
Abstract
The intestinal microbiome is a unique ecosystem that influences metabolism in humans. Experimental evidence indicates that intestinal microbiota can transfer an obese phenotype from humans to mice. Since mothers transmit intestinal microbiota to their offspring during labor, we hypothesized that among vaginal deliveries, maternal body mass index is associated with neonatal gut microbiota composition. We report the association of maternal pre-pregnancy body mass index on stool microbiota from 74 neonates, 18 born vaginally (5 to overweight or obese mothers) and 56 by elective C-section (26 to overweight or obese mothers). Compared to neonates delivered vaginally to normal weight mothers, neonates born to overweight or obese mothers had a distinct gut microbiota community structure (weighted UniFrac distance PERMANOVA, p < 0.001), enriched in Bacteroides and depleted in Enterococcus, Acinetobacter, Pseudomonas, and Hydrogenophilus. We show that these microbial signatures are predicted to result in functional differences in metabolic signaling and energy regulation. In contrast, among elective Cesarean deliveries, maternal body mass index was not associated with neonatal gut microbiota community structure (weighted UniFrac distance PERMANOVA, p = 0.628). Our findings indicate that excess maternal pre-pregnancy weight is associated with differences in neonatal acquisition of microbiota during vaginal delivery, but not Cesarean delivery. These differences may translate to altered maintenance of metabolic health in the offspring.
Similar content being viewed by others


Introduction
An increasing number of pregnant women around the world are overweight or obese1,2,3. Maternal overweight and obesity may result in 2 to 5 fold greater risk of obesity for their children4,5,6. Breaking this vicious cycle of obesity requires a better understanding of its underpinnings. The association between excess maternal weight and childhood obesity is not fully explained by known genetic polymorphisms or lifestyle factors shared between the mother and her offspring7. A novel hypothesis is that at least part of the intergenerational obesity association is due to mother-to-newborn transmission of obesogenic microbiota8,9.
As women progress through pregnancy their intestinal microbiota undergo marked shifts to promote weight gain and accommodate the growth of the fetus10. Normal pregnancy-related remodeling of the intestinal microbiota is modified by the amount of weight with which women enter pregnancy11. Specifically, compared to their normal weight counterparts, women carrying excess pre-pregnancy weight have been shown to have higher levels of Bacteroides in the third trimester11. Considering that the mother is the primary source of microbiota for the newborn9, and that transmission of obese human microbiota to germ-free mice alters the assembly of their intestinal microbiome in a manner that favors adipogenesis12,13,14, it is plausible that weight-related modifications to the maternal intestinal microbiome during pregnancy may impact assembly of the infant intestinal microbiome and, in turn, host metabolism.
Indeed, previous studies have shown that maternal pre-pregnancy body mass index (BMI) is associated with the intestinal microbial community structure of infants’ stool at 1 month, 6 months15, and 2 years of age16. But it is still unknown whether the structuring of the infant intestinal microbiome by maternal weight status is a result of mother-to-offspring transmission of microbiota during labor, after birth, or even prior to birth, as has been suggested by recent study17. In our study, we examine the role of maternal pre-pregnancy BMI in the assembly of the intestinal microbiome of neonates delivered vaginally vs. by elective Cesarean section (CS) without exposure to membrane rupture. We find that maternal pre-pregnancy BMI is associated with microbial community structure and taxomonic differences in neonatal stool if the neonate is delivered vaginally, but not if they are delivered by CS. Moreover, we show that these microbial alterations are predicted to translate into functional metagenomic differences in metabolism and energy regulation.
Results
Study population
The mean ± SD maternal pre-pregnancy BMI from our analytic sample was 24.7 ± 4.5 kg/m2, and for only the overweight or obese group it was 28.8 ± 3.7 kg/m2. The hospital from which the participants were recruited is a private hospital with high socioeconomic status patients. Almost all of the women (70 of 74) in our sample described themselves as “white” (or “branco” in Portuguese). Other clinical characteristics of the study sample are presented in Table 1.
Characterization of neonatal intestinal microbiome
To investigate the contribution of maternal body weight to the establishment of the instestinal microbiome of newborns, we undertook 16S ribosomal RNA (rRNA) sequencing on the second day stool of 56 elective CS neonates not exposed to ruptured membranes (26 from overweight or obese mothers) and 21 vaginally delivered (VD) neonates (5 from overweight or obese mothers). After quality filtering (pair-end, Phred > =Q20), we obtained 725,089 sequences from 74 neonatal stool samples; on average ± SEM, 9,799 ± 474 reads per sample (Table S1), binned to 3,717 type of OTUs. All four major bacterial phyla (Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria) were represented in the neonatal stool, accounting for ~99% of the microbial content in each sample.
Delivery mode, maternal body mass index, and neonatal intestinal microbiome
The microbial beta diversity of stool (weighted and unweighted UniFrac distance PERMANOVA, p < 0.001) was different between neonates born via CS vs. VD (Figure S1), but there were no statistical differences in measures of alpha diversity, namely phylogenetic diversity (p = 0.07), number of observed species (p = 0.29) or species richness (p = 0.25) (Figure S2).
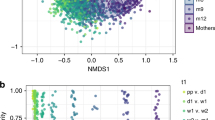
We then stratified by delivery mode (CS vs. VD) and compared the microbial communities in stool from neonates born to normal weight vs. overweight or obese (OWOB) mothers in each stratum. In VD neonates’ stool there were statistically significant differences in microbial beta diversity between those born to OWOB vs. normal weight mothers (weighted UniFrac distance PERMANOVA, p < 0.001). Consistent with the PERMANOVA results, the microbial communities in the stool of vaginally delivered neonates showed some separation according to maternal pre-pregnancy BMI in unweighted UniFrac principal coordinate analysis (PCoA) plots (Fig. 1). In contrast, maternal pre-pregnancy BMI was not associated with microbial beta diversity of stool from CS delivered neonates (weighted UniFrac distance PERMANOVA, p = 0.628). There was no evidence from PCoA that microbial communities in stools from CS-delivered neonates separated according to maternal pre-pregnancy BMI (Fig. 1). As illustrated in Figure S3, there were no differences in measures of alpha diversity, including phylogenetic diversity, number of observed species, or species richness (all p values > 0.6) between neonates born to normal weight vs. OWOB mothers in either strata of delivery mode.

Weighted UniFrac distances were used to evaluate beta diversity. PERMANOVA was used to test dissimilarity.
Next we compared the relative abundances of microbial taxa [using bacterial taxa plots and Linear Discriminant Analysis (LDA) Effect Size (LEfSe); LDA > 3.0 fold considered significant] in the stools of CS vs. VD neonates (Figure S4) and the stools of neonates born to OWOB vs. normal weight mothers, according to delivery mode strata (Figure 2 and Figure S5). Among the VD group, compared to neonates born to normal weight mothers, neonates born to OWOB mothers had stool enriched in the bacterial genus Bacteroides (primarily driven by B. vulgatus, followed by B. dorei, B. caccae, B. xylanisolvens, B. stercoris, and B. fragilis species; Table S2), but depleted in the family Xanthomonadaceae, and depleted in the genera Pseudomonas, Acinetobacter, Hydrogenophilus, Enterococcus, (Fig. 2; LDA > 3.0). Pre-pregnancy maternal BMI was not associated with taxonomic differences in the stool of CS neonates, except one minor difference in the Bacteroides genus (driven by B. vulgatus; Table S2) which, contrary to the VD group, was higher in neonates born to normal weight mothers (Fig. 2; LDA > 3.0), but at much lower abundance (relative abundance in normal weight group = 1.0% vs. OWOB group = 0.1%).

(A,B) Bacterial taxa plots at the phylum (A) and genus (B) levels. Each taxa >1% of the average relative abundance in groups is indicated by a different color. Taxa are reported at the lowest identifiable level, indicated by the letter preceding the underscore: f, family; g, genus. C. Histogram of biomarker bacteria in each group. Linear Discriminant Analysis (LDA) Effect Size (>3.0-fold) was used to determine statistically significant biomarkers.
We then used PICRUSt predictive functional profiling18 to examine if pre-pregnancy BMI was associated with altered metabolic functioning of the microbial groups (Table 2). Compared to neonates born vaginally to normal weight mothers, neonates born vaginally to OWOB mothers had higher bacterial gene content related to metabolism of several carbohydrates (fructose, mannose, galactose, and pentose). Pathways related to fatty acid metabolism were overrepresented in VD neonates born to normal weight mothers (Table 2; all comparisons use an LDA > 3.0). In contrast, but consistent with 16S rRNA results, predictive funtional profiling of stool from CS-delivered neonates showed no differences by pre-pregnancy maternal BMI (Table 2; LDA > 3.0). Results from functional profiling using PICRUSt should be interpreted conservatively because of the known intra-species variation in gene content18.
In additional analyses we examined whether the observed associations may be related to differences in prenatal antibiotics or breastfeeding. In the CS group, neither maternal use of antibiotics [taken in the first or second trimesters (n = 11) vs. no use (n = 45)] or breastfeeding [in first 24 hours after birth (n = 49) vs. not (n = 7)] were associated with neonatal fecal microbiota beta diversity (both PERMANOVA p values > 0.05; Figure S6). In the VD group, only 3 mothers used antibiotics in pregnancy, and all mothers breastfed in the first 24 hours, so comparisons were not possible.
Discussion
In this study, we first confirmed previous reports that C-section is the primary perturbation to the natural assembly of neonatal intestinal microbiome shortly after birth19,20,21. We then demonstrated, in analyses stratified by delivery mode, that excess maternal weight was associated with neonatal microbial community structure among neonates delivered vaginally, but not in those delivered by planned C-section without membrane rupture. Herein this study we show for the first time that the association between maternal pre-pregnancy body weight and infant gut microbiome15,16 may stem from vertical transmission of microbiota during labor and not before birth, as was recently suggested17.
Among vaginally delivered neonates, the most striking difference between those born to overweight or obese mothers vs. normal weight mothers was the difference in relative abundance of gram-negative Bacteroides spp. A Finnish study demonstrated overweight mothers had higher levels of Bacteroides genus in the third trimester of pregnancy11. Our findings extend these findings, suggesting that the higher levels of Bacteroides among overweight women in pregnancy may be vertically transmitted to the newborn during vaginal delivery.
Evidence from previous studies suggests that the association between maternal pre-pregnancy BMI and neonatal microbiome observed in our study may change over time. Collado et al. found that maternal pre-pregnacy BMI was negatively associated with fecal levels of Bacteroides when the infants from the mothers in the Finnish study were 1 month of age, and pre-pregnancy BMI was not associated with Bacteroides when the infants were 6 months of age15. In a separate study, Galley et al.16 demonstrated that, among children from high socioeconomic status families, maternal pre-pregnancy BMI was associated with measures of infant intestinal microbial community structure (beta diversity) and species diversity (alpha diversity) at 1 month, 6 months, and 2 years of age, but it was not associated with levels of Bacteroides.
There are several caveats that make it difficult to compare the findings from these studies to ours. First, the studies by Collado et al. and Galley et al. did not stratify by delivery mode. Our study (Figure S4) and others19,20,21,22,23,24 indicate that colonization of the intestinal tract by Bacteroides is delayed in C-section delivered offspring. As overweight mothers are more likely to deliver by C-section, not stratifying by delivery mode may make it appear that overweight mothers deliver infants with lower levels of Bacteroides. Another major difference across studies is the timing of stool specimen collection. Previous studies collected stool from 1 month onward whereas we collected at 2 days postpartum. The infant microbiota are highly variable during the first months of life25,26, and strongly influenced by breastfeeding20,27, which itself varies by maternal BMI28. In our study, all mothers who delivered vaginally initiated breastfeeding within the first 24 hours. Differences in microbiota measurement technique (FCM-FISH and qPCR15 vs. 454 sequencing16 vs. Illumina MiSeq) also makes direct comparison between previous studies and ours challenging29. Finally, it is important to note that maternal overweight is influenced by a heterogenous mix of genetic and environmental factors, each of which likely has its own effect on microbial community structure. For example, individuals who become overweight or obese consuming a high-fat, high-protein animal-based diet may have high relative abundance of bile-tolerant Bacteroides species and low relative abundance of Firmicutes species that metabolize plant polysaccharides30,31,32.
The biomarker discovery tool LEfSe also showed lower relative abundance of Enterococcus, Acinetobacter, Pseudomonas, and Hydrogenophilus in the stool of neonates delivered vaginally to overweight mothers compared to the stool of neonates delivered vaginally to normal weight mothers. Enterococcus faecalis FK-23, isolated from the feces of a healthy human subject, has been shown to inhibit development of obesity and hepatic steatosis in mice fed a high fat diet33. Double blind, placebo controlled feeding trials have shown that Enterococcus subgroups can be increased through a whole grain, prebiotic diet34,35. Acinetobacter, a genus of Gram-negative bacteria, is an antibiotic resistant infectious pathogen found in hospitals36. While this genus has been found to be positively associated with obesity in a case-control study of Taiwanese adults, the overall relative abundance in this study was low in cases and controls37. The other genera discovered by LEfSe, Pseudomonas and Hydrogenophilus, have not, to the best of our knowledge, been previously associated with overweight or obesity.
Many of the Bacteroides species (e.g., B. vulgatus/B. dorei, B. caccae, B. xylanisolvens, B. stercoris, and B. fragilis) driving the association between maternal BMI and neonatal gut micobiota in our study are commonly classified in the B. fragilis group38. Backhed et al. showed that germ-free mice incoculated with B. fragilis gained excess weight13. In prospective cohort studies, colonization with B. fragilis at 1 month of age was positively associated with BMI z-score in children following a low-fiber diet at age 4, but inversely associated with BMI z-score in children following a high-fiber diet39. Vael et al. found a positive association between B. fragilis in the feces of 3- and 26-week old infants and BMI z-score at preschool (n = 138)40. In contrast, White et al. demonstrated, that presence of Bacteroides in 30-day-old infants was associated with lower body weight z-scores at 6 months of age in males (n = 108), but not females41. Collectivley, previous studies indicate that the association of early-life Bacteroides colonization with obesity risk may depend on interactions with other factors.
Through predictive funtional profiling we found that the stool of neonates vaginally delivered by overweight or obese mothers had higher bacterial gene content related to metabolism of several sugar constituents (fructose, mannose, galactose, pentose and glucoronate), amino acids (alanine, aspartate, glutamate and histidine), methane, glycans, sphingolipids and purine, but lower gene content related to the metabolism of other amino acids (lysine, tryptophan, valine, leucine and isoleucine), fatty acids, butanoate, and benzenoate compared to the stool of neonates vaginally delivered to normal weight mothers. There were also differences in bacterial gene content related to enzyme familes and biosynthesis of other secondary metabolites (see Table 2). The enrichment of gene content related to butanoate (i.e., butyrate) metabolism in neonates born to normal weight mothers is particularly intriguing as butyrate is believed to suppress colonic inflammation related to obesity42. Functional differences in the metabolism of certain amino acids43 methane44, sphingolipids45, and purine46 have also been associated with obesity. Interrogation of these predicted functional differences using true metagenomic shotgun sequencing is warranted in future studies.
The predicted functional differences in carbohydrate metabolism, including the various sugar metabolism pathways enriched in the fecal bacterial DNA of neonates born to overweight or obese women, may be most important for the development of obesity. These differences are likely attributed to the bacterial gene content from Bacteroides spp., which have a wide capacity to use diverse types of polysaccharides47. All Bacteroides spp. can utilize frutose, sucrose, and fructo-oligosaccharides as energy48, and they are adept at using oligosaccharides including those found in human breast milk27. Bacteroides spp., except for B. vulgatus, are also able to grow efficiently using one of the long-chain fructans, inulin or levan48. B. caccae and B. fragilis can use inulin with efficiency similar to their use of glucose48. Excess of these microbiota may therefore confer more efficient extraction of energy from these non-digestable dietary components and in turn lead to excess weight gain. While it is still unclear how each of the predicted functional differences observed in our study relate to the future health and disease of these neonates, these findings do provide preliminary insight into how metabolic pathways are altered in the first days of life by delivery mode and maternal weight status, and they provide basis for future study.
There are several limitations to our study worth noting. Because we relied upon 16S rRNA sequencing data, and the mothers in our study were not sampled at delivery, we were unable to establish a direct link between the maternal fecal bacteria and the bacterial communities of the newborns’ gastrointestinal tract. However, a recent study, which sampled feces of both mother and newborn, showed that vaginally delivered newborns’ fecal microbiota resembled the fecal microbiota of their mothers49. An alternative explanation for our findings is that differences in passive immunity or breast milk oligosaccharide composition, which might select for or transfer certain bacteria to the newborn, influenced the observed association of maternal pre-pregnancy BMI with the neonatal gut microbiome. However, this alternative explanation seems unlikely, as we provide evidence that differences in neonatal microbiome by maternal weight status are confined to vaginally delivered neonates. Furthermore, we demonstrate that breastfeeding initiation within the first 24 hours was not associated with neonatal stool microbiota in our study (Figure S6), indicating that breast milk composition was not a strong determinant of the neonatal microbiome in our sample of neonates. Other limitations to our study are the lack of follow-up measures and the small number of neonates delivered vaginally to overweight or obese mothers. As such, we cannot make inferences about whether the associations extend beyond two days after birth, nor can we rule out the possibility that the associations based on the small sample of neonates delivered vaginally to overweight or obese moms (n = 5) were the result of chance.
Conclusion
In summary, the observations from our study provide pioneering evidence that maternal weight status is associated with neonatal gut microbiome assemblage differently according to the mode of delivery. Our findings lay the foundation for molecular forensic studies to test the hypothesis that maternal strains of bacteria influenced by her body weight status are transferred to her newborn during labor. Furthermore, large prospective cohort studies are needed to examine whether the differences in the newborn gut microbiota observed in our study persist beyond the first days of life and are associated with infant and childhood weight, metabolism and health outcomes.
Methods
Design and subjects
The methods of this study were carried out in accordance with the approved guidelines. The study was approved by the Research and Ethics Committee of the Hospital de Clinicas (protocol no. 11/0388) and the Hospital Mae de Deus (protocol no. 524/11) located in Porto Alegre, Brazil. Subjects for this study were recruited by trained study personnel during 2011. Written informed consent was obtained from mothers of the newborns. As part of the consent process, study personnel discussed with participants the potential risks of participation, including the risks associated with specimen collection, and the possibility that protected health information or de-identified project data stored in a public repository could be accidentally released. The protocol and consent form described precautions taken to reduce these risks. If a participant withdrew consent after providing specimens, remaining specimens and extracted nucleic acids were destroyed.
Women scheduled for either vaginal or elective C-section delivery in a private hospital in Porto Alegre, were invited to participate if they were delivering between 38 and 42 weeks (confirmed by ultrasonography that had been performed before the 20th week of gestational age); did not have HIV/AIDS, preeclampsia, other chronic metabolic diseases (e.g., diabetes, hypertension, or an autoimmune disorder); did not smoke; and were not on a restrictive diet. We excluded women who were administered oral antibiotics in 3rd trimester. Cephalosporin was the perioperative antibiotic predominantly administered for planned Cesarean deliveries in Brazil during our study. Gravidae also had to agree to provide the meconium and first stool from their newborn and to complete a standardized postnatal questionnaire adapted for this study50. For vaginal births, only women whose water broke <12 hours before delivery were included.
Of the 89 women (23 vaginal births and 66 elective C-section births) consented and enrolled in the study, 74 delivered newborns with detectable bacterial DNA in their stool. Clinical information for this analysis was derived from medical records and a investigator administed postnatal questionnaire. Information on mode of delivery, gravidity, parity, history of urinary tract infection during pregnancy, antibiotic use during pregnancy, gestational age, birth weight, birth length, head circumference, placenta weight, sex and race was derived from medical records. Pre-pregnancy weight (kg) and height (m) were self-reported on the postnatal questionnaire. Self-reported pre-pregnancy weight has been shown to be reported to be within a few pounds of actual pre-pregnancy weight51.
Body mass index (BMI) was calculated as weight in kilograms divided by the square of height in mothers. We categorized pre-pregnancy BMI status as normal weight (<25 kg/m2), overweight (≥25–30 kg/m2), and obese (≥30 kg/m2), but because there were few obese women, we combined overweight and obesity into one category for overweight or obese (OWOB).
Sample collection and processing
For the current study we used approximately 5 grams of the first neonatal stool (after meconium) collected from the diapers with sterile spatulas and placed in sterile tubes at 4 °C for up to 6 hours and stored at −80 °C until processing. The stool collection was scheduled every 3 hours according to the local routine care. DNA extraction was carried out using QIAamp DNA Stool mini kit (Qiagen Inc, Chatsworth, CA, USA) according to the instructions provided by the manufacturer. Following the DNA extraction (200 uL of final volume), samples were concentrated to 20 uL using 3M Na Acetate pH 5.2, quantified using Nano Drop 1000 Spectrophotometer (Thermo Scientific™, Life Technologies) and stored at −20 °C until analysis.
Polymerase chain reaction and 16S ribosomal RNA sequencing
The variable 4 (V4) amplicon region of 16S ribosomal RNA (rRNA) gene was sequenced on the Illumina MiSeq platform (Illumina, San Diego CA, USA) at New York University with paired-end technique using the protocol modified from Caporals et al. (2010)52. The forward primer construct (5′- AAT GAT ACG GCG ACC ACC GAG ATC TAC ACT ATG GTA ATT GTG TGC CAG CMG CCG CGG TAA-3′) contained a 5′ Illumina adapter, a forward primer pad, and the 515F primer and a two-base linker sequence (‘GT’). The reverse primer (5′- CAA GCA GAA GAC GGC ATA CGA GAT NNN NNN NNN NNN AGT CAG TCA GCC GGA CTA CHV GGG TWT CTA AT – 3′) contained the 3′ Illumina adapter, a unique 12-base error-correcting Golay barcode, a reverse primer pad, a two-base linker sequence (‘CC’) and the 806R primer. The 515F/806R V4 primers amplified the ~291 base pair region of the 16S rRNA gene.
Polymerase chain reaction (PCR) was carried out in triplicate using the Bio-Rad CFX 96 thermal cycler (Bio-Rad, Hercules CA, USA). The PCR mix contained 0.2 mM forward and reverse primers, 1 ul template DNA, 10 ul 5 Prime Hot Master Mix (5 PRIME, Gaithersburg MD, USA) and 12 ul of MoBio PCR certified water (MO BIO Laboratories, Calsbad CA, USA). Thermal cycling consisted of 94 °C for 3 minutes, followed by 35 cycles of 94 °C for 45 seconds, 50 °C for 60 seconds and 72 °C for 90 seconds, with a final extension of 10 minutes at 72 °C to confirm full amplification. Replicate amplicons were pooled and the DNA concentrations were determined using the Quant-iT PicoGreen dsDNA reagent and kit (Invitrogen, Grand Island NY, USA) based on the manufacturer’s instructions. Fluorescence was measured on the Perkin-Elmer Victor Plate reader using the 490/535 nm excitation/emission filter pair with measurement time 0.1 seconds. The amplicons were then pooled in equimolar ratios and purified using QIA quick PCR purification kit (Qiagen Inc, Chatsworth, CA, USA). The final concentration of cleaned DNA amplicon was determined using the Qubit PicoGreen dsDNA HS assay kit (Invitrogen, Grand Island, NY, USA) and was sequenced using the paired-end technique. Reagents for DNA extraction and for PCR amplification were sequenced as controls53. There were no problems to get proper DNA quantity (>100 ng per sample) by PCR amplification.
16S rRNA gene sequence analysis
We processed sequence reads using the Quantitative Insights Into Microbial Ecology (QIIME) 1.8.0 software package52 to analyze 16S rRNA gene sequence data. The quality filtrated sequences (Phred >=Q20) were used to identify and quantify Operational Taxonomic Units (OTUs), with an open-reference OTU picking method using 97% identity to the Greengenes database (v13_8)54 by UCLUST55 and PyNAST56 alignment algorithms. FastTree57 was used to construct a phylogenetic tree. ChimeraSlayer was used to identify and remove chimeric sequences.
Samples were rarefied to 1,634 reads per sample, and evaluated for alpha diversity (microbial diversity within samples) and beta diversity (community diversity divergence between samples). Alpha rarefaction was plotted using the phylogenetic distance and the detected number of species metrics. We randomly subsampled sequences over a range of specified depths and calculated alpha diversity for each sample, with 10 independent iterations at each depth. Principle coordinate analysis (PCoA) was used to measure beta diversity on normalized OTU tables using weighted and unweighted Unifrac distance58. The significance of PCoA plots were analyzed using permutational multivariate analysis of variance (PERMANOVA), which uses distance metrics to confirm the strength and statistical significance of sample groupings59. We used 999 Monte Carlo permutations in QIIME to assess statistical significance of between-group diversity metrics.
The linear discriminant analysis (LDA) effect size (LEfSe) algorithm (found in the online interface Galaxy: https://huttenhower.sph.harvard.edu) was used to identify significant differences in relative abundance of bacterial taxonomy. For the LEfSe analysis, the α value for the factorial Kruskal-Wallis test was set at 0.05. The threshold for the logarithmic LDA score was 3.0. Taxonomic predictions in species level of OTUs detected by LEfSe were carried out using BLASTN against a 16S rRNA gene reference database from NCBI at a cut-off of 97% identity.
In silico metagenome prediction
Phylogenetics Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) was used to predict functional metagenomes from the 16S rRNA gene18. Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthologs classification was used to classify predicted metagenomes60. LEfSe algorithm was used to detect significant differences of gene content between groups (LDA score > 3.0-fold).
Analysis of clinical characteristics
Descriptive analyses were performed according to the parametric or nonparametric distribution of data, as identified by the Kolmogorov–Smirnov test. We used a Student’s t test for unpaired parametric, a Mann-Whitney test for unpaired nonparametric data, and a Chi-square test to compare proportions. A p < 0.05 was considered statistically significant.
Additional Information
How to cite this article: Mueller, N. T. et al. Birth mode-dependent association between pre-pregnancy maternal weight status and the neonatal intestinal microbiome. Sci. Rep. 6, 23133; doi: 10.1038/srep23133 (2016).
References
Barros, F. C. & Victora, C. G. Maternal-child health in Pelotas, Rio Grande do Sul State, Brazil: major conclusions from comparisons of the 1982, 1993, and 2004 birth cohorts. Cad Saude Publica 24 Suppl 3, S461–467 (2008).
Heslehurst, N. et al. Trends in maternal obesity incidence rates, demographic predictors, and health inequalities in 36,821 women over a 15-year period. BJOG 114, 187–194, doi: 10.1111/j.1471-0528.2006.01199.x (2007).
Chu, S. Y., Kim, S. Y. & Bish, C. L. Prepregnancy obesity prevalence in the United States, 2004–2005. Matern Child Health J 13, 614–620, doi: 10.1007/s10995-008-0388-3 (2009).
Whitaker, K. L., Jarvis, M. J., Beeken, R. J., Boniface, D. & Wardle, J. Comparing maternal and paternal intergenerational transmission of obesity risk in a large population-based sample. Am J Clin Nutr 91, 1560–1567, doi: 10.3945/ajcn.2009.28838 (2010).
Murrin, C. M., Kelly, G. E., Tremblay, R. E. & Kelleher, C. C. Body mass index and height over three generations: evidence from the Lifeways cross-generational cohort study. BMC Public Health 12, 81, doi: 10.1186/1471-2458-12-81 (2012).
Widen, E. M. et al. Gestational weight gain and obesity, adiposity and body size in African-American and Dominican children in the Bronx and Northern Manhattan. Matern Child Nutr, doi: 10.1111/mcn.12174 (2015).
Llewellyn, C. H., Trzaskowski, M., Plomin, R. & Wardle, J. Finding the missing heritability in pediatric obesity: the contribution of genome-wide complex trait analysis. Int J Obes (Lond) 37, 1506–1509, doi: 10.1038/ijo.2013.30 (2013).
Gohir, W., Ratcliffe, E. M. & Sloboda, D. M. Of the bugs that shape us: maternal obesity, the gut microbiome, and long-term disease risk. Pediatr Res 77, 196–204, doi: 10.1038/pr.2014.169 (2015).
Mueller, N. T., Bakacs, E., Combellick, J., Grigoryan, Z. & Dominguez-Bello, M. G. The infant microbiome development: mom matters. Trends Mol Med 21, 109–117, doi: 10.1016/j.molmed.2014.12.002 (2015).
Koren, O. et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 150, 470–480, doi: 10.1016/j.cell.2012.07.008 (2012).
Collado, M. C., Isolauri, E., Laitinen, K. & Salminen, S. Distinct composition of gut microbiota during pregnancy in overweight and normal-weight women. Am J Clin Nutr 88, 894–899 (2008).
Ley, R. E. et al. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA 102, 11070–11075, doi: 10.1073/pnas.0504978102 (2005).
Backhed, F. et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA 101, 15718–15723, doi: 10.1073/pnas.0407076101 (2004).
Ridaura, V. K. et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341, 1241214, doi: 10.1126/science.1241214 (2013).
Collado, M. C., Isolauri, E., Laitinen, K. & Salminen, S. Effect of mother’s weight on infant’s microbiota acquisition, composition, and activity during early infancy: a prospective follow-up study initiated in early pregnancy. Am J Clin Nutr 92, 1023–1030, doi: 10.3945/ajcn.2010.29877 (2010).
Galley, J. D., Bailey, M., Kamp Dush, C., Schoppe-Sullivan, S. & Christian, L. M. Maternal obesity is associated with alterations in the gut microbiome in toddlers. PLoS One 9, e113026, doi: 10.1371/journal.pone.0113026 (2014).
Antony, K. M. et al. The Preterm Placental Microbiome Varies in Association with Excess Maternal Gestational Weight Gain. Am J Obstet Gynecol, doi: 10.1016/j.ajog.2014.12.041 (2014).
Langille, M. G. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31, 814–821, doi: 10.1038/nbt.2676 (2013).
Dominguez-Bello, M. G. et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci USA 107, 11971–11975, doi: 10.1073/pnas.1002601107 (2010).
Backhed, F. et al. Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe 17, 690–703, doi: 10.1016/j.chom.2015.04.004 (2015).
Jakobsson, H. E. et al. Decreased gut microbiota diversity, delayed Bacteroidetes colonisation and reduced Th1 responses in infants delivered by caesarean section. Gut 63, 559–566, doi: 10.1136/gutjnl-2012-303249 (2014).
Biasucci, G., Benenati, B., Morelli, L., Bessi, E. & Boehm, G. Cesarean delivery may affect the early biodiversity of intestinal bacteria. J Nutr 138, 1796S–1800S (2008).
Tsuji, H. et al. Molecular monitoring of the development of intestinal microbiota in Japanese infants. Benef Microbes 3, 113–125, doi: 10.3920/BM2011.0038 (2012).
Kabeerdoss, J. et al. Development of the gut microbiota in southern Indian infants from birth to 6 months: a molecular analysis. Journal of nutritional science 2, e18, doi: 10.1017/jns.2013.6 (2013).
Koenig, J. E. et al. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci USA 108, Suppl 1, 4578–4585, doi: 10.1073/pnas.1000081107 (2011).
Palmer, C., Bik, E. M., DiGiulio, D. B., Relman, D. A. & Brown, P. O. Development of the human infant intestinal microbiota. PLoS Biol 5, e177, doi: 10.1371/journal.pbio.0050177 (2007).
Marcobal, A. et al. Bacteroides in the infant gut consume milk oligosaccharides via mucus-utilization pathways. Cell Host Microbe 10, 507–514, doi: 10.1016/j.chom.2011.10.007 (2011).
Turcksin, R., Bel, S., Galjaard, S. & Devlieger, R. Maternal obesity and breastfeeding intention, initiation, intensity and duration: a systematic review. Matern Child Nutr 10, 166–183, doi: 10.1111/j.1740-8709.2012.00439.x (2014).
Nelson, M. C., Morrison, H. G., Benjamino, J., Grim, S. L. & Graf, J. Analysis, optimization and verification of Illumina-generated 16S rRNA gene amplicon surveys. PLoS One 9, e94249, doi: 10.1371/journal.pone.0094249 (2014).
Wu, G. D. et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 334, 105–108, doi: 10.1126/science.1208344 (2011).
David, L. A. et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563, doi: 10.1038/nature12820 (2014).
Graf, D. et al. Contribution of diet to the composition of the human gut microbiota. Microb Ecol Health Dis 26, 26164, doi: 10.3402/mehd.v26.26164 (2015).
Kondoh, M. et al. Beneficial effects of heat-treated Enterococcus faecalis FK-23 on high-fat diet-induced hepatic steatosis in mice. Br J Nutr 112, 868–875, doi: 10.1017/S0007114514001792 (2014).
Carvalho-Wells, A. L. et al. Determination of the in vivo prebiotic potential of a maize-based whole grain breakfast cereal: a human feeding study. Br J Nutr 104, 1353–1356, doi: 10.1017/S0007114510002084 (2010).
Costabile, A. et al. Whole-grain wheat breakfast cereal has a prebiotic effect on the human gut microbiota: a double-blind, placebo-controlled, crossover study. Br J Nutr 99, 110–120, doi: 10.1017/S0007114507793923 (2008).
Tan, C. K. et al. Correlation between antibiotic consumption and carbapenem-resistant Acinetobacter baumannii causing health care-associated infections at a hospital from 2005 to 2010. J Microbiol Immunol Infect, doi: 10.1016/j.jmii.2014.02.004 (2014).
Chiu, C. M. et al. Systematic analysis of the association between gut flora and obesity through high-throughput sequencing and bioinformatics approaches. BioMed Res Int 2014, 906168, doi: 10.1155/2014/906168 (2014).
Wexler, H. M. Bacteroides: the good, the bad, and the nitty-gritty. Clin Microbiol Rev 20, 593–621, doi: 10.1128/CMR.00008-07 (2007).
Scheepers, L. E. et al. The intestinal microbiota composition and weight development in children: the KOALA Birth Cohort Study. Int J Obes (Lond) 39, 16–25, doi: 10.1038/ijo.2014.178 (2015).
Vael, C., Verhulst, S. L., Nelen, V., Goossens, H. & Desager, K. N. Intestinal microflora and body mass index during the first three years of life: an observational study. Gut Pathog 3, 8, doi: 10.1186/1757-4749-3-8 (2011).
White, R. A. et al. Novel developmental analyses identify longitudinal patterns of early gut microbiota that affect infant growth. PLoS Comput Biol 9, e1003042, doi: 10.1371/journal.pcbi.1003042 (2013).
Le Chatelier, E. et al. Richness of human gut microbiome correlates with metabolic markers. Nature 500, 541–546, doi: 10.1038/nature12506 (2013).
Neis, E. P., Dejong, C. H. & Rensen, S. S. The role of microbial amino acid metabolism in host metabolism. Nutrients 7, 2930–2946, doi: 10.3390/nu7042930 (2015).
Basseri, R. J. et al. Intestinal methane production in obese individuals is associated with a higher body mass index. Gastroenterol Hepatol (NY) 8, 22–28 (2012).
Bikman, B. T. A role for sphingolipids in the pathophysiology of obesity-induced inflammation. Cell Mol Life Sci 69, 2135–2146, doi: 10.1007/s00018-012-0917-5 (2012).
Lima, W. G., Martins-Santos, M. E. & Chaves, V. E. Uric acid as a modulator of glucose and lipid metabolism. Biochimie 116, 17–23, doi: 10.1016/j.biochi.2015.06.025 (2015).
Xu, J. et al. Evolution of symbiotic bacteria in the distal human intestine. PLoS Biol 5, e156, doi: 10.1371/journal.pbio.0050156 (2007).
Sonnenburg, E. D. et al. Specificity of polysaccharide use in intestinal bacteroides species determines diet-induced microbiota alterations. Cell 141, 1241–1252, doi: 10.1016/j.cell.2010.05.005 (2010).
Backhed, F. et al. Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe 17, 852, doi: 10.1016/j.chom.2015.05.012 (2015).
Bernardi, J. R. et al. Impact of Perinatal Different Intrauterine Environments on Child Growth and Development in the First Six Months of Life–IVAPSA Birth Cohort: rationale, design, and methods. BMC Pregnancy Childbirth 12, 25, doi: 10.1186/1471-2393-12-25 (2012).
Russell, A., Gillespie, S., Satya, S. & Gaudet, L. M. Assessing the accuracy of pregnant women in recalling pre-pregnancy weight and gestational weight gain. J Obstet Gynaecol Can 35, 802–809 (2013).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7, 335–336, doi: 10.1038/Nmeth.F.303 (2010).
Salter, S. J. et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol 12, 87, doi: 10.1186/s12915-014-0087-z (2014).
DeSantis, T. Z. et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microb 72, 5069–5072, doi: 10.1128/AEM.03006-05 (2006).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461, doi: 10.1093/bioinformatics/btq461 (2010).
Caporaso, J. G. et al. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26, 266–267, doi: 10.1093/bioinformatics/btp636 (2010).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2–approximately maximum-likelihood trees for large alignments. PloS One 5, e9490, doi: 10.1371/journal.pone.0009490 (2010).
Lozupone, C., Hamady, M. & Knight, R. UniFrac–an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics 7, 371, doi: 10.1186/1471-2105-7-371 (2006).
Kelly, B. J. et al. Power and sample-size estimation for microbiome studies using pairwise distances and PERMANOVA. Bioinformatics 31, 2461–2468, doi: 10.1093/bioinformatics/btv183 (2015).
Kanehisa, M. et al. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res 42, D199–205, doi: 10.1093/nar/gkt1076 (2014).
Acknowledgements
This study was conducted using academic funds from the Ministry of Education (Capes), and FIPE-HCPA, UFRGS.
Author information
Authors and Affiliations
Contributions
N.T.M., M.G.D.-B. and H.A.S.G. planned the study; A.P., I.C.W., U.M. and M.Z.G. implemented the sample collection and preservation; N.T.M., H.S. and M.G.D.-B. characterized bacterial isolates; N.T.M., H.S. and M.G.D.-B. performed analyses and interpreted results; N.T.M., H.S., U.M., H.A.S.G., M.Z.G. and M.G.D.-B. wrote the paper. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Mueller, N., Shin, H., Pizoni, A. et al. Birth mode-dependent association between pre-pregnancy maternal weight status and the neonatal intestinal microbiome. Sci Rep 6, 23133 (2016). https://doi.org/10.1038/srep23133
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep23133
This article is cited by
-
The investigation of the association of pregnancy weight gain on maternal and neonatal gut microbiota composition and abundance using 16sRNA sequencing
BMC Pregnancy and Childbirth (2023)
-
Effects of paternal high-fat diet and maternal rearing environment on the gut microbiota and behavior
Scientific Reports (2022)
-
The effects of perineal disinfection on infant’s oral microflora after transvaginal examination during delivery
BMC Pregnancy and Childbirth (2019)
-
Does cesarean delivery impact infant weight gain and adiposity over the first year of life?
International Journal of Obesity (2019)
-
Microbiota signatures relating to reduced memory and exploratory behaviour in the offspring of overweight mothers in a murine model
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
