
Abstract
Two-component signal transduction systems (TCSs) are widely used by bacteria to adapt to the environment. In the present study, StoS (stress tolerance-related oxygen sensor) and SreKRS (salt response kinase, regulator, and sensor) were found to positively regulate extracellular polysaccharide (EPS) production and swarming in the rice pathogen Xanthomonas oryzae pv. oryzae (Xoo). Surprisingly, the absence of stoS or sreKRS did not attenuate virulence. To better understand the intrinsic functions of StoS and SreKRS, quantitative proteomics isobaric tags for relative and absolute quantitation (iTRAQ) was employed. Consistent with stoS and sreK mutants exhibiting a similar phenotype, the signalling circuits of StoS and SreKRS overlapped. Carbohydrate metabolism proteins and chemotaxis proteins, which could be responsible for EPS and swarming regulation, respectively, were reprogrammed in stoS and sreK mutants. Moreover, StoS and SreKRS demonstrated moderate expression of the major virulence factor, hypersensitive response and pathogenicity (Hrp) proteins through the HrpG-HrpX circuit. Most importantly, Xoo equipped with StoS and SreKRS outcompetes strains without StoS or SreKRS in co-infected rice and grows outside the host. Therefore, we propose that StoS and SreKRS adopt a novel strategy involving the moderation of Hrp protein expression and the promotion of EPS and motility to adapt to the environment.
Similar content being viewed by others


Introduction
The genus Xanthomonas comprises an important ubiquitous group of Gram-negative plant pathogenic bacteria belonging to the Gamma subdivision of Proteobacteria. Members of the genus Xanthomonas infect approximately 124 monocotyledonous and 268 dicotyledonous plants1. Bacterial blight resulting from Xanthomonas oryzae pv. oryzae (Xoo) infection is one of the most destructive rice diseases in tropical Asian countries, to which high-yielding rice cultivars are often highly susceptible. This bacteria has the potential to reduce rice yields by 20–30%, and the reduction can be as high as 50% in some areas2,3.
To survive and reproduce in the environment, bacteria require high fitness. For optimal fitness, bacteria must sense environmental changes; therefore, these microbes harbour an array of systems linking environmental stimuli to gene expression changes4. The two-component signal transduction system (TCS) is a predominant approach by which bacteria sense and adapt to changing intracellular and extracellular environments5. The prototypical TCS comprises a histidine kinase (HK) sensor protein, containing a cytoplasmic kinase core and an extrinsic membrane sensor domain, and a response regulator (RR) protein with a conserved receiver domain6. When a sensor domain recognizes a signal, the configuration of HK changes and the ATPase of the kinase core becomes activated to obtain a phosphate group from ATP7. The high-energy phosphate group of the histidine residue in HK is subsequently transferred to the aspartate residue of its cognate RR7. The activated RR regulates the downstream gene expression of its regulatory cascade as a transcription regulator or directly interacts with the target protein, resulting in an appropriate response8. To cope with various hostile stresses, Xanthomonas is equipped with a large number of TCSs9. In most bacterial genomes, dozens to hundreds of genes encode TCS proteins. In 2008, comparative genomic analyses of TCS genes in Xanthomonas were reviewed. For the six Xanthomonas strains investigated, the total number of nucleotides from TCS genes ranged from approximately 2.38% to 3.24% of the entire chromosome10.
Numerous TCS genes are present in Xanthomonas, but only some genes have been investigated. The most characterized TCS is RpfC/RpfG, which has been implicated in the perception and signal transduction of the quorum sensing diffusible signal factor11,12. Depending on the changes in the central pool of cyclic-di-GMP, a widely conserved novel second messenger implicated in the regulation of a wide range of bacterial functions, RpfC/RpfG regulates the biosynthesis of extracellular polysaccharide (EPS), extracellular cellulase, and eventually the virulence of Xanthomonas13. The OmpR-type RR, HrpG, the key regulator of hypersensitive responses and pathogenicity (hrp) genes, is an important TCS in Xanthomonas. HrpG, together with the transcriptional activator HrpX, regulates all 24 type III secretion system (T3SS) genes, the 23 T3SS effector (T3SSe) genes, and the 29 type II secretion system substrate genes14. Mutant studies have revealed that HrpG and HrpX from Xanthomonas campestris pv. vesicatoria are essential for pathogenicity15,16. Other TCSs, such as ColS/ColR and PdeK/PdeR, have also been reported to control the virulence factors of Xanthomonas17,18,19. However, the functions of many remaining TCSs in Xanthomonas remain elusive.
The present study started with a global investigation of EPS and swarming regulation applied to all HK genes in Xoo PXO99A. This investigation revealed that two TCSs, StoS (stress tolerance-related oxygen sensor) and SreKRS (salt response kinase, regulator, and sensor)19,20 regulate EPS synthesis and swarming, but not virulence in rice. To determine the intrinsic mechanisms and functions of StoS and SreKRS regulation, a quantitative proteomics analysis was performed. The global protein expression pattern revealed that StoS and SreKRS act as coordinators of virulence factors and confer high fitness to Xoo in rice and in vitro.
Results and Discussion
Systematic functional screening of two-component signal transduction systems
EPS blocks the water flow in rice xylem vessels and provides an advantage to Xoo against environmental stresses1. Swarming, bacterial group motility driven by flagellum, is considered a means for invading more territory in the natural habitats of bacteria and serves as an important virulence determinant in some species21. Here, we employed a genome-wide functional screen of TCS genes in Xoo PXO99A based on EPS production and swarming ability. All 42 unique HK genes predicted using the P2CS database9 were knocked out in-frame through homologous double-crossover events using the suicide vector pK18mobsacB. All but three genes, PXO_02837, PXO_03032, and PXO_03565, among the 42 unique HK gene mutants were obtained.
An investigation into EPS production and swarming ability was performed after systematic gene deletion. Figure 1 shows that the deletion of PXO_00069, PXO_01018, PXO_02305, PXO_03078, PXO_04304, and PXO_04306 in PXO99A significantly attenuated EPS synthesis and swarming ability. The deletion of PXO_00069 (rpfC) in PXO99A attenuated EPS synthesis and swarming ability, consistent with previous reports22,23. However, the discovery that PXO_01018 (PdeK) and PXO_02305 (ColS) contributed to EPS synthesis and swarming improved our knowledge of PdeK/PdeR and ColS/ColR, which have been implicated in the regulation of several pathogenic factors but not EPS synthesis or swarming17,18,24. The other three EPS- and swarming-related HK genes, PXO_03078, PXO_04304, and PXO_04306, were selected for further study.

(A) In the EPS production test, a lustreless and applanate colony suggests low EPS production. (B) In the swarming assay, the colony size on the 18-cm diameter Petri dish indicates the swarming ability of Xoo strains. The numbers shown beneath the colonies are abbreviations for the mutants, e.g., 00620 indicates the mutant PXOΔ00620, in which PXO_00620 was deleted.
StoS and SreKRS positively regulate EPS synthesis and swarming
PXO_03078, a hybrid HK, contains an HK kinase core, a RR receiver domain and two Per–ARNT–Sim sensor domains25. The orthologue of this gene in Xoo MAFF311018 StoS, which shares 100% coverage and 99% identity with PXO_03078, has recently been reported to be involved in stress tolerance19. Here, PXO_03078 is also referred to as stoS for consistency. Moreover, two hypothetical genes, PXO_03076 and PXO_03077, located upstream of stoS were predicted to form an operon with stoS using MicrobesOnline Operon Predictions (Supplementary Fig. S1)26. To validate the results of the bioinformatics analysis, reverse transcription polymerase chain reaction was performed on the predicted operons. Consistent with the prediction, stoS, PXO_03077, and PXO_03076 are co-transcribed (Supplementary Fig. S1).
PXO_04304 is a typical HK with a Per–ARNT–Sim domain between the transmembrane helix and the HisKA domain, whereas PXO_04306 is a hybrid HK containing two receiver domains and a HisKA domain. Between these two proteins resides the RR PXO_04305. The orthologues of PXO_04304, PXO_04305 and PXO_04306 in Xanthomonas campestris pv. campestris 8004, which share 100% coverage and ≥85% identities, have been reported to fine tune the expression kinetics of 2-amino-4-hydroxy-6-hydroxymethyldihydropteridine pyrophosphokinase (hppK), another member of the sreKRS operon responsible for folate synthesis, via a positive feedback loop during stress responses20. To maintain consistency with their orthologues, XC_0728, XC_0729, and XC_0730, we refer to PXO_04304, PXO_04305, and PXO_04306 as salt response kinase (sreK), salt response regulator (sreR), and salt response sensor (sreS), respectively.
We firstly investigated the effects of stoS and sreKRS on bacterial growth. The growth curves of PXOΔstoS and PXOΔsreK were measured. No significant difference in growth between each mutant and the wild-type strain PXO99A was observed when cultured in NB medium (Supplementary Fig. S2A). This result eliminated the possibility that the decline in EPS synthesis and swarming were caused by differences in their growth. A complementation experiment using stoS and sreK was performed to confirm that the phenotypic change resulted from the gene deletion. To restore the deleted genes under the control of their native promoters and terminators, the full ORFs of stoS or sreK and the sequences 500 bp upstream and 300 bp downstream of these operons were ligated via overlapping PCR (Supplementary Fig. S3) and cloned into the broad-host-range cloning vector pHM1, generating pHMstoS and pHMsreK, respectively. Figure 2A shows that PXOΔstoS carrying pHMstoS exhibited EPS production and swarming similar to the wild-type strain PXO99A. Similarly, the EPS synthesis and swarming of PXOΔsreK were restored through pHMsreK.

(A) Gene complementation analysis of stoS and sreK. The target genes were cloned into pHM1 and expressed in targeted mutants under the control of their native promoters and terminators. Mutants rescued with the deleted gene were applied to assay EPS production and swarming. (B) Other genes in the stoS and sreKRS operons were knocked out in-frame, and EPS production and swarming tests were performed on the mutants.
Other genes in the stoS and sreKRS operons were deleted to investigate genetic linkage within the operons. When PXO_03077 and PXO_03076 in the stoS operon were deleted, no discernible phenotypic changes were observed in the corresponding mutants compared with the wild-type strain PXO99A (Fig. 2B). This finding indicates that PXO_03077 and PXO_03076 in the stoS operon are not involved in the signal transduction responsible for EPS synthesis and swarming. The deletion of sreR or sreS resulted in a dramatic decline in EPS production and swarming ability similar to sreK deletion mutants (Fig. 2B). The gene hppK (PXO_04307) in the sreKRS operon is another vital member of the signalling cascade of SreKRS in response to salt stress20. The deletion of hppK did not affect EPS production or swarming ability, indicating that HppK is independent of the regulation of EPS synthesis and swarming, and that this signal transduction circuit differed from the regulatory mechanism involving folate synthesis previously reported20. Taken together, these results demonstrated that StoS and SreKRS positively regulate EPS synthesis and swarming.
Absence of StoS or SreKRS does not weaken the virulence of Xoo
StoS and SreKRS positively regulate EPS and swarming, which are important for the successful invasion of Xanthomonas into hosts. Furthermore, StoS and SreKRS have been previously implicated in hypoxia and high salt stress19,20, respectively. The involvement of StoS and SreKRS as stress resistance switches was further confirmed using hydrogen peroxide and osmotic pressure protection tests in the present study (Supplementary Fig. S4). To explore the final effect of the decline in the EPS production, swarming and stress tolerance in stoS and sreK mutants, a virulence assay was performed to identify pathogenicity changes. Surprisingly, when PXOΔstoS, PXOΔsreK, and wild-type PXO99A were inoculated into the leaves of susceptive cultivar MH63 using the scissor-clip method, no obvious differences in the lesion length resulting from the mutants and wild-type strain were observed (Fig. 3A).

(A) Lesion lengths on rice leaves measured at 8, 10, 12 and 14 days after inoculation with Xoo strains PXO99A, PXOΔstoS, and PXOΔsreK. (B) Pictures of representative rice leaves were obtained at 14 days past inoculation.
The plant pathogens environment is hostile and constantly changing, and TCS is a predominant means through which pathogens sense and adapt to the environment. Virulence is the most important characteristic for pathogens survival in host plants, the major natural habitats of plant pathogens. In the present study, two TCSs, StoS and SreKRS, were revealed to regulate several prominent virulence determinants but not directly attenuate virulence.
General characterization of the iTRAQ analysis
To further uncover the function of StoS and SreKRS, we identified differentially expressed proteins between PXO99A and PXOΔstoS/PXOΔsreK using the proteomics approach iTRAQ. PXO99A, PXOΔstoS, and PXOΔsreK were cultured in the hrp genes induced medium XOM2. Total proteins were isolated when Xoo was grown to the middle of the logarithmic phase (OD600 = 0.9, Supplementary Fig. S2B). PXOΔstoS and PXO99A were analysed in three biological replicates, and PXOΔsreK was analysed in two biological replicates. The proteins (100 μg) from each sample were digested with trypsin, labelled with 8-plex iTRAQ reagents, and subsequently mixed for LC-MS/MS analysis (Fig. 4A). A total of 180,948 mass spectra were generated from the eight samples, and 42,864 unique spectra were matched to specific peptides (15,123). Upon searching the genome of PXO99A, 2,270 proteins were identified using the 15,123 unique peptides (Fig. 4B). A cut-off point at ±50% variation (±0.50) generally yields 88% quantification coverage based on an analysis of the biological replicates27. Figure 4C shows that the coverage yielded at the 50% cut-off point in the iTRAQ data varied from 94.6% to 98.3%, indicating better reproducibility. Given that iTRAQ quantification might systematically underestimate ratios, a protein with ≥1.5-fold difference and P ≤ 0.05 is considered differentially expressed28,29. Compared with PXO99A, 46 proteins showed decreased levels and 36 proteins showed increased levels in PXOΔstoS (Supplementary Tables S2 and S3). In PXOΔsreK, 46 and 44 proteins showed decreased and increased levels, respectively (Supplementary Tables S2 and S3).

(A) Sample preparation process for the iTRAQ analysis. PXOΔstoS and PXOΔsreK are selected for quantitative proteomic iTRAQ analysis. Total protein was extracted from the strains cultured in XOM2, followed by trypsin digestion and labelling with 8-plex iTRAQ reagents. (B) Basic statistical information for iTRAQ. The data were acquired by LC-MS/MS and analysed using Mascot 2.3.02 against the genome of PXO99A from NCBI (NC_010717.1). (C) Analysis of the reproducibility between the iTRAQ biological replicates of each treatment. The delta error means the variation between the fold change and 1, and the fold change is calculated between every two biological replicates. The vertical axis is the ratio of the number of proteins to the delta error in the total number of proteins quantified by comparisons between two biological replicates.
StoS and SreKRS signalling circuits overlap
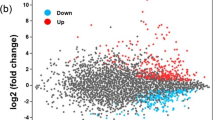
Figure 5A shows that more than half of the differentially expressed proteins (26 up-regulated proteins and 26 down-regulated proteins) in PXOΔstoS were also regulated by SreK. Moreover, these proteins were up- or down-regulated to similar degrees in the PXOΔstoS and PXOΔsreK mutants (Fig. 5B), suggesting interplay between the signal transduction and regulatory circuits associated with StoS and SreKRS in PXO99A. Notably, StoS was significantly down-regulated by 3.6-fold in the mutant PXOΔsreK compared with PXO99A (Supplementary Table S2, Fig. 5C). SreK expression was also weakened by 1.3- and 3.8-fold in PXOΔstoS and PXOΔsreK, respectively, detected by the remaining SreK peptide in iTRAQ. The transcription of the remaining segments of stoS and sreK was measured in PXOΔstoS, PXOΔsreK, and the wild-type strain PXO99A. Figure 5D shows that the transcription levels of stoS and sreK were significantly down-regulated in both mutants compared with the levels of these genes in PXO99A. This result suggests that StoS expression is positively regulated by itself and by SreKRS, and that the sreKRS operon is also activated by StoS and SreKRS.

(A) Venn diagram showing that more than half of the differential expressed proteins in PXOΔstoS and PXOΔsreK overlap. (B) The differentially expressed proteins in PXOΔstoS and PXOΔsreK compared with the wild-type strain PXO99A are shown together in a scatter diagram. (C) Relative expression levels of StoS and SreK in PXOΔstoS and PXOΔsreK compared with the wild-type strain PXO99A identified by iTRAQ. (D) mRNA abundance of stoS and sreK in PXOΔstoS and PXOΔsreK compared with the wild-type strain PXO99A. The dotted lines indicate critical 1.5-fold up- or down-regulation. *denotes a statistical difference in which P ≤ 0.05, and **denotes P ≤ 0.01. (E) The relationship between StoS and SreKRS. Results of using StoS and SreKRS as queries in the STRING database. Lines in different colours represent special relationships between two proteins. PXO_03023, methyltransferase; PXO_06172, response regulator; PXO_03077 /PXO_03076, hypothetical protein; PXO_03020, transcriptional regulator NtrC family; PXO_02019, putative signal protein with GGDEF domain; PXO_00994, response regulator.
To further explore the relationship between StoS and SreKRS, these proteins were used as queries in the STRING database, which assembles, evaluates, and disseminates information concerning protein-protein associations30. As shown in Fig. 5E, the homologous genes of stoS and sreK are neighbours in some bacterial genomes, such as baeS and baeR in Escherichia coli K12_MG1655. Putative homologues of StoS and SreK, such as ArcB and CheY in E. coli EC4115, were found to interact. Furthermore, stoS and sreKRS co-occur in many genomes, particularly in Xanthomonas group genomes. Again, this information indicates that StoS and SreKRS are closely connected and their circuits overlap. The overlapped signalling circuits of StoS and SreKRS elucidate similar phenotypes in stoS and sreK mutants.
Genome-wide microarray analysis of all TCS mutants in Escherichia coli suggests that signal circuits of numerous TCSs overlap31,32. Cross-regulation facilitates the elicitation of some of the same responses through distinct pathways31,32. The domain architectures of StoS and SreKRS are markedly different from each other, suggesting that StoS and SreKRS might be activated through distinct signals. We proposed that StoS and SreKRS could sense different stimuli to express similar virulence factors, resulting from overlapping pathways. This pathogen takes advantage of the connection between diverse functional modules for survival in hostile environments.
The EPS and swarming regulatory circuit of StoS and SreKRS
EPS produced by Xanthomonas consists of D-glucosyl, D-mannosyl, and D-glucuronyl acid residues at a molar ratio of 2:2:1 and variable proportions of O-acetyl and pyruvyl residues33. This EPS is assembled stepwise from UDP-glucose, GDP-mannose, and UDP-glucuronic acid, which are also important substrates of many carbohydrate metabolic pathways, such as starch and sucrose metabolism and galactose metabolism (http://www.genome.jp/kegg/kegg2.html). The synthesis of EPS could also be affected by other carbohydrate metabolic pathways. The pathway enrichment analysis showed that the Xoo mutants might reprogram starch and sucrose metabolism through the regulation of four proteins in PXOΔstoS and three proteins in PXOΔsreK (P = 0.0033 and 0.0401, respectively) (Fig. 6A; Supplementary Tables S4 and S5). In addition to starch and sucrose metabolism enrichment, the galactose metabolism pathway was enriched by differentially expressed proteins in PXOΔstoS (P = 0.0016) and PXOΔsreK (P = 0.0341) (Supplementary Tables S4 and S5). The differential expression of the glycogen debranching protein GlgX (PXO_01859) in the starch and sucrose metabolism pathways was verified at the mRNA level by qRT-PCR (Fig. 6B). Because EPS production was tested on NA medium, the expression differences of glgX were further confirmed by measuring the mRNA abundance in bacteria incubated on NA medium. Regardless of culture in XOM2 or on NA medium, glgX expression differences between mutants and wild type were consistent with the results of the iTRAQ analysis (Fig. 6B). These results indicated that the carbohydrate metabolism in PXOΔstoS and PXOΔsreK was reprogrammed, which might be involved in the attenuation of EPS synthesis. In Xanthomonas, gum gene cluster proteins were recognized to be response for the biosynthesis of EPS and were extremely important in EPS production34. The gum gene cluster comprises a tandem array of 12 ORFs, from gumB to gumM. All 12 proteins were identified by iTRAQ (data not shown), and two of these proteins, GumB and GumE, were up-regulated in PXOΔstoS and PXOΔsreK compared with the wild-type strain PXO99A (Supplementary Table S3). GumB and GumE might be required for the polymerization of EPS subunits or the export of the polymer33. The overexpression of GumB and GumE in PXOΔstoS and PXOΔsreK might be a compensatory measure for reduced EPS production.

(A) Several carbohydrate metabolic pathway proteins (GlgX, glycogen debranching protein; TreY (PXO_03372), malto-oligosyltrehalose synthase; PXO_03668, alpha-amylase family protein; OtsA (PXO_04869), alpha, alpha-trehalose-phosphate synthase and PXO_03299, endoglucanase) were differentially expressed in PXOΔstoS and PXOΔsreK compared with the wild-type strain PXO99A, as identified by iTRAQ. (B) The expression difference in glycogen debranching protein GlgX was confirmed at the mRNA level. (C) Three chemotaxis proteins, PXO_00047, PXO_00050, and PXO_00057, were down-regulated in PXOΔstoS and PXOΔsreK compared with the wild-type strain PXO99A, as identified by iTRAQ. (D) The expression difference for PXO_00057 was confirmed by qRT-PCR. The dotted lines indicate a critical 1.5-fold down-regulation. *denotes a statistical difference in which P ≤ 0.05, and **denotes P ≤ 0.01. (E) PXO_00050 and PXO_00057 were deleted, generating PXOΔ00050 and PXOΔ00057. The swarming ability of these mutants was measured by colony size in semi-solid medium.
Swarming, an important virulence determinant, was sharply attenuated in PXOΔstoS and PXOΔsreK. The iTRAQ data showed that the expression levels of the three chemotaxis genes, PXO_00047, PXO_00050, and PXO_00057, declined more than 1.5-fold in PXOΔstoS and 1.2-fold in PXOΔsreK compared with the wild-type strain PXO99A (Fig. 6C). qRT-PCR was employed to detect the mRNA abundance in the Xoo sample cultured in XOM2 and on TYGS medium, consistent with the culture conditions used for the swarming ability test. Figure 6D shows that PXO_00057 expression at the mRNA level decreased more than 1.5-fold in PXOΔstoS and PXOΔsreK. To further explore the role of chemotaxis proteins in swarming, PXO_00050 and PXO_00057 were deleted in-frame. Although the absence of chemotaxis-specific methylesterase PXO_00057 does not affect swarming, the chemotaxis signal transduction protein PXO_00050 contributes to the motility of Xanthomonas (Fig. 6E). These results indicated that chemotaxis could be one of the circuits to regulate the swarming of Xoo.
StoS and SreKRS fine-tune hrp and T3SSe genes expression
The hrp genes are extremely important for pathogenicity. Hrp proteins include T3SS regulon regulatory proteins, pilus components, and proteins functioning at different stages of injectisome assembly35. These hrp genes were clustered in a 28-kb pathogenicity island in PXO99A. T3SSe can be injected into the host cell cytosol to manipulate plant cellular processes, such as basal defences, to benefit the pathogen1. Typically, T3SSe encoding genes are scattered throughout the chromosome. Among the up-regulated proteins, Hrp proteins together with the T3SSe accounted for 22% in PXOΔstoS and 27% in PXOΔsreK (Fig. 7A). As shown in Fig. 7B, eight hrp genes or T3SSe encoding genes in PXOΔstoS and 12 hrp genes or T3SSe encoding genes in PXOΔsreK were up-regulated more than 1.5-fold compared with PXO99A. Seven identical genes were both up-regulated in PXOΔstoS and PXOΔsreK. A total of 13 up-regulated unique hrp genes or T3SSe encoding genes were identified in the two mutants. Eight of the 13 genes were clustered in a hrp pathogenicity island. The other five genes encoding T3SSe were scattered throughout the chromosome (Fig. 7B). The differential expression of the type III secretion apparatus protein HrpD1 and the type III effector XopF1 in mutant and wild-type bacteria was confirmed by qRT-PCR (Fig. 7C). These results suggest that StoS and SreKRS modulate the expression of hrp and T3SSe genes.

(A) The proportion of hrp and T3SSe genes in the list of up-regulated proteins of PXOΔstoS and PXOΔsreK compared with wild-type PXO99A. (B) iTRAQ data shows that Hrp proteins and T3SSe were negatively regulated by StoS and SreKRS. (C) The relative expression levels of type III secretion apparatus protein HrpD1 and XopF1 effectors were measured by qRT-PCR. The relative expression of hrpX and hrpG in PXOΔstoS and PXOΔsreK compared with the wild-type PXO99A was also determined by qRT-PCR. (D) HrpG quantitative analysis at the post-transcriptional level by western blot analysis. The results of Coomassie brilliant blue (CBB) staining show similar quantities in different lanes. Western blot and CBB staining were under the same experimental conditions for protein sample preparation and SDS-PAGE. Lane 1, hrpGHis/PXOΔhrpG. Lane 2, hrpGHis/PXOΔhrpG&stoS Lane 3, hrpGHis/PXOΔhrpG&sreK. The dotted lines indicate the critical 1.5-fold up-regulation. *denotes a statistical difference in which P ≤ 0.05, and **denotes P ≤ 0.01.
To minimize metabolic consumption, hrp and T3SSe genes are not constitutively expressed but rather become activated when Xanthomonas enters the host or is cultured in certain minimal media36. All genes, except hrpA in Xanthomonas, are under the direct transcriptional control of HrpX (PXO_01953), a transcription regulator that is transcriptionally activated by the global response regulator, HrpG35. Therefore, the expression levels of hrpX and hrpG in PXOΔstoS and PXOΔsreK were detected using qRT-PCR. The experimental results verified the hypothesis that the mRNA abundance of hrpX and hrpG was significantly increased in PXOΔstoS and PXOΔsreK compared with that in PXO99A, indicating that the negative regulation of hrp and T3SSe genes occurs through the HrpG-HrpX circuit.
Because none of the SreKRS and StoS members possess the helix-turn-helix domain, the essential domain of a typical transcriptional regulator, StoS and SreKRS might not directly regulate hrpG at the transcriptional level. HrpG plays global roles in the signalling network of Xanthomonas. Interestingly, hrpG exists in the list of HrpG positively regulated gene14, suggesting that HrpG potentially possesses self-regulation activity. Therefore, we hypothesized that the differential expression of hrp and T3SSe encoding genes resulting from StoS and SreKRS was implemented by HrpG self-regulation. The quantitative analysis of HrpG protein at the post-transcriptional level was performed in mutants and wild type. HrpG fused with a 6*His tag was under the control of the constitutive lac promoter in a broad-host-range cloning vectors pBBR1MCS4_START37. HrpGHis was subsequently trans-expressed in the hrpG mutant, the hrpG and stoS double mutant, and the hrpG and sreK double mutant. The HrpGHis quantitation on different genetic backgrounds was detected by western blot with monoclonal antibody against 6*His tag. As shown in Fig. 7D, HrpG is sharply up-regulated because of the absence of stoS or sreK. This result shows that StoS and SreKRS regulate HrpG at the post-transcriptional level. Taken together, StoS and SreKRS decrease HrpG at the post-transcriptional level, subsequently reduce hrpG at the transcriptional level by feedback regulation, and moderate the expression of all hrp and T3SSe genes.
StoS and SreKRS contribute to the full fitness of Xoo in vivo and in vitro
EPS produced by Xanthomonas is highly hydrated and anionic consistency, and protects bacteria from environmental stresses1. Swarming is a powerful means of invading more territory in the natural habitat and accelerating the biomass production of bacteria38. Hrp proteins, major virulence factors of Xanthomonas, are extremely important for pathogenicity. However, virulence traits carry a significant cost to the pathogen39, and redundant virulence factors induce enormous metabolic consumption and are disadvantageous to bacterial fitness40,41,42. StoS and SreKRS finely orchestrated these virulence factors and responded to stress tolerance as previously reported19,20. Moreover, the growth of the stoS and the sreK mutants are significantly inferior to that of the wild-type strain growth in XOM2 (Supplementary Fig. S2). Based on these facts, we hypothesized that StoS and SreKRS are essential for Xoo fitness in rice, although StoS and SreKRS do not directly control virulence.
To verify this hypothesis, we measured the relative fitness of PXOΔstoS and PXOΔsreK in rice and in XOM2 using a competition assay43. The mutants and wild-type strains were mixed at a ratio of 1:1 and inoculated into rice leaves and XOM2. The relative fitness indices of the mutants were calculated by dividing mutants colonies number by wild-types colonies number. As shown in Fig. 8, the relative fitness index of PXOΔstoS was 0.28 when co-infected with PXO99A in rice. Similarly, the absence of sreK reduced the fitness in rice to 0.25. When co-cultured in XOM2 with the wild-type strain, stoS and sreK mutants were outcompeted, with relative fitness indices of 0.29 and 0.33, respectively. This finding shows that StoS and SreKRS confer Xoo fitness for infection rice and growth independent of host plants.

PXO99A and mutants were mixed at a ratio of 1:1 and inoculated into rice leaves and XOM2 medium. The ratio between mutant and PXO99A colony forming units was measured at 10 days after inoculating to rice and 60 hours after inoculating to XOM2. This ratio was defined as the relative fitness of PXOΔstoS and PXOΔsreK
Genotypes with higher fitness will tend to produce more offspring and thereby increase in frequency over time compared with less-fit competitors43. Although stoS and sreK mutants did not show obvious virulence weakness under laboratory condition, as which is constant and benefit to Xoo infection, Xoo strains without StoS or SreKRS might be eliminated in hostile natural habitats evolved with competitors and environmental stresses.
Conclusion
The TCS is a prevalent strategy for coupling environmental stimuli to adaptive responses in bacteria6. In most bacterial genomes, dozens to hundreds of genes encode TCS proteins. However, most of these TCSs remain functionally unknown. In the present study, we revealed that StoS and SreKRS confer fitness to Xoo by contributing to EPS production, motility and stress tolerance, and moderating the expression of Hrp proteins. This mechanism is supposed to be a novel strategy to adapt to changing environments. These findings show the roles of StoS and SreKRS in the network of TCS-mediated environmental adaptation. StoS, SreKRS, and the previously characterized TCSs form an intricate signalling network, providing Xanthomonas with an economic and effective gene expression regulation mechanism to adapt to changing intra- and extracellular environments. Figure 9 shows an updated model of the TCS network, in which the signalling circuits uncovered in the present study are highlighted in blue. These results improved the current knowledge of the TCS network in Xanthomonas. However, the exact details of StoS and SreKRS regulatory mechanisms remain unknown. The negative regulation of Hrp proteins has drawn much attention, and the regulatory mechanism is worthy of further exploration.

All of the functionally characterized TCSs in Xanthomonas are updated in this figure. The signalling circuits depicted in blue were identified in the present study, and other circuits were summarized from previous studies (RpfC/G13, HpaS/HrpG53, RavS/R54, ColS/R18,24, PhoQ/P55, RaxH/R56,57, LOV58, PdeK/R17, TctE/D59, and VemR48). The characterized SreKRS circuit is represented by dotted lines, because which component of the SreKRS is in charge of the signal output is still unknown. The differentially expressed genes might be not directly regulated by StoS or SreKRS but are regulated by other unknown transcriptional regulator (TR?). Lines with arrowheads represent signal transduction or positive regulation. “T” lines indicate negative regulation.
Methods
Strains, plasmids, and culture conditions
Xoo PXO99A, 5-azacytidine-resistant derivative of Philippine race 6 PXO99, was kindly presented by Dr. Ya-Wen He44. The plasmid used for gene knock-out pK18mobsacB was offered by Dr. Jin He. The broad-host-range cosmid used for gene rescue pHM1 was a gift from Dr. Gong-You Chen in Shanghai Jiao Tong University45. Mutants obtained in this study are listed in Supplementary Table S1. Xanthomonas strains were cultivated at 28 °C in NB medium (beef extract, 3 g/L; yeast extract, 1 g/L; polypeptone, 5 g/L; and sucrose, 10 g/L) or on solid NB medium added with 1.5% agar (NA) except for special circumstances. When required, growth media were supplemented with antibiotics at the following final concentrations: kanamycin, 25 μg/mL; spectinomycin, 50 μg/mL; ampicillin, 100 μg/ml.
Gene deletion in Xoo
All the HK genes in PXO99A were designed to generate mutants via allelic homologous recombination. In brief, two flanking regions of the target gene were cloned to the suicide plasmid pK18mobsacB by three appropriate restriction sites46 (Supplementary Table S1). For the convenience of the clone and efficiency of recombination, the lengths of homologous DNA fragments were between 500 and 900 bp. The recombinant plasmid verified by sequencing was subsequently imported into the wild-type strain PXO99A via electroporation to generate one cross-over mutant. This cross-over mutant was then cultured in NAS medium (NA containing 10% sucrose) to generate a second cross-over. Strains which are able to grow on NAS lost the plasmid because of the second cross-over event that either restored the wild-type situation or led to the gene deletion mutant, which could be sorted by PCR.
Gene complementation
For complementation analysis, the ORF region of the target gene, up-stream 500 bp segment which was predicted as promoter, and down-stream 300 bp segment which was predicted as terminator were amplified and ligated by overlapping PCR (Supplementary Table S1). The target gene under the control of its native promoter and terminator was then cloned to a broad-host-range cosmid pHM1. The recombinant vector was electroporated into the mutant to analyze the complementation17.
EPS production and swarming ability determination
An effective and visualized method was applied to analyze EPS production47. In brief, strains were cultured in NB medium and adjusted to the same concentration. EPS production was detected by the appearance of colonies arising in NA medium after 48 to 72 hours from 2 μL of culture drops. Low EPS production would lead to a lustreless and applanate colony. Swarming ability was measured by colony dimension arising in semi-solid medium TYGS (tryptone, 1 g/L; yeast extract, 0.5 g/L; glucose, 10 g/L; NaCl, 1 g/L; agar 0.6%) from 2 μL drops of the equivalent concentration cultures48,49. The experiments were repeated at least three times.
Virulence assay
The susceptible rice breed MH63 was utilized to analyze the virulence of the Xoo strains. In brief, five rice plants were grown per circular pot (about 80 mm2) in a tray containing standing water for 4–5 weeks, while Xoo strains were cultured in NB medium for 24–48 hours to late logarithmic phase. All the strains were adjusted to OD600 = 1.0. Two youngest fully expanded leaves of the prepared plants were clipped about 2cm from the tip with scissors that have been immersed in a bacterial suspension immediately prior to each clipping. All the five plants in one pot were used for inoculation of each Xoo strain. The inoculated rice plants were grown in the green house under 28–35 °C with 75% relative humidity. Lesion lengths of the diseased leaf were measured at 14 days after inoculation50. The experiments were repeated at least three times.
Protein preparation for isobaric tags for relative and absolute quantitation
Xoo strains were grown in XOM251 (D-xylose, 0.18%; D, L-methionine, 670 μM; sodium L(+)-glutamate, 10 mM; KH2PO4, 14.7 mM; MnSO4, 40 μM; Fe(III)-EDTA, 240 μM; and 5 mM MgCl2) to the middle logarithmic phase (OD600 = 0.9). The harvested cultures were washed twice with PBS (137 mM NaCl; 2.7 mM KCl; 10 mM Na2HPO4; and 2 mM KH2PO4, pH 7.4). The pellets were subsequently resuspended in 10 mL of PBS containing a tablet of protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany) per 100 mL of culture, followed by cell disruption using a low temperature ultra-high pressure continuous cell disrupter. The disrupted cells were centrifuged at 12,000 rpm and 4 °C for 20 min to remove cell debris, and the supernatant containing soluble proteins was precipitated with precooling acetone (25 mL per 5 mL of supernatant) overnight at −20 °C. The precipitates were washed twice with cold acetone, and the proteins were dissolved in 7 M urea and 2 M thiourea. A reductive alkylation reaction was subsequently applied to each protein sample, followed by acetone precipitation and dissolution in 0.5 M tetraethylammonium bromide. Finally, the protein concentration was determined using the Bradford method.
Isobaric peptide labeling and Strong Cation Exchange fractionation
The proteins (100 μg) from each sample were digested with trypsin and labelled with 8-plex isobaric tags for relative and absolute quantitation (iTRAQ) reagents according to the manufacturer’s protocol. All samples were labelled as follows: PXO99A1#_113, PXO99A2#_116, PXO99A3#_119, PXOΔstoS1#_114, PXOΔstoS2#_117, PXOΔstoS3#_121, PXOΔsreK1#_115, and PXOΔsreK2#_118. All labelled samples were pooled and dissolved in 4 mL of buffer A (25 mM NaH2PO4 in 25% ACN, pH 2.7). Peptides were fractioned using a 4.6 mm × 250 mm UltremexSCX column on a Prominence LC-20AB HPLC system with a constant flow rate of 1 mL/min. A 22 min gradient consisting of 100% buffer A for 10 min, 5% to 35% buffer B (25 mM NaH2PO4, 1 M KCl in 25% ACN, pH 2.7) for 11 min, and 35% to 80% buffer B for 1 min was performed. The chromatogram was monitored under 214 nm. A total of 12 fractions were obtained from the Strong Cation Exchange Choematography separation, and each fraction was desalted and dried.
LC–MS/MS
The LC–MS/MS package and system parameters were similar to those previously described28. Briefly, peptides from Strong Cation Exchange fractions were enriched and desalted using Symmetry C18 Trap column (5 μm, 180 μm × 20 mm), and separated via BEH130 C18 column performing on nanoACQuity (Waters, USA) liquid chromatography system. Data was acquired by Triple TOF 5600 (AB SCIEX, Concord, ON, USA) fitted with a Nanospray III source (AB SCIEX, USA) and a pulled quartz tip as the emitter (New Objectives, USA).
Data analysis
Protein identification and quantification were carried out using Mascot 2.3.02 against the genome of PXO99A from NCBI (RefSeq accession number NC_010717.1). Other search parameters were as follows: type of search, MS/MS ion search; enzyme, trypsin; fragment mass tolerance, ±0.05 Da; mass values, monoisotopic; variable modifications, Gln->pyro-Glu (N-term Q), oxidation (M), iTRAQ8plex (Y); peptide mass tolerance, ±0.1 Da; instrument type, default; max missed cleavages, 1; and fixed modifications, carbamidomethyl (C), iTRAQ8plex (N-term), and iTRAQ8plex (K). For protein identification and quantification, P ≤ 0.05 (with 95% confidence) was considered statistically significant. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium52 via the PRIDE partner repository with the dataset identifier PXD002625. The results obtained were exported as a Microsoft Excel file for further analysis.
Pathway enrichment analysis was performed to identify the significantly enriched pathways. Briefly, all the differential expressed proteins were mapped to Kyoto Encyclopedia of Genes and Genomes (KEGG) database, and the number of every mapped pathway term was acquired. Hypergeometric test was then employed to find the enriched pathways using all the proteins identified by iTRAQ as background data.
Real-time quantitative reverse transcription PCR
To verify gene expression differences identified by iTRAQ, Xoo were cultured in XOM2 to the middle logarithmic phase (OD600 = 0.9) for RNA extraction. Consistent with the culture conditions used for the EPS production and swarming tests, Xoo strains were also cultured on NA (NB added with 1.5% agar) or TYGS semi-solid medium for approximately 60 hours, and the bacteria were collected from NA or TYGS for RNA extraction. RNA was extracted using the RNApure Bacteria Kit (Beijing CoWin Biotech, Beijing, China) according to the manufacturer’s instructions. RNA integrity was confirmed by electrophoresis on 0.8% agarose gels. The RNA purity and concentration were determined by NanoDrop 2000 (Thermo Fisher Scientific, Waltham, MA, USA). cDNA was synthesized from total RNA (2 μg) using a SuperRT cDNA Kit (Beijing CoWin Biotech, Beijing, China). The primers used for real-time quantitative reverse transcription PCR (qRT-PCR) are listed in Supplementary Table S1. The qRT-PCR reaction was performed using SYBR® Select Master Mix (Applied Biosystems, Foster City, CA, USA) on a ViiA 7 PCR system (Applied Biosystems, Foster City, CA, USA), and 16s rRNA was used as an endogenous control for gene expression analysis by ΔΔCT. All genes were investigated with three technological repeats, and ANOVA for differences in CT values (ΔCT) was performed to assess whether a statistical difference exists in gene expression level between the mutant and wild-type strains. The experiments were repeated at least three times.
Quantitative analysis of HrpG protein by western blotting
The hrpG and stoS/sreK double mutants, PXOΔhrpG&stoS and PXOΔhrpG&sreK, were generated by knocking out stoS or sreK in the hrpG mutant PXOΔhrpG. The hrpG ORF was amplified by the primers in Supplementary Table S1, and the sequence encoding the 6*His tag was added to the 5′ terminal of the reverse primer to fuse the 6*His tag to the C terminal of the HrpG protein. hrpGHis was subsequently cloned into pBBR1MCS4_START37, generating pBBRhrpG. pBBRhrpG was imported into PXOΔhrpG, PXOΔhrpG&stoS and PXOΔhrpG&sreK to quantitatively analyse HrpGHis protein under different genetic backgrounds.
Xoo strains were cultured in XOM2 to the middle logarithmic phase (OD600 = 0.9) and harvested by centrifugation. The pellets were subsequently resuspended in PBS and disrupted using an ultrasonic processor. Supernatants containing soluble proteins were used for SDS-PAGE and western blot analyses. A monoclonal antibody against 6*His and the HRP-conjugated secondary antibody were commercially purchased from Proteintech (Wuhan, China). Western blot analysis was performed using a general method, and the proteins were detected by chemiluminescent detector system using Pierce ECL (Thermo Fisher Scientific, Waltham, MA, USA) as a HRP Substrate.
Relative fitness assay
Wild-type and mutant Xoo strains were cultured in NB medium for approximately 24 hours to the middle logarithmic phase and adjusted to OD600 = 1.0. The adjusted wild-type and mutant Xoo strains were mixed at a ratio of 1:1. The mixed culture was subsequently inoculated into rice leaves and XOM2 medium as described above to assay the relative fitness of Xoo in rice and in vitro, respectively. For the in vivo assay, the diseased rice leaves were cut from the plants at 10 days after inoculation, sterilized in 70% ethanol for 30 seconds, rinsed three times in sterile water, and subsequently homogenized and plated on NA medium after serial dilution. Similarly, culture in XOM2 was serially diluted and plated on NA medium at 60 hours after inoculation. Xoo colonies on NA were counted at 72 hours after plating. The stoS and sreK mutant colonies were lustreless and applanate, whereas the colonies from the wild-type strain PXO99A had lustre and were raised. The relative fitness of the mutant was defined as the ratio of mutant colonies and PXO99A colonies. The experiments were repeated at least three times.
Additional Information
How to cite this article: Zheng, D. et al. Two overlapping two-component systems in Xanthomonas oryzae pv. oryzae contribute to full fitness in rice by regulating virulence factors expression. Sci. Rep. 6, 22768; doi: 10.1038/srep22768 (2016).
References
Büttner, D. & Bonas, U. Regulation and secretion of Xanthomonas virulence factors. FEMS Microbiol. Rev. 34, 107–133 (2010).
Mew, T. W. Current Status and Future Prospects of Research on Bacterial Blight of Rice. Annu. Rev. Phytopathol. 25, 359–382 (1987).
Verdier, V., Vera Cruz, C. & Leach, J. E. Controlling rice bacterial blight in Africa: Needs and prospects. J. Biotechnol. 159, 320–328 (2012).
Mitchell, S. L., Ismail, A. M., Kenrick, S. A. & Camilli, A. The VieB auxiliary protein negatively regulates the VieSA signal transduction system in Vibrio cholerae . BMC Microbiol. 15, 59 (2015).
Capra, E. J. & Laub, M. T. Evolution of Two-Component Signal Transduction Systems. Annu. Rev. Microbiol. 66, 325–347 (2012).
Gao, R. & Stock, A. M. Biological Insights from Structures of Two-Component Proteins. Annu. Rev. Microbiol. 63, 133–154 (2009).
Casino, P., Rubio, V. & Marina, A. The mechanism of signal transduction by two-component systems. Curr. Opin. Struc. Biol. 20, 763–771 (2010).
Stock, A. M., Robinson, V. L. & Goudreau, P. N. Two-component signal transduction. Annu. Rev. Biochem. 69, 183–215 (2000).
Barakat, M. et al. P2CS: a two-component system resource for prokaryotic signal transduction research. BMC Genomics 10, 315 (2009).
Qian, W., Han, Z. J. & He, C. Two-component signal transduction systems of Xanthomonas spp.: a lesson from genomics. Mol. Plant Microbe Interact. 21, 151–61 (2008).
He, Y.-W. et al. Genome scale analysis of diffusible signal factor regulon in Xanthomonas campestris pv. campestris: identification of novel cell-cell communication-dependent genes and functions. Mol. Microbiol. 59, 610–622 (2006).
An, S. Q. et al. The PAS domain-containing histidine kinase RpfS is a second sensor for the diffusible signal factor of Xanthomonas campestris . Mol. Microbiol. 92, 586–597 (2014).
He, Y.-W. & Zhang, L.-H. Quorum sensing and virulence regulation in Xanthomonas campestris. FEMS Microbiol. Rev. 32, 842–857 (2008).
Guo, Y., Figueiredo, F., Jones, J. & Wang, N. HrpG and HrpX play global roles in coordinating different virulence traits of Xanthomonas axonopodis pv. citri . Mol. Plant Microbe Interact. 24, 649–61 (2011).
Wengelnik, K., Van den Ackerveken, G. & Bonas, U. HrpG, a key hrp regulatory protein of Xanthomonas campestris pv. vesicatoria is homologous to two-component response regulators. Mol. Plant Microbe Interact. 9, 704–12 (1996).
Wengelnik, K., Rossier, O. & Bonas, U. Mutations in the regulatory gene hrpG of Xanthomonas campestris pv. vesicatoria result in constitutive expression of all hrp genes. J. Bacteriol. 181, 6828–31 (1999).
Yang, F. et al. A novel two-component system PdeK/PdeR regulates c-di-GMP turnover and virulence of Xanthomonas oryzae pv. oryzae . Mol. Plant Microbe Interact. 25, 1361–1369 (2012).
Yan, Q. & Wang, N. The ColR/ColS Two-Component System Plays Multiple Roles in the Pathogenicity of the Citrus Canker Pathogen Xanthomonas citri subsp. citri . J. Bacteriol. 193, 1590–1599 (2011).
Ikawa, Y., Furutani, A., Ochiai, H. & Tsuge, S. StoS, a hybrid histidine kinase sensor of Xanthomonas oryzae pv. oryzae, is activated by sensing low O concentration and is involved in stress tolerance and virulence. Mol. Plant Microbe Interact. 27, 537–545 (2014).
Wang, F. F. et al. A three-component signaling system finetunes expression kinetics of HPPK responsible for folate synthesis by positive feedback loop during stress response of Xanthomonas campestris . Environ. Microbiol. 16, 2126–44 (2014).
Kim, W. & Surette, M. G. Metabolic differentiation in actively swarming Salmonella . Mol. Microbiol. 54, 702–14 (2004).
Slater, H., Alvarez-Morales, A., Barber, C. E., Daniels, M. J. & Dow, J. M. A two-component system involving an HD-GYP domain protein links cell–cell signalling to pathogenicity gene expression in Xanthomonas campestris . Mol. Microbiol. 38, 986–1003 (2000).
Zhao, Y. et al. Function of a two-component system RpfCxoc/RpfGxoc in Xanthomonas oryzae pv. oryzicola . Wei Sheng Wu Xue Bao 52, 449–56 (2012).
Subramoni, S., Pandey, A., Vishnu Priya, M. R., Patel, H. K. & Sonti, R. V. The ColRS system of Xanthomonas oryzae pv. oryzae is required for virulence and growth in iron-limiting conditions. Mol. Plant Pathol. 13, 690–703 (2012).
Möglich, A., Ayers, R. A. & Moffat, K. Addition at the Molecular Level: Signal Integration in Designed Per–ARNT–Sim Receptor Proteins. J. Mol. Biol. 400, 477–486 (2010).
Price, M. N. A novel method for accurate operon predictions in all sequenced prokaryotes. Nucleic Acids Res. 33, 880–892 (2005).
Gan, C. S., Chong, P. K., Pham, T. K. & Wright, P. C. Technical, experimental, and biological variations in isobaric tags for relative and absolute quantitation (iTRAQ). J. Proteome Res. 6, 821–7 (2007).
Wang, J. et al. The metabolic regulation of sporulation and parasporal crystal formation in Bacillus thuringiensis revealed by transcriptomics and proteomics. Mol. Cell Proteomics 12, 1363–1376 (2013).
Karp, N. A. et al. Addressing accuracy and precision issues in iTRAQ quantitation. Mol. Cell Proteomics 9, 1885–97 (2010).
Franceschini, A. et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 41, D808–15 (2013).
Laub, M. T. & Goulian, M. Specificity in Two-Component Signal Transduction Pathways. Annu. Rev. Genet. 41, 121–145 (2007).
Oshima, T. et al. Transcriptome analysis of all two-component regulatory system mutants of Escherichia coli K-12. Mol. Microbiol. 46, 281–291 (2002).
Becker, A., Katzen, F., Puhler, A. & Ielpi, L. Xanthan gum biosynthesis and application: a biochemical/genetic perspective. Appl. Microbiol. Biotechnol. 50, 145–52 (1998).
Kim, S.-Y., Kim, J.-G., Lee, B.-M. & Cho, J.-Y. Mutational analysis of the gum gene cluster required for xanthan biosynthesis in Xanthomonas oryzae pv oryzae . Biotechnol. Lett. 31, 265–270 (2008).
Tampakaki, A. P. et al. Playing the “Harp”: Evolution of Our Understanding of hrp/hrc Genes. Annu. Rev. Phytopathol. 48, 347–370 (2010).
Song, C. Mutagenesis of 18 Type III Effectors Reveals Virulence Function of XopZ PXO99 in Xanthomonas oryzae pv. oryzae Mol. Plant Microbe Interact. 23, 893–902 (2010).
Obranić, S., Babić, F. & Maravić-Vlahoviček, G. Improvement of pBBR1MCS plasmids, a very useful series of broad-host-range cloning vectors. Plasmid 70, 263–267 (2013).
Fraser, G. M. & Hughes, C. Swarming motility. Curr. Opin. Microbiol. 2, 630–5 (1999).
Vasanthakrishnan, R. B. et al. PrfA regulation offsets the cost of Listeria virulence outside the host. Environ. Microbiol. 17, 4566–79 (2015).
Raymond, B., West, S. A., Griffin, A. S. & Bonsall, M. B. The dynamics of cooperative bacterial virulence in the field. Science 337, 85–8 (2012).
Deng, C. et al. Division of labour and terminal differentiation in a novel Bacillus thuringiensis strain. ISME J. 9, 286–96 (2015).
Sturm, A. et al. The cost of virulence: retarded growth of Salmonella Typhimurium cells expressing type III secretion system 1. PLoS Pathog. 7, e1002143 (2011).
Wiser, M. J. & Lenski, R. E. A Comparison of Methods to Measure Fitness in Escherichia coli . PLoS One 10, e0126210 (2015).
Salzberg, S. L. et al. Genome sequence and rapid evolution of the rice pathogen Xanthomonas oryzae pv. oryzae PXO99A. BMC Genomics 9, 204 (2008).
Zou, H. et al. Identification of an avirulence gene, avrxa5, from the rice pathogen Xanthomonas oryzae pv. oryzae. Sci. China Life Sci. 53, 1440–1449 (2010).
Schäfer, A. et al. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145, 69–73 (1994).
Tang, J. L. et al. Genetic and molecular analysis of a cluster of rpf genes involved in positive regulation of synthesis of extracellular enzymes and polysaccharide in Xanthomonas campestris pathovar campestris . Mol. Gen. Genet. 226, 409–17 (1991).
Tao, J. & He, C. Response regulator, VemR, positively regulates the virulence and adaptation of Xanthomonas campestris pv. campestris . FEMS Microbiol. Lett. 304, 20–28 (2010).
Skerker, J. M., Prasol, M. S., Perchuk, B. S., Biondi, E. G. & Laub, M. T. Two-component signal transduction pathways regulating growth and cell cycle progression in a bacterium: a system-level analysis. PLoS Biol. 3, e334 (2005).
Yang, B. & Bogdanove, A. Inoculation and virulence assay for bacterial blight and bacterial leaf streak of rice. Methods Mol. Biol. 956, 249–55 (2013).
Tsuge, S. et al. Expression of Xanthomonas oryzae pv. oryzae hrp Genes in XOM2, a Novel Synthetic Medium. J. Gen. Plant Pathol. 68, 363–371 (2002).
Vizcaino, J. A. et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–6 (2014).
Li, R.-F. et al. Identification of a putative cognate sensor kinase for the two-component response regulator HrpG, a key regulator controlling the expression of the hrp genes in Xanthomonas campestris pv. campestris . Environ. Microbiol. 16, 2053–2071 (2013).
He, Y.-W., Boon, C., Zhou, L. & Zhang, L.-H. Co-regulation of Xanthomonas campestris virulence by quorum sensing and a novel two-component regulatory system RavS/RavR. Mol. Microbiol. 71, 1464–1476 (2009).
Lee, S. W. et al. The Xanthomonas oryzae pv. oryzae PhoPQ Two-Component System Is Required for AvrXA21 Activity, hrpG Expression, and Virulence. J. Bacteriol. 190, 2183–2197 (2008).
Burdman, S., Shen, Y., Lee, S. W., Xue, Q. & Ronald, P. RaxH/RaxR: a two-component regulatory system in Xanthomonas oryzae pv. oryzae required for AvrXa21 activity. Mol. Plant Microbe Interact. 17, 602–12 (2004).
Stolov, A., Valverde, A., Ronald, P. & Burdman, S. Purification of soluble and active RaxH, a transmembrane histidine protein kinase from Xanthomonas oryzae pv. oryzae required for AvrXa21 activity. Mol. Plant Pathol. 8, 93–101 (2007).
Corrochano, L. M. et al. A LOV Protein Modulates the Physiological Attributes of Xanthomonas axonopodis pv. citri Relevant for Host Plant Colonization. PLoS One 7, e38226 (2012).
Tamir-Ariel, D., Rosenberg, T. & Burdman, S. The Xanthomonas campestris pv. vesicatoria citH gene is expressed early in the infection process of tomato and is positively regulated by the TctDE two-component regulatory system. Mol. Plant Pathol. 12, 57–71 (2011).
Acknowledgements
We are grateful to Dr. Ya-Wen He, Dr. Jin He, and Dr. Gong-You Chen for providing the experimental materials. We also thank Prof. Jacques Mahillo for his helpful suggestions on the manuscript, and Prof. Margaret McFall-Ngai, Prof. Edward (Ned) Ruby, and Dr. Yanling Wang for their constructive advice on this study. This study was supported by National Natural Science Foundation of China (Project J1103510, http://www.nsfc.gov.cn/).
Author information
Authors and Affiliations
Contributions
D.H.Z., M.S. and L.F.R. conceived of and designed the study. D.H.Z., X.Y.Y., M.D., Y.F.L., B.L. and P.Y.Q. performed the experiments. D.H.Z., M.S. and L.F.R. performed the data analyses. D.H.Z. and L.F.R. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zheng, D., Yao, X., Duan, M. et al. Two overlapping two-component systems in Xanthomonas oryzae pv. oryzae contribute to full fitness in rice by regulating virulence factors expression. Sci Rep 6, 22768 (2016). https://doi.org/10.1038/srep22768
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep22768
This article is cited by
-
A transcriptional Regulator Gar Regulates the Growth and Virulence of Xanthomonas oryzae pv. oryzae
Current Microbiology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
