
Abstract
Climate change and slow yield gains pose a major threat to global wheat production. Underutilized genetic resources including landraces and wild relatives are key elements for developing high-yielding and climate-resilient wheat varieties. Landraces introduced into Mexico from Europe, also known as Creole wheats, are adapted to a wide range of climatic regimes and represent a unique genetic resource. Eight thousand four hundred and sixteen wheat landraces representing all dimensions of Mexico were characterized through genotyping-by-sequencing technology. Results revealed sub-groups adapted to specific environments of Mexico. Broadly, accessions from north and south of Mexico showed considerable genetic differentiation. However, a large percentage of landrace accessions were genetically very close, although belonged to different regions most likely due to the recent (nearly five centuries before) introduction of wheat in Mexico. Some of the groups adapted to extreme environments and accumulated high number of rare alleles. Core reference sets were assembled simultaneously using multiple variables, capturing 89% of the rare alleles present in the complete set. Genetic information about Mexican wheat landraces and core reference set can be effectively utilized in next generation wheat varietal improvement.
Similar content being viewed by others


Introduction
Grain yield of wheat, the world’s staple food crop, is estimated to decrease by ~6% for each °C elevation in temperature1. The earth’s average global temperature is increasing at a rate of 0.1 °C per decade2, indicating the magnitude of climatic variation. More than one-third of wheat yield variability in the global breadbasket is explained by climate variation3. According to an estimate, average wheat yield loss due to climate change during 1981–2002 was 19 Mt/year4. In view of these challenges, Food and Agriculture Organization launched its Climate-Smart Agriculture (FAO-CSA) initiative to ensure food security5. Furthermore, meeting the expected wheat demand to feed a human population of 9.6 billion by 20506, in the current era of climate change and slow yield gains7, would require development of high yielding and climate smart ‘next generation varieties’. A multifaceted approach is required for developing such varieties, with a key role being played by unexploited germplasm including landraces long preserved in gene banks8.
Landraces that have been selected over many generations by farmers are the repositories of crop’s genetic diversity, and their value to modern plant breeding is massive9. Non-elite natural populations such as landraces harbour rare alleles besides other high- and low-frequency alleles. Alleles present at high frequencies are most likely responsible for the wide adaptation of landraces. On the other hand, crop domestication10 and improvement11,12 is accompanied by a selective advantage of alleles present at a very low frequency in those populations. Classic examples of such rare alleles are Green Revolution genes Rht1 and sd1 in wheat and rice, respectively13. Landraces may also have rare alleles for disease resistance and tolerance to environmental stresses. The pressing need to cope with climate change and maximize yield gains can be addressed in part through obtaining a combination of both types of alleles from landraces.
Adaptation of wheat landraces in their native environments has resulted in the accumulation of favourable alleles for domestication traits14,15,16. Mexican landraces, also known as Creole wheats, were brought to the Americas from the 16th through 18th centuries and gradually became adapted to the local environments. Their genetic diversity is believed to be depleted in the germplasm collections of Spain and Europe17. Mexico has climatic diversity because of its large variety of landscapes, from tropical and temperate forests to desert areas18. Broadly dry and tropical climatic regimes prevail in the south-central and northern parts of Mexico, respectively. During the cropping season in some of the northern states (e.g., Durango), temperatures can reach up to 40 °C (Supplementary Figures 1 and 2). Landrace accessions adapted to the varying climates of Mexico should thus have useful genetic variation for stress tolerance. The large-scale introduction into breeding pipelines of the genetic diversity available in these landraces could greatly help in developing next generation varieties, leading to increased global wheat production.
Characterization of a large collection of landraces adapted to wide climatic regimes is an urgent need of the wheat breeders worldwide. An ambitious project of the International Maize and Wheat Improvement Center (CIMMYT), Seeds of Discovery19, aims to characterize all the accessions in the Wheat Germplasm Bank (~120,000 accessions) and move the unexploited variation into the breeding pipelines20. This is being followed by genotyping using the state-of-the-art technology, genotyping-by-sequencing (GBS) which provides a cost effective platform for genotyping thousands of accessions. Numerous platforms are available for assessing single nucleotide polymorphisms (SNPs) and cost per data point is least in GBS rendering it a suitable platform for large scale germplasm characterization. A total of 9811 accessions collected from different Mexican states during the 1990s are maintained in the CIMMYT wheat germplasm bank21. An in-depth and systematic characterization is required to capture the full potential of these valuable genetic resources.
An important step in utilizing gene banks is defining a manageable core reference set as a subset of the larger germplasm collection22. A manageable core reference set is the one which breeders can precisely evaluate for their trait (s) of interest. Core reference sets have been established in the past mainly based on one variable23,24,25, for example genotypic data or phenotype measures or geographical distribution. Simultaneous use of multiple types of variables (genotype, phenotype, geography etc.) while classifying a germplasm set should provide a robust diversity estimate for its application in plant breeding. However, merging different types of variables in one matrix is a challenge and researchers are often reluctant to do such analyses because of the limitations of the software tools or methods26. Till date there is no report available in wheat wherein core set is developed by using multiple variables. A strategy for utilizing both discrete and continuous variables, as well as combining hierarchical multiple-factor analysis (HMFA) and the two-stage Ward Modified Location Model (Ward-MLM), has been proposed by Franco et al.27. Gower28 proposed a method for simultaneously analyzing continuous and categorical variables by transforming each to a 0-to-1 scale, irrespective of the type of variables. This approach was followed in the present study to define a core reference set of Mexican wheat landraces.
The objectives of this study were to: (1) characterize Mexican wheat landraces conserved in the CIMMYT wheat germplasm bank and (2) develop a core reference set using multiple variables simultaneously.
Results
Of 8,416 Mexican landraces subjected to analysis, 7,986 were found to be hexaploid based on their genotypic profiles (Supplementary Figure 3), phenotypic evaluation and grain characteristics. Total number of high quality and filtered SNPs used for the study was 20,526 out of which 8,297 were present in frequency less than 0.05. All these markers showed heterozygosity in range of 0–30.5%. The percentage of GBS-based SNPs with allele frequencies ranging from 0.05 to 0.95 was 59.6. The remaining 40.4% had allele frequency <0.05 or >0.95 which enabled us to identify useful genetic variation presented in different sections onwards in this report. Means and variances of phenotypic evaluation and grain characteristics have been presented in Table 1.
Genetic classification of Mexican hexaploid wheat landraces
The principal component analysis (PCA) explicitly revealed broad separation of the northern (Durango, Chihuahua and Coahuila) hexaploid landraces from the accessions of southern Mexico (Oaxaca) and central valley (Mexico, Puebla, Tlaxcala, Queretaro, Toluca, Guanajuato, Hidalgo and Michoacán) with little overlaps (Fig. 1). Genetic classification revealed 15 groups of hexaploid landraces. Thirteen of the 15 groups included accessions from just one region: central, southern, or northern Mexico. Some of the groups had accessions from specific places in Mexico, such as Durango (Group 5), Oaxaca (Group 8), Mexico (Group 9), Coahuila (Group 11), Michoacán (Group 13), Chihuahua (Groups 6 and 14) and Guanajuato (Group 15). Group 3 had accessions from Chihuahua and Oaxaca, located in the extreme northern and southern parts of the country respectively, whereas, Group 7 had lines from the central and southern regions with maximum overlap (Fig. 2). Group 1 showed highest Shannon’s and Nei’s diversity indices followed closely by groups 4, 7, 8, 12 and 13 containing accessions from southern and/or central Mexico. In contrast, group 5 had very low diversity (Table 2). The genetic differentiation (Fst) among these 15 groups ranged from 0.041 to 0.277. Groups 3, 5, 6 and 14 were genetically the most divergent (Supplementary Table 1).

There were a total of 15 groups that correspond to different Mexican states. 1 = Yellow (MEXICO, PUEBLA), 2 = Light blue (MEXICO, QUERETARO), 3 = Dark blue (CHIHUAHUA, OAXACA), 4 = Orange (MEXICO, PUEBLA, QUERETARO, HIDALGO), 5 = Light green (DURANGO), 6 = Dark green (CHIHUAHUA 95.5), 7 = Pink (OAXACA, TLAXCALA, TOLUCA, PUEBLA), 8 = Purple (OAXACA), 9 = Turquoise (MEXICO), 10 = Brown (MEXICO, MICHOACAN), 11 = Red (COAHUILA), 12 = Gray (TLAXCALA, MEXICO, MICHOACAN), 13 = Maroon (MICHOACAN), 14 = Beige (CHIHUAHUA 95.5), 15 = Black (GUANAJUATO). The PC1, PC2 and PC3 contribute 10.5%, 8.2% and 6.9% of the total variation respectively.

1 = Yellow (MEXICO, PUEBLA), 2 = Light blue (MEXICO, QUERETARO), 3 = Dark blue (CHIHUAHUA, OAXACA), 4 = Orange (MEXICO, PUEBLA, QUERETARO, HIDALGO), 5 = Light green (DURANGO), 6 = Dark green (CHIHUAHUA), 7 = Pink (OAXACA, TLAXCALA, TOLUCA, PUEBLA), 8 = Purple (OAXACA), 9 = Turquoise (MEXICO), 10 = Brown (MEXICO, MICHOACAN), 11 = Red (COAHUILA), 12 = Gray (TLAXCALA, MEXICO, MICHOACAN), 13 = Maroon (MICHOACAN), 14 = Beige (CHIHUAHUA), 15 = Black (GUANAJUATO).Map was made using ESRI’s ArcGIS Desktop ArcMap 10.2.2 software. On the blank political map, groups were pointed manually using Microsoft power point application.
The genetic diversity analysis of above mentioned 15 groups clustered them into six, referred to as clusters 1–6 (Supplementary Figure 4). Cluster 1 was very distinct from the others and had just one group that contained lines from the northern and southern regions. Cluster 2 contained the largest number of accessions and consisted of lines only from the central region. Clusters 3 and 4 had accessions from two or more regions, while Clusters 5 and 6 had just one homogeneous group each. Accessions from the north appeared in four of six clusters and in five groups. Accessions from the central region appeared in four clusters and nine groups. Accessions from the south appeared in two clusters and just three groups, including Group 8.
Characterization of the core reference sets
A core reference set of 1,133 landrace accessions was selected from the complete population (Supplementary Table 2). The mean Gower’s distances of the core reference set and the complete set were 0.093 and 0.088, respectively, showing a genetic diversity gain of 5.5%. Frequency of 158 rare alleles (rare alleles of complete population) increased above 0.05 in the core population (Supplementary Figure 5). Marker allele frequencies (Supplementary Figure 6) and diversity indexes of complete and core sets were comparable (Table 3). Phenotypic means and variances were compared using ratio of complete set/core set (Supplementary Table 3). Range for means and variances were 0.92–1.02 and 0.81–1.04, respectively. Finally, representativeness of core reference set has been shown in multidimensional scaling (MDS) graph in Fig. 3. Overall, comparative analysis of phenotypic variances, diversity measures and allele frequencies among the complete and core reference sets shows that the core reference is a representative of the complete set. The core reference set was also subjected to evaluation for yellow rust disease which led to identify seven resistant landrace accessions with disease severity of 20% or less through screening across two locations- Punjab Agriculture University, India and Toluca, Mexico (Supplementary Table 4).

Complete and core set entries represented by pink and black dots respectively in the graph.
Characterization of rare alleles
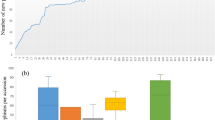
Marker alleles, with minor allele frequency (MAF) less than 0.05 were considered as rare alleles. Of 20,526 SNPs, 8,539 had frequencies ranging [0, 0.05) and 7,775 alleles had a frequency within (0, 0.05), which were considered as rare alleles (Supplementary Figure 7). The total numbers of rare alleles in the complete and core reference sets were 7,775 (18.94%) and 6,876 (16.74%), respectively. Comparative analysis of the two populations revealed that the core reference set captured 88.75% of the rare alleles of the complete set (Table 3). The average number of rare alleles per accession was estimated for each of the 15 genetic groups. The highest number of rare alleles per accession occurred in the genetic groups belonging to Guanajuato (Group 12) followed by Durango (Group 5), Chihuahua (Group 14), Oaxaca (Group 8) and Michoacán (Group 13), as shown in Table 4 and Fig. 4. Interestingly, a very high percentage of unique rare alleles were observed in the landrace accessions of Michoacán (Group 13).

Unique rare allele ratio = Number of unique rare allele in a group/total number of rare alleles in the group. Calculation of the average temperature and rainfall was made using climate data of collection site of each accession. Precipitation and temperature data of 50 years (i.e. from 1951 to 2000) was used for estimating the average. The 15 genetic groups have been plotted on ‘X’ axis. Precipitation (ppt), maximum (tmax) and minimum (tmn) and unique rare allele have been presented on ‘Y’ axis. For precipitation data a correction factor of 0.1 was used (e.g 769 mm = 76.9 mm). Units for precipitation and temperature were Millimeter and Fahrenheit.
The rare allele analysis carried out for identifying fixed marker alleles (not segregating) revealed genomic regions dispersed throughout the twenty one chromosomes (Supplementary Figure 8). Maximum fixed alleles were found in accessions from Chihuahua followed by the ones the central valley. List of the marker alleles is presented in Supplementary Table 5. Clustering based on longitude, latitude and altitude, grouped Chihuahua accessions in to one which also corresponding genetic groups 6 and 14 (Supplementary Table 6).
Discussion
The Green Revolution has been associated with the replacement of traditional varieties and landraces by high-yielding, input-responsive, semi-dwarf varieties of cereal crops29,30, which significantly contributed to global food security. However, this also led to increased monoculture and the depletion of on-farm varietal diversity, which today poses a serious threat to food production under climate change scenarios. Reinforcement of the genetic variation from underutilized landraces into modern varieties should provide impetus to achieve the targets of proposed climate smart agriculture5. Efforts made in the present study are an example of large-scale characterization of germplasm that remained unexploited in gene banks. At the global level, similar approaches for mobilizing gene bank and on-farm diversity to breeding pipelines will make it possible to accomplish the goals of FAO-CSA initiative.
Mexican wheat landraces have been used by researchers in breeding for abiotic stress tolerance and grain quality improvement. Small sets were formed based on the available information (e.g. collection site) and therefore only 25% of entire Mexican landrace collection was utilized31,32,33. Three-fourths of the Mexican landrace collection remained uncharacterized. The present study provides a thorough genetic understanding of Mexican wheat landraces for researchers intending to utilize them. PCA, genetic classification and cluster analysis showed a broad differentiation of landraces belonging to northern and southern Mexico while some of the groups, being adapted to specific regions (Figs 1 and 2). They therefore have more potential for adaptation to a wide range of environments compared to the landraces that are adapted to specific ecosystems. Contrarily, some other landrace groups showed different patterns, for example, regional groups from Oaxaca, Guanajuato, Michoacán, Durango, Coahuila and Chihuahua (Fig. 2). For example, group 3 has accessions from extreme north and south Mexico but group 8 represents only Oaxaca, the southern province but diversity index of group 8 was significantly higher than that of group 3. Above mentioned distinct groups belonging to specific regions showed presence of relatively higher number of rare alleles per accession as compared to other groups (Table 4). Possible reasons for their adaptation could be either their early introduction or development of strong environmental imprints in them. One of the genetic groups representing a specific collection site in Mexico (Michoacán) showed an exceptionally high frequency of unique rare alleles (Fig. 4). Temperature and rainfall were at the higher end of the scale for this site, representing a unique climatic regime. The unique allelic diversity of the Michoacán landraces (Group 13) may be associated with their adaptation to this peculiar climate. Landraces from Durango (Group 5), a region characterized by very high annual average temperatures and low precipitation, had a very high number of rare alleles per accession. Combinations of different rare alleles may be the contributing factor to the adaptation of landraces to dry climates where heat stress and droughts are frequent. An intensive analysis of these two groups (Groups 5 and 13) could unveil the relevance of allelic diversity to climatic adaptation. Additionally, landrace accessions adapted to specific environments could be utilized efficiently in developing varieties for similar target ecosystems. Sources for heat stress tolerance as well as other stresses have been identified from these landraces and being used through large scale pre-breeding efforts (Sukhwinder Singh, CIMMYT, Unpublished). Nevertheless, Mexico is not the center of origin for wheat; landraces are adapted to a wide range of the climatic regimes which render them fit candidates for climate resilient wheat genetic improvement. Further, comparison of their diversity profile with landraces from the Fertile Crescent and Southern Europe will generate useful information regarding their evolutionary history and practical deployment in climate resilience breeding.
Another interesting pattern was observed in landraces from Chihuahua which is situated in extreme north part of the country. Maximum number of accessions with fixed marker alleles was found in Chihuahua followed by Central Valley (Supplementary Figure 8). Gene flow analysis further indicated towards their divergence; however, a definitive test would confirm this (Supplementary Figure 9). There might be two different possibilities, either wheat landraces from different sources would have been introduced to Chihuahua and Central Valley or genomic regions harboring such alleles could have undergone natural selection. Since wheat is introduced almost 400–500 years ago into Mexico by Spanish, the former possibility seems more likely. However, an in-depth analysis of phylogenetic history would confirm about ‘introduction’ and ‘selection imprint’. Adaptation of genetic groups to diverse climatic regions of Mexico, the diversity pattern of these genetic groups (reflected in their diversity indices) and the distribution of rare alleles have provided first-hand information that will allow wheat researchers worldwide to efficiently utilize Mexican landraces in breeding pipelines.
The systematic utilization of the Mexican landrace diversity requires a manageable representative germplasm set. Natural, non-elite landrace populations are known to harbor agronomically important alleles in very low frequencies13. Therefore, a core reference set of natural populations with maximized rare alleles will likely unveil genetic variation useful for crop improvement. In the past, wheat core reference sets have been developed using one variable at a time23,24, resulting in significant reduction of rare allele diversity24. In this study, a unique core reference set development strategy was followed, i.e., using an integrated data matrix of both continuous and discrete variables (phenotypic and genotypic), thereby maximizing overall diversity for analysis. In this strategy, we reduced the dimensions of the marker data to 2,000 principal components explaining 84% of the total variance and then merged them to 23 phenotypic variables to form a data matrix in such a way that genotypic and phenotypic contributions were 75% and 25%, respectively. This arbitrary ratio was chosen for enhancing the role of phenotypic variation to core development. Detailed methodology has been explained in ‘materials and methods’ section. Wingen et al.24 concluded that the core reference set strategy is not useful for discovering very rare alleles, perhaps due to the sample size or low-throughput marker platform. We report a significant enrichment of rare alleles in the core reference set. The percentage of rare alleles in the original landrace population was 18.94%, whereas it was 16.74% in the core reference set, suggesting an 88.75% rare allele recovery in the latter. Furthermore, frequencies of 158 rare alleles (in the complete set) rose above 0.05 in the core reference set (Supplementary Figure 5). This enrichment of rare alleles in the study was due to the base population (7,986 genotypes), high-throughput SNP data ( >20,000 markers), coupled with a unique core reference set development strategy, which simultaneously uses multiple variables. Minor allele frequency (MAF), geographical representation and phenotypic variance of the core reference set were comparable to those of the complete set (Supplementary Figure 6, Tables 3 and Supplementary Table 3).
Characterization of the core reference set enriched with rare alleles is a valuable germplasm resource for trait dissection and gene discovery. This representative population can be efficiently utilized by wheat researchers globally. Core reference set has already been distributed to researchers in Africa, South Asia, and the USA. Its evaluation in two geographically divergent environments, India and Mexico for yellow rust disease has identified resistant genotypes (Supplementary Table 4). Information presented in this study about Mexican wheat landraces will serve as a high value resource base for wheat breeders world-wide to develop high-yielding and climate-resilient next-generation wheat varieties.
Online Methods
Description of plant material
Wheat landraces used in this study were collected from Mexico. A total of 9,811 Mexican wheat landraces were collected from 16 Mexican states as part of a project sponsored by Mexico’s National Commission for the Study and Use of Biodiversity21. After eliminating duplicate accessions based on phenotypic information, a total of 8,416 Mexican wheat landraces, including hexaploid and diploid accessions as well as other species, were used in the analysis. A total of 7986 hexaploid landraces were used for genetic analysis and their details have been provided in Supplementary Table 7.
Genotypic analysis of Mexican wheat landraces
Seed of a single plant from each accession was used for DNA extraction and seed of the same plant was used for the phenotypic evaluation. Extraction of genomic DNA was carried out by a modified CTAB (cetyltrimethylammonium bromide) method34, followed by quantification using NanoDrop 8000 spectrophotometer V 2.1.0. Genotyping was performed through DArT-seq GBS technology (called DArTseq™) at DArT Pyt Ltd, Canberra, Australia35. This method follows two-step complexity reductions. In this approach, two enzymes, PstI_ad/TaqI/HpaII and PstI_ad/TaqI/HhaI_ad, along-with TaqI restriction enzyme were used to eliminate subsets of PstI -HpaII and PstI-HhaI fragments, respectively. To encode the DNA samples in plates which were ligated into small restriction fragments, PstI-specific adapters were tagged with 96 different barcodes. These PstI adapters were characterized by a sequencing primer and tags generated after sequencing were read with the help of PstI sites. Restriction products were amplified, quality was checked and then all 96 samples in a plate were pooled. Pooled DNA was run in a single lane on an Illumina Hiseq 2000 instrument for sequencing. To obtain the DArT score tables and SNP tables, a proprietary analytical pipeline developed by DArT P/L was used.
Marker data were filtered based on reproducibility, call rate and the average read depth using the pipeline. Reproducibility was determined by assaying approximately 60% of samples twice. The minimum threshold values for completeness, reproducibility and call rate were kept at 50%, 95% and 85%, respectively. The average read depth was 7. Variants were called within the data by clustering sequences by sequence similarity, thereby avoiding the use of an external reference genome. In this approach, either the most common sequence in the population or a wheat sequence previously recorded by DArT genotyping protocol was considered as the reference. This approach of recalling GBS samples has been used in recent wheat studies with the DArT-seq markers20.
A total of 20526 SNPs were recalled from the raw GBS data (Supplementary Data File). These markers had varying numbers of missing scores (Supplementary Figure 10). Of these, 20039 markers with less than 50% missing scores were used in the analysis. Stringency was kept to this level to (1) include a high number of minor/rare alleles in the analysis, and (2) minimize the risk of under-representing a genomic region because the chromosome locations of all GBS tags were not known. Missing scores could represent biological data points (presence and absence variations-PAV). A total of 20039 SNP markers were used in the diversity analysis, in core reference set development and in marker-trait association analysis. For determining the genomic position of markers, two different consensus genetic linkage maps were referred (20, DArT, Australia, unpublished). In order to estimate diversity indices and genetic differentiation (Fst values), we select 301 markers as equally spaced as possible in the chromosome (Supplementary Figure 11). Selection was performed following the next steps: (1) Calculate the percentage of markers per chromosome and determine the number of markers of 301 to preserve the representation of the chromosomes; (2) Calculate the distance between the first and the last marker in each chromosome and divide this amount by the number of markers obtained in 1; (3) Starting from the first marker, calculate the points in the chromosome to have equally spaced markers; and (4) Identify the markers as closest to each point. Those markers were selected in the sample. This analysis was performed using a custom code in SAS v9.4. The set of 301 markers was used. Map in Fig. 2 was made using ESRI’s ArcGIS Desktop ArcMap 10.2.2 software36. The dataset used to make the maps on rainfall and temperature was Worldclim 1.437,38. 30 s resolution (ca 1 km) long term (1950–2000) monthly average grids for rainfall and minimum, maximum temperature were used to generate annual average grids for rainfall and average temp and mapped subsequently. Geo-referenced locations of origin of land races were converted to vector data and integrated in the maps.
Phenotypic analysis of Mexican wheat landraces
Phenotypic characterization of all 8,416 Mexican wheat landraces was carried out in three different environments: well irrigated, drought stress and heat stress. Trials were conducted at the CIMMYT Experiment Station near Ciudad Obregon, Mexico (27 20° N, 109 54° W, 38 m ASL) during the 2011–2012 crop season. The well-irrigated and drought trials were conducted during the spring wheat season (November 2011–April 2012). Trials under heat stress were planted in April 2012 so they were exposed to high temperature stress at anthesis. Each trial was conducted as an augmented design with 0.3 m2 plot size; check varieties ‘Vorobey’ and ‘Baj’ were repeated multiple times in the experiment. Appropriate measures were used to fertilize and control weeds, diseases, and pests. In the well-irrigated trial, the plots were watered so that approximately 600 mm of water were applied during the complete wheat cycle. Irrigation was provided to plots whenever approximately 50% of available soil moisture was depleted according to gravimetric scales. Approximately 200 mm of total soil moisture was available during the growing season for the drought treatment. The heat stress treatment was watered the same as the irrigated treatment to avoid the confounding effect of drought.
Seeds for the grain quality analysis were obtained from the irrigated treatment; grain morphological characteristics were evaluated with the digital image system SeedCount SC5000 (Next Instruments, Australia) and thousand-kernel weight (TKW, g), test weight (TW, kg/hl), average grain length and width (mm), as well as percentage of grains affected by yellow berry (%) were determined. Grain size distribution was measured using sieves of 2.8 mm (Screen 1), 2.5 mm (Screen 2) and 2.2 mm (Screen 3). Near-infrared spectroscopy (NIRS, Antaris II FT-Analyzer, Thermo Scientific, USA) was used to determine grain hardness (GH, %) and grain protein content (GP, %). The near-infrared spectroscopy instrument was calibrated based on AACC methods39 for particle size index (AACC Method 55–30) and protein (AACC Method 46–11A). Grain protein was adjusted to 12.5% moisture content. Whole-meal flour samples were obtained using a UDY Cyclone mill (0.5 mm sieve). Only one gram of whole-meal flour was used to perform the SDS-sedimentation (SDS, ml) test40.
Phenotypic screening of Mexican core reference set for yellow rust
Evaluation of the core reference sets for yellow rust was performed under field conditions at CIMMYT’s Experiment Station in Toluca, Mexico (May 2014 to October 2014) and at Punjab Agriculture University, India (October 2014 to March 2015). Accessions were planted in a randomized complete block design with two replicates, in a plot consisting of two one-meter rows. Approximately 60–70 seeds were sown in each plot. Yellow rust susceptible variety Avocet was planted around the whole experimental block. Inoculation of spreader/border rows was done according to the method explained by Hao et al.41. Disease assessment was performed when the susceptible check variety Avocet showed 100% yellow rust severity (during the mid-dough stage of plant growth). Assessment of percent disease severity was performed according to the modified Cobbs Scale42.
Cluster and diversity analysis of Mexican landrace accessions
A three-step approach was followed for classifying Mexican wheat landraces. First, we performed principal component analysis (PCA) using only genetic data from all the accessions, including hexaploid, tetraploid and other species. Next, we conducted a genetic diversity analysis of just the hexaploid landraces to define their characteristics with respect to their geographic collection sites. Finally, we used classification to develop a core reference set of hexaploid accessions combining different types of variables (genotypic and phenotypic) using the Hierarchical Multiple Factor Analysis43,44; and Gower’s distance was used as a measure of genotypic and phenotypic diversity28.
Principal component analysis of Mexican landrace accessions
Principal component analysis was done with the GBS data using the “princomp” function of R-project version 3.1.145.
Cluster and diversity analysis of Mexican hexaploid wheat landrace accessions
Genetic distances were calculated using SAS PROC DISTANCE and cluster analysis was done with the SAS PROC CLUSTER procedures of SAS v9.446. Classification of landrace accessions was done using hierarchical multiple-factor analysis (HMFA) in a step-wise manner, and in every step, one group was split into two groups. We identified final working groups that allowed representation of heterogeneity between individuals and as homogenous as possible considering geographical distribution. A simple matching coefficient transformed into squared Euclidean distance was used to measure the similarity between landraces. Formula used for Euclidean distances was:

DEU = Euclidean distance; u = individual landraces and Xu and Yu are different individuals. The genetic distance between two landraces x and y, denoted as d(x, y), was calculated by one minus the ratio between the number of matches (M) and the total number of non-missing pairs (N), that is,

A complete algorithm (used for clustering) was used to estimate the distance between groups based on the greatest distance between any two individuals in different groups. The distance between groups a and b, denoted as d(a, b), was estimated as the maximum distance among all pairs of individuals in different groups:

where xai represents the individual i in group a, and xbj represents the individual j in group b. Allele frequencies were determined with Tassel version 547. Nei’s and Shannon’s distances were calculated to estimate the extent of diversity within the entire population, within each group, and within the core reference set based on Euclidian distances. For Nei’s index, the following formula was used,

Shannon’s index was calculated as,

where Pi is the frequency of the major allele of the ith marker and t is the number of markers.
For estimation of gene flow, the coefficient of gene differentiation (GST) was calculated as:

Where HT is the total genetic diversity and HS is the intra-population genetic diversity, both determined through Nei’s genetic diversity statistics48. Gene flow (Nm) was estimated as:

For population differentiation, Wright’s Fst values49 were calculated using Genepop software50. We also performed correlation analysis between sample size and diversity indices to see the effect of sample size on diversity statistics and concluded that in our study sample sizes do not significantly affect the diversity indices (Supplementary Figure 12).
Rare allele characterization in different populations
Rare alleles were defined for different purposes with respect to populations and presented in qualitative ways i.e. (1) rare allele per unit accession, (2) unique rare alleles and (3) fixed rare alleles in accessions belonging to specific regions.
-
1
For comparing allele richness in core and complete set complete set of Mexican landrace (7986 individuals) was considered as a population and rare alleles were determined with respect to this population. Richness of these ‘rare alleles’ were then observed in the core set population comprising 1133 individuals (Table 3, Supplementary Figure 5).
-
2
Secondly, rare alleles were estimated in each of the genetic groups. Each group was considered as a population and rare alleles were defined based on the respective population. These genetic groups were defined based on genetic similarity matrix. Further, in order to explain rare alleles in a more un-biased way we have estimated the, “Unique rare alleles” which is a qualitative measure-not affected by size of the groups (Fig. 4).
-
3
Further, to identify alleles fixed in groups (groups based on genetic similarity matrix or geology) we first selected list of markers with at least 60% data points showing allele frequency of less than 0.05. Clusters were formed using latitude, longitude and altitude informations with the k-means method. Euclidean distance and least square method were followed for this analysis. A table with cross classification between cluster based on geographical data and 15 genetic groups was created (Supplementary Table 6). Allele frequency was analyzed in environmental clusters to identify the alleles fixed in particular locations. Markers with fixed allele in particular locations (Chihuahua and central valley) were then mapped to the wheat genome using Jim Kent’s blast-like alignment tool (BLAT)51. The version 2.26 of the International Wheat Genome Sequencing Consortium was downloaded from Gramene website52. All the markers where aligned to each of the chromosomes and filtered using the highest match percentage. Markers showing identity of 95% and above were used for presentation. Based on this information genomic regions have been determined and presented in Supplementary Figure 8.
Simulation of optimum population size for core set selection
Optimum size of the core reference set was determined through Monte Carlo simulations by 1000 replications. Twenty levels (i.e., 5–100% with step size 5%) of population size were randomly selected from the whole population. For each level of sub-population size, the genetic variance was calculated by the following equations:


Thus

where fST is the statistic to test for allele frequency differences among populations; p and q are allele frequencies satisfying p + q = 1; var (p) is the variance of allele frequency p; n is the number of populations, which is equal to the number of simulations in this study;  and
and  are the mean allele frequencies of p and q, respectively, across simulation runs, and
are the mean allele frequencies of p and q, respectively, across simulation runs, and  is the genetic variance53. A core reference set that contained approximately 15% of the complete set could reach the highest genetic variance among the twenty sub-populations (Supplementary Figure 13). Therefore, 1,133 landrace accessions (14.18%) out of 7,986 accessions in the complete population were selected as the core set.
is the genetic variance53. A core reference set that contained approximately 15% of the complete set could reach the highest genetic variance among the twenty sub-populations (Supplementary Figure 13). Therefore, 1,133 landrace accessions (14.18%) out of 7,986 accessions in the complete population were selected as the core set.
Classification of Mexican landraces for core reference set selection
To select the core reference sets, Mexican landraces were classified using the data matrix containing genotypic and phenotypic variables. We first used PCA by the marker data to reduce the dimensions of the marker data to 2,000 principal components (from 20,039) that explained 84% of the total variance. Thereafter the scores of the 2,000 PCA components and the 23 phenotypic variables were merged to form a data matrix that was then used in an HMFA43,44. Six principal axes (PA) were selected from HMFA in such a way that genotypic and phenotypic contributions to the total variance explained by the six axes were 75% and 25%, respectively. Using this data matrix we estimated the contributions on genotype and phenotype for principal axes (PA) of 1 to 25. The first dimension (or principal axis) explained phenotype: genotype variability ratio of 50:50% whereas, 25th dimension correspond to 8:92% ratio. Six PA explained 25% and 75% phenotype and genotype variability respectively. Similarly, another landrace data set (2403 Iranian landraces) was analyzed which also suggested that 6 PA explain 30% and 70% phenotype and genotype variation respectively (CIMMYT, Unpublished data). Total number of PC dimensions representing 20,526 markers was 2000, whereas only 23 phenotype variables were present and to provide weight to phenotype 25% phenotype contribution was decided. Therefore, the 25:75 ratio (phenotype: Genotype) was not exactly an arbitrary criterion but based on two data sets of two independent genetic populations (Mexican and Iranian wheat landraces). For the HMFA analysis, the FactoMineR library was used in R software54. The ratio of genotypic and phenotypic variables was more than a thousand to one, so increasing the phenotype representation ratio of 75:25 was chosen to achieve some equilibrium. The coordinates for each accession on each PA were then used to group accessions following the mixture of normal distributions methodology and the optimum number of groups was selected by maximum likelihood27. A total of 21 groups were defined. A predefined number of accessions were selected from each group. The number of accessions per group was estimated by the D-method, proportional to group diversity55. Following the stratified random sampling method, 1000 candidate reference sets were created and their diversity was estimated using the average Gower’s distance28; the subset showing the maximum average Gower’s distance was selected to be the core reference set. Analyses were performed using different libraries from the open source software R45. To visualize whether the core reference set is representative of the complete set, we used a graphical multidimensional scaling (MDS) method. Secondly, we determined the variance of the phenotypic traits in both populations.
Additional Information
How to cite this article: Vikram, P. et al. Unlocking the genetic diversity of Creole wheats. Sci. Rep. 6, 23092; doi: 10.1038/srep23092 (2016).
Change history
20 May 2016
A correction has been published and is appended to both the HTML and PDF versions of this paper. The error has been fixed in the paper.
References
Asseng, S. et al. Rising temperatures reduce global wheat production Nature Climate Change 5, 143–147 (2015).
IPCC 2007: IPCC Fourth Assessment Report: Climate Change 2007 (AR4), Working Group I Report “The Physical Science Basis”. Available at: https://www.ipcc.ch/publications_and_data/ar4/wg1/en/spmsspm-projections-of.html (Accessed: 15th July 2015).
Ray, D. K., Gerber, J. S., MacDonald, G. K. & West, P. C. Climate variation explains a third of global crop yield variability. Nature Commun 6, 1–9 (2015).
Lobell, D. B. & Field, C. B. Global scale climate-crop yield relationships and the impacts of recent warming. Environ Res Lett 2, 014002 (2007).
FAO-CSA Sourcebook, available on FAO’s (2013). website: http://www.fao.org/climatechange/37491-0c425f2caa2f5e6f3b9162d39c8507fa3.pdf (Accessed: 15th July 2015).
United Nations, Department of economic and social afairs., World population prospects: The 2012 revision, Technical Report. (2013) Available at: http://esa.un.org/unpd/wpp/publications/Files/WPP2012_HIGHLIGHTS.pdf (Accessed: 14th February 2016).
Ray, D. K., Mueller, N. D., West, P. C. & Foley, J. A. Yield trends are insufficient to double global crop production by 2050. PLoS ONE 8(6), e66428 (2013).
FAO. Coping with climate change–the roles of genetic resources for food and agriculture. Rome, available at http://www.fao.org/3/a-i3866e.pdf (2015). (Accessed: 15th July 2015).
Lopes, M. S. et al. Exploiting genetic diversity from landraces in wheat breeding for adaptation to climate change. J Exp Bot 66, 3477–3486 (2015).
Xu, Y. B. Molecular Plant Breeding (CABI, 2010).
Gale, M. D. & Marshall, G. A. Chromosomal location of Gai-1 and Rht-1, genes for gibberellin insensitivity and semi-dwarfism, in a derivative of Norin-10 wheat. Heredity 37, 283–289 (1976).
Murai, M., Takamure, I., Sato, S., Tokutome, T. & Sato, Y. Effects of the dwarfing gene originating from Dee-geo-woo-gen on yield and its related traits in rice. Breed Sci 52, 95–100 (2002).
Lyu, J. et al. Analysis of elite variety tag SNPs reveals an important allele in upland rice. Nat Commun 4, 2138 (2013).
Fuller, D. Q. Contrasting patterns in crop domestication and domestication rates: Recent archaeobotanical insights from the Old World. Ann Bot 100, 903–924 (2007).
Charmet, G. Wheat domestication: Lessons for the future. Comptes Rendus Biologies 334, 212–220 (2011).
Peng, J. H., Sun, D. & Nevo, E. Domestication evolution, genetics and genomics in wheat. Mol Breed 28, 281–301 (2011).
Guzman, C., Ortega, R., Yamamori, M., Peña, R. & Alvarez, J. Molecular characterization of two novel null waxy alleles in Mexican bread wheat landraces. J Cereal Sci 62, 8e14 (2015).
Magaña, V. Evaluación de escenarios regionales de clima actual y de cambio climático futuro para México. Retrieved 04 13, 2015, from http://www.atmosfera.unam.mx/cclimat/publicaciones/cambio_climatico/escena.pdf (2012). (Accessed: 15th July 2015).
“Seeds of discovery”, CIMMYT, Mexico: http://seedsofdiscovery.org/en/. (Accessed: 15th July 2015).
Li, H. et al. A high density GBS map of bread wheat and its application for dissecting complex disease resistance traits. BMC Genomics 16, 216 (2015).
Skovmand, B., Fox, P. N., Varughese, G. & Gonzalez-de-Leon, D. International activities in wheat germplasm: CIMMYT’s perspective. In: Duncan, R.R. (ed.). International Germplasm Transfer: Past and Present, pp. 135–148, CSSA Special Publication Number 23. American Society of Agronomy, Inc., Soil Science Society of America, Inc., Madison, Wisconsin, USA (1995).CONABIO report available at CIMMYTs data repository, www.cimmyt.org. (Accessed: 15th July 2015).
Brown, A. H. D. Core collections: a practical approach to genetic resources management. Genome 31, 818–824 (1989).
Balfourier, F., Roussel, V., Strelchenko, P., Exbrayat-Vinson, F. & Sourdille-Boutet, G. A. Worldwide bread wheat core collection arrayed in a 384-well plate. Theor Appl Genet 114, 1265–1275 (2007).
Wingen, L. U. et al. Establishing the A.E. Watkins landrace cultivar collection as a resource for systematic gene discovery in wheat. Theor Appl Genet 127(8), 1831–42 (2014).
Martynov, S. P. et al. Genealogical approach to the formation of the winter wheat core collection. Genetika 39, 1091–1098 (2003).
Vieira, E. A., Carvalho, F. I. F., Bertan, I., Kopp, M. M., Zimmer, P., Benin, G. et al. Association between genetic distances in wheat (Triticum aestivum L.) as estimated by AFLP and morphological markers. Genet Mol Biol 30, 392–399 (2007).
Franco, J., Crossa, J. & Desphande, S. Hierarchical Multiple-Factor Analysis for classifying genotypes based on phenotypic and genetic data. Crop Sci 50, 105–117 (2010).
Gower, J. C. A general coefficient of similarity and some of its properties. Biometrics 27, 857–874 (1971).
Frei, M. & Becker, K. Agro-biodiversity in subsistence-oriented farming systems in a Philippine upland region: nutritional considerations. Biodivers Conserv 13, 1591–1610 (2004).
Vikram, P. et al. Drought susceptibility of the modern rice varieties: an effect of linkage of drought tolerance with undesirable traits. Nat Sci Rep 5, 14799 (2015)
Hede, A. R. et al. Evaluating genetic diversity for heat tolerance traits in Mexican wheat landraces. Genet Resour Crop Ev 46, 37–45 (1999).
DeLacy, I. H., Skovmand, B. & Huerta, J. Characterization of Mexican wheat landraces using agronomically useful attributes. Genet Resour Crop Ev 47, 591–602 (2000).
Reynolds, M., Dreccer, F. & Trethowan, R. Drought-adaptive traits derived from wheat wild relatives and landraces. J Exp Bot 58, 177–186 (2007).
Hoisington, D., Khairallah, M. & Gonzalez-de-Leon, D. Laboratory protocols, CIMMYT Applied Molecular Genetics Laboratory, 2nd ed. CIMMYT, Mexico, DF (1994).
Diversity Arrays Technology, Canberra, Australia: http://www.diversityarrays.com/. (Accessed: 15th July 2015).
ESRI (2015). ArcGIS Desktop Help 10.2.2. URL: http://resources.arcgis.com/en/help/.
Hijmans, R. J., Cameron, S. E., Parra, J. L., Jones, P. G. & Jarvis, A. Very high resolution interpolated climate surfaces for global land areas. Int J Climatol 25, 1965–1978 (2005).
Hijmans, R. J., Cameron, S. E., Parra, J. L., Jones, P. G. & Jarvis, A. Worldclim Version 1.4 (release 3). 30s Monthly temperature and rainfall grids (2014). URL: www.worldclim.org. (Accessed: 7th March 2015).
AACC International. Approved methods of the American Association of Cereal Chemists, 10th edition. The Association, St. Paul, MN (2000).
Peña, R. J., Amaya, A., Rajaram, S. & Mujeeb-Kazi, A. Variation in quality characteristics associated with some spring 1B/1R translocation wheats. J Cereal Sci 12, 105–112 (1990).
Hao, Y. et al. Characterization of a major QTL for adult plant resistance to stripe rust in US soft red winter wheat. Theor Appl Genet 123, 1401–1411 (2011).
Peterson, R. F., Campbell, A. B. & Hannah, A. E. A diagrammatic scale for rust intensity on leaves and stems of cereals. Can J Res 26, 496–500 (1948).
Escoffier, B. & Pages, J. Analyses factorielles simpleset multiples. 3rd edition. Paris: Dunod (1998).
Le-Dien, S. & Pagès, J. Hierarchical Multiple Factor Analysis: application to the comparison of sensory profiles. Food Qual Prefer 14, 397–403 (2003).
R Core Team (2013). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL http://www.R-project.org/.
SAS® 9.4 (2013). Cary, NC: SAS Institute Inc.
Bradbury, P. J. et al. TASSEL: Software for association mapping of complex traits in diverse samples. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635 (2007).
Nei, M. Analysis of genetic diversity in subdivided populations. Proc Natl Acad Sci USA 70, 3321–3323 (1973).
Wright, S. The genetical structure of populations. Ann Eugen 15, 323–354 (1951).
Raymond, M. & Rousset, F. GENEPOP Version 1.2: population genetics software for exact tests and ecumenicism. J Hered 86, 248–249 (1995).
Kent, W. J. BLAT—The BLAST-Like Alignment Tool. Genome Res 12(4), 656–664 (2002). URL: http://doi.org/10.1101/gr.229202.
Monaco, N. K. et al. Gramene 2013: comparative plant genomics resources. Nucleic Acids Res 42(D1), D1193–D1199 (2014).
Weir, B. S. Genetic data analysis. Methods for discrete population genetic data[M]. Sinauer Associates, Inc. Publishers (1990).
Josse, L. S. J. & Husson, F. FactoMineR: An R package for multivariate analysis. J Stat Soft 25, 1–18 (2008).
Franco, J., Crossa, J., Taba, S. & Shands, H. A. Sampling strategy for conserving genetic diversity when forming core subsets. Crop Sci 45, 1035–1044 (2005).
Acknowledgements
The authors duly acknowledge the financial support received from Mexico’s Secretariat of Agriculture, Livestock, Rural Development, Fisheries and Food (SAGARPA) through the Seeds of Discovery-Sustainable Modernization of Traditional Agriculture project (Mas-Agro). We acknowledge Diversity Array Technology (DArT), Canberra, Australia, for the genotyping service provided. Authors extend sincere thanks to Drs Kanwarpal Singh Dhugga, Julio Huerta-Espino, Ky Mathews, Dave Marshall. The direct and indirect support of research technicians is duly acknowledged. We extend our special thanks to Seeds of Discovery project leader and Director of the Genetic Resources Program, Dr. Kevin Pixley, for his valuable support and encouragement to the team.
Author information
Authors and Affiliations
Contributions
S.S. and P.V. conceived and designed the experiments; J.F., J.B., H.L., P.V., D.S. and C.S. performed the diversity, statistical and association analyses; C.S.P., M.R. and M.T. carried out large scale evaluation for heat-drought; C.G. evaluated landraces for grain quality; S.S., C.O., C.P.S. M.E and P.W. contributed to Genotyping; K.S. performed climate data analysis and prepared maps; G.F.D., P.S., A.S., N.S.B., G.P.S., P.V. and S.S. screened core set for diseases; T.P. provided seed material from gene bank; J.C. and S.S. were in-charge to oversee the data collection and analyses; P.V. and S.S. wrote the manuscript and other authors contributed later on; and all authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Vikram, P., Franco, J., Burgueño-Ferreira, J. et al. Unlocking the genetic diversity of Creole wheats. Sci Rep 6, 23092 (2016). https://doi.org/10.1038/srep23092
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep23092
This article is cited by
-
Characterization of Mexican wheat landraces for drought and salt stress tolerance potential for future breeding
Cereal Research Communications (2023)
-
Genome-wide association mapping in elite winter wheat breeding for yield improvement
Journal of Applied Genetics (2023)
-
Genetic mapping of adult-plant resistance genes to powdery mildew in triticale
Journal of Applied Genetics (2022)
-
K-seq, an affordable, reliable, and open Klenow NGS-based genotyping technology
Plant Methods (2021)
-
Weed competitive ability in wheat: a peek through in its functional significance, present status and future prospects
Physiology and Molecular Biology of Plants (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
