate of right precuneus. Nonetheless, these associations did not reach significant after FDR correction. Interestingly, we found slower atrophy rate of right posterior cingulate in individuals with variations of rs561655 (G allele), rs543293 (A allele), rs592297 (C allele), rs1237999 (G allele) and 7941541 (G allele) and faster atrophy rate of the same region in individuals with variation of rs642949 (C allele) The associations were still significant after FDR correction (Fig. 3A–F).

We identified six loci which associations were still significant after FDR correction in the mixed population. The (A–F) depicted that variations of rs1237999, rs642949, rs561655, rs543293, rs7941541 and rs592297 were associated with one-year atrophy rate of posterior cingulate.
After two years follow-up, no loci showed significant association with atrophy rate of hippocampus or hippocampal CA1 region. Atrophy rate of middle temporal area showed trend of association with variation of rs642949. Atrophy rate of posterior cingulate showed trend of association with variation of rs3851179, rs543293, rs7941541, and rs642949. Atrophy rate of precuneus showed trend of association with variation of rs561655 and rs642949. Atrophy rate of parahippocampal area showed trend of association with variation of rs642949. Nonetheless, none of these reached significance after FDR correction, possibly due to the shrunken sample size after two years follow-up (Fig. 2 and Supplementary Table 2).
Altogether, we can infer that posterior cingulate may be the pivotal region on which PICALM variations target. Further, we selected the posterior cingulate as our sole ROI and independently tested its association with PICALM variations in NC and MCI individuals, respectively.
Posterior cingulate and PICALM genotypes in NC individuals
The associations of variations in four PICALM loci (rs561655, rs1237999, rs543293 and rs592297) with slower one-year atrophy rate of posterior cingulate were further validated in the NC population (Fig. 4B–F). Interestingly, we found that one PICALM variation (C allele of rs642949) was associated with larger thickness of posterior cingulate at baseline (Fig. 4A). We found significant association of rs561655, rs7941541 and rs3851179 with slower two years atrophy rate of posterior cingulate (Fig. 4G–I). This is expectable since the major contributors to brain atrophy and atrophy rate differ between NC individuals and MCI/AD individuals, such that the overall atrophy rate of posterior cingulate in the mixed population (NC+MCI+AD) was faster than that in the NC population (Fig. 4L).

(A) Depicted that rs642949 (C allele) was associated with larger thickness of posterior cingulate in NC population; (B–I) Depicted that variations of rs561655, rs1237999, rs543293, rs592297, rs7941541 were associated with slower atrophy rate of posterior cingulate in NC population; (J,K) Depicted that rs543293 and rs592297 were associated with slower atrophy rate of posterior cingulate in MCI population. (L) Depicted that the contributors to brain atrophy majorly included normal aging and pathological insults. The proportion of the latter would arise constantly as the stage progresses (from NC to MCI to AD) and finally become the predominant factor. This may explain the difference of association which PICALM genetic variations showed in NC and MCI population. Abbreviations: NC = normal cognition; MCI = mild cognition impairment; AD = Alzheimer’s disease.
Posterior cingulate and PICALM genotypes in MCI individuals
Compared to NC population, the trend of associations of PICALM variations with atrophy rate of posterior cingulate in MCI population were consistent (Fig. 2). We found A allele of rs543293 and C allele of rs592297 were associated with slower atrophy rate (after one year and two years) of posterior cingulate, respectively (Fig. 4J,K).
Posterior cingulate and PICALM genotypes in AD individuals
We failed to identify any significant associations of reserve capacities of posterior cingulate with PICALM variations in AD population, possibly due to the constrained sample size.
Discussion
We present here an explorative study about how single nucleotide polymorphisms of PICALM impart influences on brain reserve capacity of AD-associated brain regions. Seven SNPs were finally included in the analysis. We found six loci (all except rs3851179) in mixed population, two loci (rs592297 and rs543293) in MCI population and all seven loci in NC population, which were significantly associated with higher baseline thickness and/or slower atrophy rate of posterior cingulate, both of which would be favorable in fighting against AD insults, despite in a passive manner (Fig. 1B) These findings were significant given that 1) our study further revealed the potential pathways by which these genetic variations act in protecting brain from AD; 2) Our findings confirmed the protective roles of certain loci of PICALM gene, which is consistent with previous meta-analysis results of association of AD with rs561655 (odd ratio [OR] = 0.87; 95% confidence interval [95% CI] = 0.83–0.92) and rs543293 (OR = 0.89; 95% CI = 0.85–0.94). (http://www.alzgene.org/meta.asp?geneID=636)
Though our study showed that PICALM variations were associated with higher brain reserve capacities of posterior cingulate, the mechanism was still a mystery. Generally, the major contributors to brain atrophy include normal aging in which the process is relatively slower and stable, as well as abnormal pathological insults in which the process is relatively faster and changeable. In NC individuals, cerebral resistance power is enough to tolerate these two adverse contributors, thus contributing to the normal cognitive functions. Although we know little about which contributor held a dominant position in inducing brain atrophy in the NC stage, it is reasonably inferred that the role of abnormal pathologies is increasingly rising and would finally surpass that of normal aging as the stage further progresses (for example, from NC to MCI/AD) (Fig. 4L). In the present analysis, we found that PICALM genetic variations were more inclined to be associated with atrophy rate in NC individuals. Given that normal aging might play a more important role in causing brain atrophy in the stage of NC than MCI/AD, it can be thus inferred that the potential pathways by which PICALM variations act may be possibly associated with fighting against normal aging of posterior cingulate. More researches warrant to validate this hypothesis.
On the other hand, posterior cingulate cortex is located in the medial part of the inferior parietal lobe and lies within the posteromedial cortex. This specific brain area is highly anatomically connected and is known as a pivotal part of the default mode network (DMN), which is a resting-state functional networks and is particularly active in healthy people when they do not think about anything (for review see23,24). Previous cross-sectional analysis suggested that both AD and MCI subjects showed significant difference of posterior cingulate when compared with the health25,26,27. Also, disorder of DMN was a characteristic feature seen in early AD24. All these findings were suggestive of an impellent role of neurodegeneration of posterior cingulate in the very early stage of AD28. Our study provided the first evidence linking PICALM genetic variations with slower atrophy rate of posterior cingulate, leading to a reasonable postulation that individuals carrying these specific variations would be less vulnerable to lower reserve capacity of posterior cingluate and be possibly thus more powerful in fighting neurodegenerative insults.
Furthermore, as a critical network of brain, DMN was composed of large amounts of communication hubs named “synapses”. A very recent study proposed that DMN correlated with the orchestrated activity of dozens of genes linked to ion channel activity and synaptic function29, emphasizing the importance of synapses in maintaining normal functions of this network. On the other hand, it was previously reported that abnormalities of synapses in posterior cingulate occurred in the early stage of AD30. Therefore, PICALM may protect the normal operations of synapse by facilitating neurotransmitter delivering, which is against the negative impacts derived from normal aging or pathological insults such as Aβ. (for review see31)
Several limitations exist in our study. First of all, the sample size in the present analysis was smaller than that in the traditional large GWAS studies (n > 10,000); Second, the follow-up was relatively short. Both of these lead to restricted power and thus restrain our making definite conclusions. Therefore, this study is only a preliminary investigation and future replication with larger sample size and longer follow-up is necessary. Third, not all SNPs of PICALM gene were included due to the restriction of ADNI database. Fourth, associations of PICALM gene with posterior cingulate in AD population need more work given the AD sample in our study is obviously constrained. These may lead to insufficient digging of influences of PICALM genetic variations and future research warrant. Fifth, it is noteworthy that the results from sub-population (NC or MCI or AD) may be more informative than those from mixed population, which lead to, again, necessities of future efforts with larger sample.
In summary, this study provided preliminary evidences supporting that PICALM variations render protections by facilitating reserve capacities of posterior cingulate in non-demented elderly.
Materials and Methods
Definition of brain reserve (BR)
Brain reserve (BR) can be metaphorized as cerebral pre-existing troops (such as brain/specific brain region size, neuron/synaptic count, and dendritic branching, etc.), which are wholeheartedly responsible for maintaining a normal cognitive function by passively defending against attacks from pathological insults (for example, Alzheimer’s disease) as well as normal aging. However, once the loss of these troops achieved a certain level (so-called threshold model), cognitive impairments occurred.
ADNI database
Alzheimer’s Disease Neuroimaging Initiative (ADNI) is a large, multicenter, longitudinal neuroimaging study, initiated in 2003 by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, the Food and Drug Administration, private pharmaceutical companies, and nonprofit organizations32. The initial goal of ADNI was to recruit 800 subjects but the ADNI has been further followed by ADNI-GO and ADNI-2. To date, the three protocols have recruited over 1,500 adults (aged 55 to 90), consisting of cognitively normal older individuals, people with early or late MCI, and people with AD. The study was approved by the institutional review boards of all participating centers, and written informed consent was obtained from all participants or authorized representatives after extensive description of the ADNI according to the 1975 Declaration of Helsinki33. The study was approved by the institutional review boards of all participating centers (Ocean University of China, Qingdao Municipal Hospital, Nanjing First Hospital, Memory and Aging Center in University of California, and ADNI) and written informed consent was obtained from all participants or authorized representatives. In addition, the methods were carried out in accordance with the approved guidelines.
Participants
The data used in this study were obtained from the ADNI database (http://adni.loni.usc.edu) Inclusion criteria for AD subjects is National Institute of Neurological and Communication Disorders/Alzheimer’s Disease and Related Disorders Association (NINCDS/ADRDA) criteria for probable AD, with a Mini Mental State Examination (MMSE) score between 20 and 26, a global Clinical Dementia Rating (CDR) of 0.5 or 1, a sum-of-boxes CDR of 1.0 to 9.0. All amnestic MCI subjects fulfilled a MMSE score of 24 to 30 and a Memory Box score of at least 0.5. Otherwise, the subjects who had any serious neurological disease other than possible AD, or any history of brain lesions or head trauma, or were psychoactive medication user (including antidepressants, neuroleptics, chronic anxiolytics, or sedative hypnotics) were excluded. More details concerning the ADNI cohort were reported elsewhere32,34. The final dataset for the present analysis comprised 812 individuals, including 281 health controls (normal cognition, NC), 483 MCI and 48 AD at baseline. The basic data of subjects in our analysis was downloaded from the ADNI website in 2015.
Genetic data and SNP selection
Bead Studio 3.2 software and a recent Genome Studio v2009.1 (Illumina) were successively used to generate SNP genotypes from bead intensity data35. Additionally, the widely used PLINK data format was accessible to facilitate analysis by other groups. In our study, PICALM genotypes were extracted from the ADNI PLINK data format and the quality control procedures were performed using PLINK software. Filtering criteria applied to individuals and SNPs were as follows: minimum call rates >90%, minimum minor allele frequencies (MAF) >0.05, Hardy-Weinberg equilibrium test P > 0.001 (Table 2).
SNPs reported to be significantly associated with AD by GWASs9,10,12 were preferentially selected for analysis. As supplementary strategy, we further searched the potentially promising PICALM SNPs from meta-analysis and replication studies15,36,37,38,39. A total of 22 SNPs (Supplementary Table 1) were initially identified in the initial screening, among which 15 SNPs were further excluded, including 12 not found in ADNI and 3 with a MAF < 0.05 (Fig. S1). Finally, we chose the remaining 7 loci as our target SNPs in this study (Table 2).
MRI structure
ADNI MRIs were acquired at multiple sites with a GE Healthcare (Buckinghamshire, England), Siemens Medical Solutions USA (Atlanta, Georgia), or Philips Electronics 3.0 T system (Philips Electronics North America; Sunnyvale, California)40. These analyses utilized the dataset of UCSF FreeSurfer to conduct association test of PICALM genotypes with brain structure. We processed the cerebral image segmentation and analysis using the FreeSurfer version 5.1.0 software package (http://surfer.nmr.mgh.harvard.edu/) based on the 2010 Desikan-Killany atlas41. The main work contained that motion correction and averaging of multiple volumetric T1-weighted images (when more than one is available)42, removal of non-brain tissue using a hybrid watershed/deformable surface algorithm43, automated Talairach transformation, segmentation of the subcortical white matter and deep gray matter volumetric structures (including hippocampus, amygdala, caudate, putamen, ventricles)44,45, intensity normalization46, tessellation of the gray matter white matter boundary, automated topology correction47, and surface deformation following intensity gradients to optimally place the gray/white and gray/cerebrospinal fluid (CSF) borders at the location where the greatest shift in intensity defines the transition to the other tissue class48. More detailed technical procedures were available in previous study48.
Here, we defined seven brain regions, including hippocampus, hippocampus CA1 subregion, middle temporal area, entorhinal area, posterior cingulate, precuneus and parahippocampal area, as regions of interest (ROIs). These regions were known to be affected by AD and their atrophy in AD has been previously validated via MRI studies49,50,51,52,53. In the present analysis, there were 812 (NC = 281, MCI = 483, AD = 48) individuals included in the regional volume/thickness analysis (Table 1).
Statistical analysis
Differences in continuous variables were examined using one-way analysis of variance (ANOVA), and categorical data were tested using chi-square test. Furthermore, a multiple linear regression model which considered age, gender, education, intracranial volume and ApoE4 status as covariates was used to estimate the possible correlation between volume/thickness (baseline data and follow-up changes) and PICALM genotypes. All statistical analyses were performed by R 3.12 (http://www.r-project.org/) and PLINK 1.07 (http://pngu.mgh.harvard.edu/wpurcell/plink/). As Bonferroni correction was inappropriate owing to the nonindependence of tests40, we used the false discovery rate (FDR), the method developed by Hochberg and Benjamini54, to control for multiple hypothesis testing. The criterion for significant difference was P < 0.05 according to FDR correction.
We first screened significant brain regions associated with PICALM loci in the mixed population comprising individuals with normal cognition (NC), mild cognitive impairment (MCI) and Alzheimer’s disease. To further validate the hereditary susceptibility in different population, we then repeated the test independently using sub-population, including NC and MCI and AD individuals.
Additional Information
How to cite this article: Xu, W. et al. The impact of PICALM genetic variations on reserve capacity of posterior cingulate in AD continuum. Sci. Rep. 6, 24480; doi: 10.1038/srep24480 (2016).
References
Prince, M. et al. The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement. 9, 63–75 e62, 10.1016/j.jalz.2012.11.007 (2013).
Chan, K. Y. et al. Epidemiology of Alzheimer’s disease and other forms of dementia in China, 1990–2010: a systematic review and analysis. Lancet 381, 2016–2023, 10.1016/S0140-6736(13)60221-4 (2013).
Wimo, A. et al. The worldwide economic impact of dementia 2010. Alzheimers Dement. 9, 1–11 e13, 10.1016/j.jalz.2012.11.006 (2013).
Murray, C. J. et al. UK health performance: findings of the Global Burden of Disease Study 2010. Lancet 381, 997–1020, 10.1016/S0140-6736(13)60355-4 (2013).
Murray, C. J. et al. The state of US health, 1990–2010: burden of diseases, injuries, and risk factors. Jama-J Am Med Assoc. 310, 591–608, 10.1001/jama.2013.13805 (2013).
Alzheimer’s, A. Alzheimer’s disease facts and figures. Alzheimers Dement. 11, 332–384 (2015).
Jiang, T., Yu, J. T., Tian, Y. & Tan, L. Epidemiology and etiology of Alzheimer’s disease: from genetic to non-genetic factors. Curr Alzheimer Res. 10, 852–867 (2013).
Xu, W., Yu, J. T., Tan, M. S. & Tan, L. Cognitive reserve and Alzheimer’s disease. Mol Neurobiol. 51, 187–208, 10.1007/s12035-014-8720-y (2015).
Harold, D. et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 41, 1088–1093, 10.1038/ng.440 (2009).
Seshadri, S. et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. Jama 303, 1832–1840, 10.1001/jama.2010.574 (2010).
Lambert, J. C. et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 45, 1452–1458, 10.1038/ng.2802 (2013).
Naj, A. C. et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 43, 436–441, 10.1038/ng.801 (2011).
Carrasquillo, M. M. et al. Replication of CLU, CR1, and PICALM associations with alzheimer disease. Arch Neurol-Chicago. 67, 961–964, 10.1001/archneurol.2010.147 (2010).
Corneveaux, J. J. et al. Association of CR1, CLU and PICALM with Alzheimer’s disease in a cohort of clinically characterized and neuropathologically verified individuals. Hum Mol Genet. 19, 3295–3301, 10.1093/hmg/ddq221 (2010).
Jun, G. et al. Meta-analysis confirms CR1, CLU, and PICALM as alzheimer disease risk loci and reveals interactions with APOE genotypes. Arch Neurol-Chicago. 67, 1473–1484, 10.1001/archneurol.2010.201 (2010).
Lambert, J. C. et al. Evidence of the association of BIN1 and PICALM with the AD risk in contrasting European populations. Neurobiol Aging. 32, 756 e711–755, 10.1016/j.neurobiolaging.2010.11.022 (2011).
Kamboh, M. I. et al. Association of CLU and PICALM variants with Alzheimer’s disease. Neurobiol Aging. 33, 518–521, 10.1016/j.neurobiolaging.2010.04.015 (2012).
Liu, G. et al. PICALM gene rs3851179 polymorphism contributes to Alzheimer’s disease in an Asian population. Neuromol Med. 15, 384–388, 10.1007/s12017-013-8225-2 (2013).
Yu, J. T. et al. Genetic association of PICALM polymorphisms with Alzheimer’s disease in Han Chinese. J Neurol Sci. 300, 78–80, 10.1016/j.jns.2010.09.027 (2011).
Stern, Y. Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol. 11, 1006–1012, 10.1016/S1474-4422(12)70191-6 (2012).
Stern, Y. What is cognitive reserve? Theory and research application of the reserve concept. J Int Neuropsych Soc. 8, 448–460 (2002).
Barulli, D. & Stern, Y. Efficiency, capacity, compensation, maintenance, plasticity: emerging concepts in cognitive reserve. Trends Cogn Sci. 17, 502–509, 10.1016/j.tics.2013.08.012 (2013).
Leech, R. & Sharp, D. J. The role of the posterior cingulate cortex in cognition and disease. Brain: a journal of neurology 137, 12–32, 10.1093/brain/awt162 (2014).
Huang, Y. & Mucke, L. Alzheimer mechanisms and therapeutic strategies. Cell 148, 1204–1222, 10.1016/j.cell.2012.02.040 (2012).
Alves, G. S. et al. Different patterns of white matter degeneration using multiple diffusion indices and volumetric data in mild cognitive impairment and Alzheimer patients. PloS one 7, e52859, 10.1371/journal.pone.0052859 (2012).
Kiuchi, K. et al. Abnormalities of the uncinate fasciculus and posterior cingulate fasciculus in mild cognitive impairment and early Alzheimer’s disease: a diffusion tensor tractography study. Brain Res. 1287, 184–191, 10.1016/j.brainres.2009.06.052 (2009).
Choo, I. H. et al. Posterior cingulate cortex atrophy and regional cingulum disruption in mild cognitive impairment and Alzheimer’s disease. Neurobiol Aging. 31, 772–779, 10.1016/j.neurobiolaging.2008.06.015 (2010).
Pengas, G., Hodges, J. R., Watson, P. & Nestor, P. J. Focal posterior cingulate atrophy in incipient Alzheimer’s disease. Neurobiol Aging. 31, 25–33, 10.1016/j.neurobiolaging.2008.03.014 (2010).
Richiardi, J. et al. Correlated gene expression supports synchronous activity in brain networks. Science 348, 1241–1244, 10.1126/science.1255905 (2015).
Scheff, S. W. et al. Synaptic change in the posterior cingulate gyrus in the progression of Alzheimer’s disease. J Alzheimers Dis. 43, 1073–1090, 10.3233/JAD-141518 (2015).
Xu, W., Tan, L. & Yu, J. T. The Role of PICALM in Alzheimer’s Disease. Mol Neurobiol. 10.1007/s12035-014-8878-3 (2014).
Mueller, S. G. et al. The Alzheimer’s disease neuroimaging initiative. Neuroimag clin n am. 15, 869–877, xi-xii, 10.1016/j.nic.2005.09.008 (2005).
Carrillo, M. C., Bain, L. J., Frisoni, G. B. & Weiner, M. W. Worldwide Alzheimer’s disease neuroimaging initiative. Alzheimers Dement. 8, 337–342, 10.1016/j.jalz.2012.04.007 (2012).
Petersen, R. C. et al. Alzheimer’s Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology 74, 201–209, 10.1212/WNL.0b013e3181cb3e25 (2010).
Saykin, A. J. et al. Alzheimer’s Disease Neuroimaging Initiative biomarkers as quantitative phenotypes: Genetics core aims, progress, and plans. Alzheimers Dement. 6, 265–273, 10.1016/j.jalz.2010.03.013 (2010).
Lee, J. H. et al. Identification of novel loci for Alzheimer disease and replication of CLU, PICALM, and BIN1 in Caribbean Hispanic individuals. Arch Neurol. 68, 320–328, 10.1001/archneurol.2010.292 (2011).
Parikh, I., Medway, C., Younkin, S., Fardo, D. W. & Estus, S. An intronic PICALM polymorphism, rs588076, is associated with allelic expression of a PICALM isoform. Mol Neurodegener. 9, 32, 10.1186/1750-1326-9-32 (2014).
Schnetz-Boutaud, N. C. et al. Identification and confirmation of an exonic splicing enhancer variation in exon 5 of the Alzheimer disease associated PICALM gene. Ann Hum Genet. 76, 448–453, 10.1111/j.1469-1809.2012.00727.x (2012).
Furney, S. J. et al. Genome-wide association with MRI atrophy measures as a quantitative trait locus for Alzheimer’s disease. Mol Psychiatr. 16, 1130–1138, 10.1038/mp.2010.123 (2011).
Biffi, A. et al. Genetic variation and neuroimaging measures in Alzheimer disease. Arch Neurol. 67, 677–685, 10.1001/archneurol.2010.108 (2010).
Desikan, R. S. et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. NeuroImage 31, 968–980, 10.1016/j.neuroimage.2006.01.021 (2006).
Reuter, M., Rosas, H. D. & Fischl, B. Highly accurate inverse consistent registration: a robust approach. NeuroImage 53, 1181–1196, 10.1016/j.neuroimage.2010.07.020 (2010).
Segonne, F. et al. A hybrid approach to the skull stripping problem in MRI. NeuroImage 22, 1060–1075, 10.1016/j.neuroimage.2004.03.032 (2004).
Fischl, B. et al. Sequence-independent segmentation of magnetic resonance images. NeuroImage 23 Suppl 1, S69–84, 10.1016/j.neuroimage.2004.07.016 (2004).
Fischl, B. et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron 33, 341–355 (2002).
Sled, J. G., Zijdenbos, A. P. & Evans, A. C. A nonparametric method for automatic correction of intensity nonuniformity in MRI data. Ieee T Med Imaging. 17, 87–97, 10.1109/42.668698 (1998).
Segonne, F., Pacheco, J. & Fischl, B. Geometrically accurate topology-correction of cortical surfaces using nonseparating loops. Ieee T Med Imaging. 26, 518–529, 10.1109/TMI.2006.887364 (2007).
Fischl, B., Liu, A. & Dale, A. M. Automated manifold surgery: constructing geometrically accurate and topologically correct models of the human cerebral cortex. Ieee T Med Imaging. 20, 70–80, 10.1109/42.906426 (2001).
Simmons, A. et al. MRI measures of Alzheimer’s disease and the AddNeuroMed study. Ann Ny Acad Sci. 1180, 47–55, 10.1111/j.1749-6632.2009.05063.x (2009).
Kesslak, J. P., Nalcioglu, O. & Cotman, C. W. Quantification of magnetic resonance scans for hippocampal and parahippocampal atrophy in Alzheimer’s disease. Neurology 41, 51–54 (1991).
Convit, A. et al. Specific hippocampal volume reductions in individuals at risk for Alzheimer’s disease. Neurobiol Aging. 18, 131–138 (1997).
Jack, C. R. Jr. et al. Rate of medial temporal lobe atrophy in typical aging and Alzheimer’s disease. Neurology 51, 993–999 (1998).
Risacher, S. L. et al. Baseline MRI predictors of conversion from MCI to probable AD in the ADNI cohort. Curr Alzheimer Res. 6, 347–361 (2009).
Hochberg, Y. & Benjamini, Y. More powerful procedures for multiple significance testing. Stat Med. 9, 811–818 (1990).
Acknowledgements
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen Idec Inc.; Bristol-Myers Squibb Company; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Medpace, Inc.; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Synarc Inc.; and Takeda Pharmaceutical Company. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuroimaging at the University of Southern California. This work was also supported by grants from the National Natural Science Foundation of China (81471309, 81171209, 81371406, 81571245, 81501103).
Author information
Authors and Affiliations
Consortia
Contributions
J.T.Y. and Lan.T. design the whole study. X.W. analyzed the data, wrote the main manuscript text and prepared all figures. H.F.W. collected the data from ADNI database and prepared the tables. Lin.T., M.S.T., C.C.T. and X.C.Z. helped analyze the data. D.M. and W.J.Y. helped collect the data from ADNI database. T.J. helped to revise the manuscript. All authors reviewed the manuscript. Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Xu, W., Wang, HF., Tan, L. et al. The impact of PICALM genetic variations on reserve capacity of posterior cingulate in AD continuum. Sci Rep 6, 24480 (2016). https://doi.org/10.1038/srep24480
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep24480
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
