
Abstract
Time-lapse imaging is a fundamental tool for studying cellular behaviours, however studies of primary cells in complex co-culture environments often requires fluorescent labelling and significant light exposure that can perturb their natural function over time. Here, we describe ptychographic phase imaging that permits prolonged label-free time-lapse imaging of microglia in the presence of neurons and astrocytes, which better resembles in vivo microenvironments. We demonstrate the use of ptychography as an assay to study the phenotypic behaviour of microglial cells in primary neuronal co-cultures through the addition of cyclosporine A, a potent immune-modulator.
Similar content being viewed by others

Introduction
Observing cells over time utilising either brightfield or fluorescence based techniques is a valuable tool to study cellular behaviour in co-culture models1. Whilst brightfield microscopy does not require cell labelling, the suitability of this approach in mixed cell co-cultures is limited by lack of contrast and presence of imaging artefacts making it difficult to easily distinguish different cell types. Fluorescence labelling of living cells is therefore frequently used to overcome these limitations, where cells are either labelled using organic dyes or as expression of fluorescently encoded reporter genes. Although fluorescence labelling provides the contrast needed to differentiate between cells it has the potential to alter normal cell function. Furthermore extended time-lapse imaging of fluorescently labelled cells can cause significant phototoxicity2, which limits the duration of continuous imaging. Focal drift is an additional problem associated with prolonged time-lapse imaging as specimen movement from the focal plane can result in experimental failure as images become uninterpretable. Although autofocusing hardware and algorithms are available, these require additional light exposure and are susceptible to culture artefacts. To overcome these limitations, there has been a steady rise in the development of label-free imaging platforms that exploit phase shifted information from specimens3,4,5,6,7,8. These label-free technologies are ideally suited to cell cultures and stem cells where labelling of cells may be undesirable or not possible (e.g. primary human cells). We demonstrate the use of ptychography as a novel label-free imaging assay that permits continuous and prolonged time-lapse imaging of complex primary co-cultures. Ptychography also allows virtual post-acquisition focusing, preventing loss of images due to focal drift during image collection.
Through the use of an iterative phase retrieval algorithm, ptychography generates label-free, high contrast quantitative phase images3. In this technique the specimen is moved through a coherent illumination probe yielding an array of overlapping diffraction patterns that are recorded onto a CCD camera. The collected diffraction patterns are then processed by the ptychographic algorithm to produce an image that has both amplitude and phase information that relate to the refractive index and the thickness of the specimen3. Recently, ptychography has been used to quantify key cellular and biological events such as mitosis, cell cycle6 and angiogenesis9. In these studies, ptychographic imaging only required very low illumination power densities in the region of pW/μm2 at 635 nm, which permits the near continuous imaging of cells over many days.
Results and Discussion
Previously we demonstrated the advantages of ptychography to image commonly used immortalised cell lines6. Here we reveal a new application of ptychography for long-term imaging of complex primary neuronal cultures10, where phenotypic behaviour of microglia can be studied. Microglia are the resident macrophages of the mammalian brain where they exhibit either a surveillance role11,12 in the healthy brain or respond to neurological insults by adopting an activated state13. In many studies immortalised microglial cell lines are used to examine the function of activated microglia14. However, the use of such cell lines lacks the contextual environment within which therapeutics for treating neurological conditions would need to act to be effective in vivo. Microglia behaviour has been shown to be dynamically linked to interactions with both neurons15 and astrocytes16. Neurons in particular are able to provide on or off signals that can manipulate microglial activity15 and conversely microglia have been shown to modulate neuronal plasticity17. Here we have developed and demonstrated the potential of a novel phenotypic assay using ptychography for label-free imaging of primary microglia activity/behaviour over prolonged time periods in the presence of both astrocytes and neurons.
We began by comparing the capability of ptychography and established label-free imaging techniques to image cells in a co-culture environment. Differences between ptychographic phase and classic bright field imaging, are highlighted in Fig. 1a; although cells could be identified in all cases, brightfield (BF)/differential interference contrast (DIC) and phase contrast microscopy possess inherent problems and artefacts that undermine their suitability to image and track cells over time. Brightfield microscopy, as expected, lacked sufficient contrast to discern cellular details and cellular boundaries were not readily identified. DIC similarly struggled with the grey levels varying relatively little between different cell types. Imaging using Zernike phase contrast effectively displayed the neurites, however it suffered from the classic halo artefacts around neuronal cell soma, which can become detrimental at higher cell densities or where cells are in close proximity. Imaging of primary neuronal cultures using ptychography offered superior contrast without generating imaging and post-processing artefacts, enabling cellular identification even when cells were in direct contact in the co-cultures.

(a) Brightfield, Zernike phase contrast, DIC and ptychographic phase image showing the same field of view of primary neuronal cultures. (b) Brightfield images captured on the VL21 microscope acquired at various focal positions by moving objective in the z axis. At each focal position a ptychographic phase image was obtained and successfully refocused post-acquisition. (c) Maturation of primary hippocampal neuron cultures imaged in time-lapse starting at day 1 in-vitro. A 550 × 550 μm field of view was acquired every 6 minutes for a total period of 6 days (144 hours). Representative images show 24 hour cropped snapshots from a continuous time lapse sequence (scale bar = 100 μm, full time lapse sequence available as supplementary video S1).
Maintaining the specimen in the focal plane is a crucial requirement for successful time-lapse imaging. We tested the post-acquisition refocusing capability of ptychography by defocusing the specimen by 50, 100 and 200 μm (Fig. 1b). Brightfield images of the sample when moved 50 μm from the imaging plane are incomprehensible. Strikingly, due to its inherent post-acquisition refocusing capability, ptychographic phase images taken at the same z positions as the brightfield images maintain contrast and quality.
We next sought to test the non-invasive properties of ptychography by imaging dissociated primary hippocampal cultures in prolonged time-lapse experiments. A large (550 × 550 μm) field of view of the primary cultures starting at day one in vitro, was imaged every six minutes for six days (Fig. 1c). Over the duration of the time-lapse, maturation of these cultures can be seen clearly as the hippocampal neurons develop extensive neurite connectivity together with the proliferation of both microglia and astrocytes (supplementary video S1).
Having demonstrated the long-term label-free imaging capability of ptychography, we next investigated the extent to which individual cell types within the culture could be distinguished. To achieve this a hippocampal culture specimen was imaged using ptychography (Fig. 2a) followed by confocal microscopy of the identical field of view, where cells were fluorescently labelled with antibodies raised against microglia (Fig. 2b) or neurons (Fig. 2c). Hippocampal neurons were easily identified due to their distinct morphology in the ptychographic phase images. The cell soma were small, yet their phase shift was relatively large due to their thickness giving the cells a unique bright characteristic in the ptychographic phase images. Neurites were also easily distinguishable against the background. Conversely astrocytes were relatively thin and therefore did not appear as bright in terms of phase intensity. The morphology of the astrocytes and microglia in the culture was also very different from the hippocampal neurons; whilst the shape of the microglia was similar to that of astrocytes, they differed greatly with regards to intracellular morphology. Microglial cells have a granular cytoplasmic structure and often appear vacuolated.

(a) Ptychographic phase image of the live primary hippocampal culture was acquired prior to fixation, where microglia (green arrows), neurons (red stars) and astrocytes (yellow triangles) are easily distinguished. Following fixation correlative confocal images from the same field of view show (b) microglia labelled with Iba1, (c) hippocampal neurons labelled with β-tubulin and (d) a merged image of Iba1, β-tubulin, DAPI labelled cells overlaid with the ptychographic phase image. The remaining unlabelled population of cells represent astrocytes in the culture. (e) Segmentation of the individual cell types (microglia-green, neurons-red and astrocytes-yellow). (f) Standard deviation of pixel intensity, (g) mean intensity and (h) and the relationship between the two parameters for each segmented cell (one way ANOVA with Tukey’s post-hoc test, ****p < 0.0001, ***p < 0.001, **p < 0.01). (i) Ptychographic phase images from a time-lapse sequence clearly showing the phagocytic activity of microglia (indicated by a green arrow) towards an apoptotic neuron (indicated by an asterisk). Healthy neurons are indicated by a red star. Further examples of phagocytic activity in supplementary video S2.
Quantitative analysis was then undertaken on the segmented cells of the ptychographic phase image shown in Fig. 2e. Here we measured the standard deviation of pixel intensity and the mean intensity within each cell. Microglia, astrocytes and neurons were individually distinguished based on these two parameters (Fig. 2e–h) where the individual cell types had a clear subset of characteristics. Microglia displayed a significantly higher standard deviation of pixel intensity (4.55 × 103 ± 361) than astrocytes (1.20 × 103 ± 117), although their mean pixel intensity was similar (10.6 × 103 ± 407, 8.89 × 103 ± 274, respectively). This likely reflects the differences in intracellular granularity between the cell types. Neurons on the other hand displayed a significantly higher mean intensity (17.0 × 103 ± 426) than either microglia (10.6 × 103 ± 407) or astrocytes (8.89 × 103 ± 274).
Furthermore by viewing the culture in time-lapse the individual cells types could be identified due to their motion and behaviour. Microglia cells are very motile in comparison to astrocytes and also exhibited characteristic phagocytic activity (Fig. 2i and supplementary video S2). This ability to distinguish the individual cells without labels provides a platform to assess microglial cell behaviour and activity. The dynamic interplay that exists between the three cell types15,16,17 clearly identifies a requirement to study them in co-cultures without the introduction of labels that have the potential of perturbing the underlying cell behaviours.
To exploit the ability of long-term imaging together with the capability to identify individual cell types within neuronal cultures, we assessed biologically relevant aspects of microglial behaviour and its pharmacological modulation. Cyclosporine A (CsA) has previously been reported to reduce microglia activation and activity using biochemical and immuno-fluorescent assays18,19. Here we investigated the effects of CsA on phenotypic traits of microglia in vitro, without the addition of any fluorescent labels and therefore monitoring their activity in an un-perturbed system.
Following application of 1 μM CsA, microglia motility was tracked in the time-lapse videos over a 72 hour period. The effects of CsA on microglial speed, Euclidean distance and meandering index was calculated for each 24-hour time-window over the 72 hours of continuous imaging. Microglia tracks are presented as migration plots from their point of origin at each time window (Fig. 3a). The application of 1 μM CsA significantly reduced microglia speed by 32.3%, 20.7% and 27.2% at the time points of 0–24, 24–48 and 48–72 hours, respectively when compared to controls (Fig. 3c,f). However, CsA had no effect on the Euclidean distance (Fig. 3d,f) or meandering index (Fig. 3e,f). Moreover, cultures treated with CsA for 72 hours (Fig. 3b) displayed evidence of more cellular debris and the neuronal processes were less pronounced. Taken together, our data indicate that CsA reduces microglia speed without affecting the size of the domain within which they migrate. This reduction in speed defines a novel phenotypic trait of microglial behaviour that is influenced by CsA; this is in addition to the already established effects of CsA on cytokine release19. Such behavioural traits could be used for low or medium throughput screens for candidate compounds that could be used to treat neurological conditions14.

(a) Activity of microglia from 3 independent rodent neuronal cultures was measured over a period of 72 hours following treatment with 1 μM cyclosporine A and compared to control conditions. Tracks of individual microglia are plotted from their point of origin (μm) in each 24 hour window over the 72 hour period. Tracks in black represent microglia that possess a mean speed that is less than 0.5 μm/min whilst those in green have a mean speed greater than 0.5 μm/min. (b) Representative images at each 24 hour time point show the development and the maturation of both control and cyclosporine A treated neuronal cultures (full time-lapse videos available as supplementary videos S3 and S4). Quantitative parameters relating to the microglia activity such as; (c) speed; (d) Euclidean distance and (e) meandering index were calculated for multiple microglia from both the control and cyclosporine A treated neuronal cultures during each time period. (f) These values are summarised in a table form (Mann-Whitney non-parametric test of significance, **p < 0.01, *p < 0.05).
Cells in culture and in particular co-culture are heterogeneous in nature, and averaging these populations in assays undermines the properties of subpopulations20,21. Imaging studies are often limited to end-point assays that remove critical behavioural information. Here we have demonstrated the use of ptychography for prolonged time-lapse imaging of multiple cell types in co-cultured environments, revealing behavioural and phenotypic information of microglia.
The use of ptychography in the present study offers significant advantages over conventional label-free imaging techniques such as Zernike phase contrast microscopy. The superior contrast alone enables greater confidence and robustness in tracking the microglia, especially in confluent complex cell cultures (Fig. 1a). Ptychography also has the inherent ability to re-focus images post acquisition, without the need for additional autofocusing hardware (Fig. 1b). This capability is crucial to overcome focal drift when performing time-lapse imaging studies. Furthermore, ptychography offers quantitative outputs that relate to physical properties of the cell itself (Fig. 2e–h), thus demonstrating the future applicability of this type of imaging to quantitatively differentiate between multiple cell types. Taken together, these features are a notable advantage over conventional label-free imaging.
We anticipate that the label-free, non-invasive and quantitative features of ptychography will be widely applicable across many live-cell imaging studies providing subtle behavioural and phenotypic information that has not before been studied.
Methods
Primary hippocampal cell culture and preparation
Mixed cultures of hippocampal neurons, astrocytes and microglia were prepared from new born Wistar rats as described in Belfield et al. (2006)10 except that the growth medium was Neurobasal medium (Invitrogen, Carlsbad, CA) containing 2% B27 (Invitrogen), 5% foetal calf serum (PAA Lab, Pasching, Austria), 1 mM L-glutamine, 35 mM glucose (Sigma, St. Louis, MO), 100 U/ml of penicillin, and 0.1 mg/ml streptomycin (Invitrogen).
Cells were plated in poly-l-lysine treated 8 well Lab-Tek II chambered coverslips (Nalge Nunc Int, Rochester, NY). Immediately prior to imaging the wells were filled with conditioned media and sealed with a coverslip (22 × 50 mm, no. 1.5, SLS, Hessle, UK) and high vacuum grease (Dow Corning Corp, Midland, MI). This eliminated the meniscus and provided an airtight seal so that only the temperature was to be maintained.
Ptychographic phase imaging
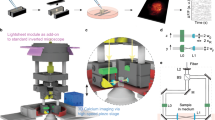
Ptychographic phase images were acquired as described previously6, however in the current study we used the inverted VL21 microscope (Phasefocus, Sheffield, UK). Briefly the specimen was moved in the x-y plane through the 635 nm diode laser illumination. The resulting diffraction pattern was collected using a 16-bit, 1megapixel Pike AVT (F-100B) CCD camera set in the detector plane. An Olympus LMPlanFLN 20x/0.40 objective was used to collect the exit wave leaving the specimen.
Time-lapse imaging was performed by placing the chamber slide on the VL21 microscope surrounded by a Solent Scientific environmental chamber (Solent Scientific Limited, UK) maintaining the cells and the microscope at 37 °C. A 22 × 22 tile scan was selected that generated 484 overlapping diffraction patterns through movement of the x-y stage, with an acquisition time of approximately 90 s. Images were acquired in this manner by sequentially visiting 4 wells of the chamber slide with a time of 6 minutes between imaging the same well. Image reconstruction was performed post acquisition using the extended Ptychographic Iterative Engine (ePIE) algorithm (The Phase Focus Virtual Lens®, Phasefocus, UK, www.phasefocus.com). Time-lapse video sequences and 16-bit Tiff images were created using the Cell Analysis Toolbox (CAT) software package (Phasefocus, UK, www.phasefocus.com).
Correlative DIC and Zernike phase contrast images were acquired using Zeiss LSM 510 confocal microscope equipped with a Zeiss AxioCam HRm CCD camera and Axiovision software.
Correlative confocal microscopy
Neurons were fixed in 3% paraformaldehyde in PBS supplemented with 4% sucrose and processed for immunocytochemistry as described in Belfield et al. (2006). The following primary antibodies were used at the indicated dilutions. Anti-Iba1 rabbit polyclonal antibody (Wako Chemicals, USA) was used at 1:200 to detect microglial cells. Anti-β-tubulin III (TuJ1) mouse monoclonal antibody (Sigma, St Louis, MO) was used at 1:500 to detect neurons. Anti-GFAP raised in guinea pig (Advanced Immunochemical, Long Beach, CA) was used at 1:200 to detect astrocytes. Images were acquired using a Zeiss LSM 780 confocal microscope.
Quantitative analysis of ptychographic phase images
Ptychographic phase images were imported into ImageJ(Fiji), where manual segmentation was performed. The standard deviation of pixel intensity and mean intensity were measured for each segmented cell.
Cell tracking and analysis
Time-lapse videos were generated using the CAT software (Phasefocus, UK) and loaded in to ImageJ(Fiji). The MtrackJ plugin was then used to track individual microglia over the 72 hour time-lapse. Quantitative cell motility parameters such as speed, meandering index and Euclidean distance were measured. Briefly the mean speed was calculated by dividing the total distance travelled by the microglia by the total time of the track. The meandering index was calculated as the final displacement of the microglia from the point of origin divided by the total distance travelled by the microglia. The Euclidean distance was calculated as the final displacement in a straight line from the point of origin of the microglia. The data was presented as rose-plots by importing the tracking co-ordinates into the Chemotaxis tool software (Ibidi GmbH, Germany).
Statistical analysis
Data are represented as mean values ± SEM. Statistical significance was determined using one way ANOVA with Tukey’s post-hoc test or Mann-Whitney non-parametric test of significance, where appropriate. All statistical analysis was performed using Graphpad Prism Version 6.0.
Additional Information
How to cite this article: Suman, R. et al. Label-free imaging to study phenotypic behavioural traits of cells in complex co-cultures. Sci. Rep. 6, 22032; doi: 10.1038/srep22032 (2016).
Change history
24 March 2016
The HTML version of this paper was updated shortly after publication, following a technical error that resulted in the link to the Supplementary Videos being omitted. This has now been corrected; the PDF version of the paper was correct from the time of publication.
References
Song, Y. et al. Stem cell traits in long-term co-culture revealed by time-lapse imaging. Leukemia 24, 153–161 (2009).
Dixit, R. & Cyr, R. Cell damage and reactive oxygen species production induced by fluorescence microscopy: effect on mitosis and guidelines for non-invasive fluorescence microscopy. Plant. J. 36, 280–290 (2003).
Maiden, A. M. & Rodenburg, J. M. An improved ptychographical phase retrieval algorithm for diffractive imaging. Ultramicroscopy 109, 1256–1262 (2009).
Kemper, B. et al. Label-free quantitative cell division monitoring of endothelial cells by digital holographic microscopy. J. Biom. Opt. 15, 036009-036009-6 (2010).
Greenbaum, A. et al. Imaging without lenses: achievements and remaining challenges of wide-field on-chip microscopy. Nat. Meth. 9, 889–895 (2012).
Marrison, J., Raty, L., Marriott, P. & O’Toole, P. Ptychography-a label free, high-contrast imaging technique for live cells using quantitative phase information. Sci. Rep. 3, 2369 (2013).
Godden, T. M., Suman, R., Humphry, M. J., Rodenburg, J. M. & Maiden, A. M. Ptychographic microscope for three-dimensional imaging. Opt. Express 22, 12513–12523 (2014).
Mir, M. et al. Label-Free Characterization of Emerging Human Neuronal Networks. Sci. Rep. 4 (2014).
Roberts, A. C. et al. Aberrant phenotype in human endothelial cells of diabetic origin: implications for saphenous vein graft failure? J. Diabetes. Res. 2015, 409432 (2015).
Belfield, J. L., Whittaker, C., Cader, M. Z. & Chawla, S. Differential Effects of Ca2+ and cAMP on Transcription Mediated by MEF2D and cAMP-response Element-binding Protein in Hippocampal Neurons. J. Biol. Chem. 281, 27724–27732 (2006).
Nimmerjahn, A., Kirchhoff, F. & Helmchen, F. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo . Science 308, 1314–1318 (2005).
Paolicelli, R. C. et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 333, 1456–1458 (2011).
Hanisch, U.-K. & Kettenmann, H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10, 1387–1394 (2007).
Figuera-Losada, M., Rojas, C. & Slusher, B. S. Inhibition of Microglia Activation as a Phenotypic Assay in Early Drug Discovery. J. Biomol. Screen. 19, 17–31 (2014).
Harrison, J. K. et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. USA 95, 10896–10901 (1998).
Pascual, O., Ben Achour, S., Rostaing, P., Triller, A. & Bessis, A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc. Natl. Acad. Sci. USA 109, E197–E205 (2012).
Ji, K., Akgul, G., Wollmuth, L. P. & Tsirka, S. E. Microglia Actively Regulate the Number of Functional Synapses. PLoS ONE 8, e56293 (2013).
Gabrusiewicz, K. et al. Characteristics of the Alternative Phenotype of Microglia/Macrophages and its Modulation in Experimental Gliomas. PLoS ONE 6, e23902 (2011).
Tribouillard-Tanvier, D., Carroll, J. A., Moore, R. A., Striebel, J. F. & Chesebro, B. Role of Cyclophilin A from Brains of Prion-infected Mice in Stimulation of Cytokine Release by Microglia and Astroglia in Vitro . J. Biol. Chem. 287, 4628–4639 (2012).
Spiller, D. G., Wood, C. D., Rand, D. A. & White, M. R. H. Measurement of single-cell dynamics. Nature 465, 736–745 (2010).
Huang, S. Non-genetic heterogeneity of cells in development: more than just noise. Development 136, 3853–3862 (2009).
Acknowledgements
We thank Gareth Evans, Jo Marrison and Ian Morrison for helpful insight and practical expertise to resolve experimental issues. This work was supported by Innovate UK and EPSRC in the form of a Knowledge Transfer Partnership (KTP No. 009111) between Phasefocus and The University of York, and pump priming funds from the Biological Physical Sciences Institute (BPSI), University of York.
Author information
Authors and Affiliations
Contributions
P.O.T., R.S., M.C. and S.C. conceived and designed the study. R.S., S.C., G.S., K.E.A.H. and R.K. performed experiments and analysed data. R.S. wrote the manuscript with comments from all authors.
Corresponding authors
Ethics declarations
Competing interests
Yes there are potential competing financial interests. RS performed experiments and wrote the manuscript as a Knowledge Transfer Partnership (KTP) Associate (Innovate UK) that was part funded by Phasefocus Ltd. R.S. and R.K. are now employees of Phasefocus Ltd. P.O.T., S.C., M.C., G.S. and K.E.A.H. declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Suman, R., Smith, G., Hazel, K. et al. Label-free imaging to study phenotypic behavioural traits of cells in complex co-cultures. Sci Rep 6, 22032 (2016). https://doi.org/10.1038/srep22032
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep22032
This article is cited by
-
The CellPhe toolkit for cell phenotyping using time-lapse imaging and pattern recognition
Nature Communications (2023)
-
Metastatic breast cancer cells induce altered microglial morphology and electrical excitability in vivo
Journal of Neuroinflammation (2020)
-
Quadriwave lateral shearing interferometric microscopy with wideband sensitivity enhancement for quantitative phase imaging in real time
Scientific Reports (2017)
-
Classification of M1/M2-polarized human macrophages by label-free hyperspectral reflectance confocal microscopy and multivariate analysis
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
