
Abstract
True flies are insects of the order Diptera and encompass one of the most diverse groups of animals on Earth. Within dipterans, Schizophora represents a recent radiation of insects that was used as a model to develop a pipeline for generating complete mitogenomes using various sequencing platforms and strategies. 91 mitogenomes from 32 different species were sequenced and assembled with high fidelity, using amplicon, whole genome shotgun or single molecule sequencing approaches. Based on the novel mitogenomes, we estimate the origin of Schizophora within the Cretaceous-Paleogene (K-Pg) boundary, about 68.3 Ma. Detailed analyses of the blowfly family (Calliphoridae) place its origin at 22 Ma, concomitant with the radiation of grazing mammals. The emergence of ectoparasitism within calliphorids was dated 6.95 Ma for the screwworm fly and 2.3 Ma for the Australian sheep blowfly. Varying population histories were observed for the blowfly Chrysomya megacephala and the housefly Musca domestica samples in our dataset. Whereas blowflies (n = 50) appear to have undergone selective sweeps and/or severe bottlenecks in the New World, houseflies (n = 14) display variation among populations from different zoogeographical zones and low levels of gene flow. The reported high-throughput mitogenomics approach for insects enables new insights into schizophoran diversity and population history of flies.
Similar content being viewed by others

Introduction
True flies are insects that undergo complete metamorphosis and belong to the two-winged insects order Diptera. They represent one of the most diverse groups in the Kingdom Animalia1 and are structured into two major sub-orders: Lower Diptera (mosquitoes) and Brachycera (short-horned flies). Worldwide, more than 150,000 species have been described2, making them one of the most successful groups on Earth that occupy almost every terrestrial niche. They parasitize plants and animals, act as biological and mechanical vectors of diseases, serve as biological control agents, as well as model organisms for science3. The clade Schizophora contains the majority of the family level diversity3,4 among Dipterans and represents a recent rapid radiation of lineages. The resulting relationships among Schizophora families remain a challenge for fly phylogeny2,3. The rapid radiation in combination with a low extinction rate has led to a diversity that surpasses even the number of all terrestrial species of vertebrates2,3,4. The scarcity of fossil records and the sparse availability of genetic data make Schizophora an interesting target for large-scale molecular evolutionary analyses. These analyses were historically based on mitochondrial markers consisting of single genes or short sequence intervals. More recently, complete mitochondrial genomes have become commonly used for in-depth phylogenetic and population studies5. In particular for vertebrates, generation of complete mitochondrial genomes is well established6, including those of extinct species7,8,9,10. Sequencing of complete mitochondrial genomes has been proven to refine resolution of relationships both between and within species11,12,13,14,15,16, in addition to improving the characterization of genome evolution17,18 and patterns of substitution rate19.
In contrast, the sequencing and assembly of complete mitochondrial genomes of invertebrate species has progressed at a much slower pace. This is especially true for insects, whose mitochondrial genomes have been obtained by isolation from whole cell lysates in combination with restriction fragment or shotgun sequencing. Those initial datasets served as references for the further development of universal primers17,20,21,22,23. Interestingly, even a decade after the emergence of next-generation sequencing (NGS), most invertebrate mitochondrial genomes are still based on primer walking and Sanger sequencing16,24,25,26, resulting in an underrepresentation of molecular studies on invertebrate evolution, compared to other taxa.
An alternative approach uses large amplicons (several kilobases in length) generated by long-range PCR in a shotgun approach to assemble these mitogenomes. A number of arthropods mitochondrial genomes have been successfully amplified by this method using sets of conserved primers27,28,29 and sequenced on current versions of NGS platforms. Most of the generated data has been focused on the description of a species’ mtDNA sequence30,31,32,33,34,35 and in a few instances on population studies29,36. This approach is limited to closely related species, as larger evolutionary distances often disrupt the annealing sites of the universal primers.
Studies targeting a wide range of insect diversity therefore depend on more universally applicable methods. One such approach is the whole genome shotgun (WGS) sequencing of total DNA. This approach can be applied to whole animals or specific body parts, allowing for morphological preservation and molecular analysis of the same specimen. In addition, due to the short DNA read length of NGS data, partially degraded samples may be analyzed successfully.
In order to assess the applicability of the above approaches, we generated a total of 91 complete mitochondrial genomes for 32 different species of flies from the Schizophora radiation. Mitochondrial genomes were sequenced from whole insects, body parts and ethanol-preserved specimens using three different techniques: (i) a combination of long-range PCR and shotgun sequencing; (ii) WGS sequencing of genomic DNA using Illumina short reads; and (iii) WGS sequencing of genomic DNA using long reads from single molecule real time (SMRT) sequencing. These strategies allowed us to increase the taxonomic breadth of dipteran phylogenies and calculate divergence dates for major Schizophora clades. In addition, we inferred the population structure of two important mechanical vectors of diseases in different continents, the Oriental latrine blowfly Chrysomya megacephala and the housefly Musca domestica. We show that genetic information of a wide-variety of species can be recovered, even if only small body parts or degraded DNA are available. Our results contribute to the comparative mitogenomics and population-based analyses of invertebrates and enable the generation of molecular data in an automated high-throughput fashion for insects in general.
Results
mtDNA sequencing strategies and coverage
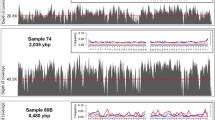
For the 32 schizophoran species sequenced, 16 mtDNAs were assembled using a combination of long-range PCR and shotgun sequencing and 16 were recovered using the WGS technique. The number of reads generated and mapped against the mtDNA reference is in Supplementary Table S1. For amplicon sequencing, the mtDNA coverage ranged from 2492-fold (Morellia lopesae) to 8830-fold (Chloroprocta idioidea; Supplementary Table S1). On average, 80.16% (±10.55) of the reads were aligned to the reference. As expected, the strategy using WGS provided a lower percentage of the total reads aligned to the reference (1.43% ± 1.22), since the nuclear genome and the metagenome are also generated. However, mtDNA-assigned reads were sufficient to generate high coverage assemblies, ranging from 12-fold (Muscina levida) to 13,169-fold (Phormia regina; Supplementary Table S1), dependent on the total number of reads generated for each library. The WGS using SMRT platform yielded a total of 72,445 pre-assembled reads (p-reads) that contain the full mtDNA sequence of the species C. megacephala. This strategy generated a mitogenome with 21,488-fold coverage (sample F03, Supplementary Table S2), with 99.94% identity to the reference genome (NC_019633.126). Also, the PacBio SMRT platform was the only technique that allowed for the complete recovery of mtDNA, including the full control region (CR). The mitochondrial coverage of short-read assemblies is shown in Fig. 1A,B for all 32 schizophoran species and the assembly with long reads is in Fig. 1C. The coverage of remaining samples used for population analyses is in Supplementary Fig. S1 and in Supplementary Table S2.

Coverage plots.
(A) Coverage of mitogenomes from 16 species of Schizophora generated by long-range PCR and shotgun sequencing. Highlighted regions indicate an overlap (green) and the 16S gap (orange) region between the two amplicons. Gaps in 16S sequence were further closed through standard PCR and Sanger sequencing. (B) 16 mitogenomes assembled from short reads generated by whole genome sequencing. Both strategies used short reads from MiSeq or HiSeq Illumina platforms to generate high-quality assemblies. (C) Coverage plot of the complete mtDNA assembled with long reads generated with SMRT sequencing technology. The scheme shows the complete mtDNA of C. megacephala (sample F03) assembled with 15,835 bp. First track shows the low GC content (23.5%). Orange bars on the second track refer to the coverage. The innermost track shows gene order in each mtDNA strand. Yellow arrows denote PCGs, green arrows show rRNA subunits and orange arrows refer to tRNAs.
Phylogenetic analyses
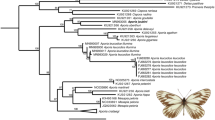
A total of 48 mitochondrial genomes, comprising 13 families and 8 superfamilies of Schizophora (see Supplementary Table S3 for full phylogenetic dataset and Fig. S2 for a map with sampling sites) were used for phylogenetic inferences under maximum likelihood (ML) and Bayesian inference (BI) methods. Phylogenetic trees inferred under different methods and partitioning strategies were concordant, with a few exceptions regarding the relationships among some Acalyptratae lineages. Although a major topological difference was not observed among analyses, differences in Bayes factor model comparisons were statistically significant (>200 in likelihood values; Supplementary Table S4), always favoring more complex partitioning strategies. The profiles of intra and inter-familial genetic distances (Supplementary Fig. S3) suggest different evolutionary rates among protein coding genes (PCGs), justifying the use of more complex strategies. The topology inferred using the Bayes Factor-favored strategy (mtDNA11PF) was the most recovered topology in our analyses and is shown in Fig. 2.

Phylogenetic tree inferred for the Schizophora clade.
Phylogenetic trees were generated for the 48 mitochondrial genomes analysed through the favoured partition scheme mtDNA11PF (see Methods and Supplementary Table S4 for details) under BI and ML methods. The ML tree topology is identical to the BI tree (shown) with exception of relationships among some Acalyptratae lineages. Values at branches refer to node supports of BI posterior probabilities and ML bootstrap proportions among 1000 replicates, respectively. Bootstrap supports below 50 were omitted (−). In red we highlight the new mitogenomes sequenced in this work.
Schizophora was recovered as a monophyletic group, containing the two subsections Acalyptratae and Calyptratae. Both subsections were traditionally classified based on the size of the lower calypter as a synapomorphy, which showed to be unreliable. The Acalyptratae is a large subsection that lacks a comprehensive phylogenetic analyses and show contradictory synapormorphies3,37. The Drosophilidae family (superfamily Ephydroidea) was recovered as the sister-clade of Calyptratae in both BI and ML inferences, although modestly supported by bootstrap values (Posterior Probability [PP] = 0.99 and bootstrap [BS] = 65, Fig. 2). A monophyletic Calyptratae clade (PP = 1.0, BS = 100) was nested inside the paraphyletic Acalyptratae grade.
Calyptratae is one of the most diverse and successful fly groups37, classically divided into three superfamilies: Hippoboscoidea, Muscoidea and Oestroidea, all sampled in our analyses. Within Calyptratae, the Hippoboscoidea Glossina morsitans was recovered as the sister-taxon of a clade composed of a paraphyletic Muscoidea grade and a monophyletic Oestroidea, with the Muscoidea clade (Anthomyiidae + Scathophagidae) as sister group of Oestroidea (PP = 1.0, BS = 80). These relationships are in accordance with recently published molecular phylogenies based on combined datasets of nuclear and mitochondrial genes2,38,39. The monophyly of Calyptratae is well supported in literature3,4, but its position within the main Schizophora clade remains controversial. The search for the sister-taxon of Calyptratae shows a close relationship with both the Acalyptratae superfamilies Ephydroidea (including Drosophilidae) and Tephritoidea (including Tephritidae)2,40, as also observed in this study. A complete split between Calyptratae and Acalyptratae, both being monophyletic, has also been proposed4 but little support for this hypothesis has been found.
Oestroidea interfamilial relationships were also consistent among BI and ML analyses, with a monophyletic core-Calliphoridae (sensu38,39,41), excluding Mesembrinellinae (PP = 1.0, BS = 100). This group was recovered as sister-group of a clade composed of (Sarcophagidae + (Oestridae + (Tachinidae + Mesembrinella sp.))) (PP = 0.99, BS = 66). In literature, Oestroidea interfamilial relationships have been contentious, with few agreements among different studies4,38,39,41,42,43,44. The exclusion of Mesembrinella sp. from the core-Calliphoridae lineage and its placement as sister group of Tachinidae (PP = 1.0, BS = 75) is particularly interesting, as recurrent studies show support for the creation of a distinct family named Mesembrinellidae within Oestroidea. Our results support this taxonomical revision and the creation of Mesembrinellidae, comprising Mesembrinella and other small related genera of the subfamily Mesembrinellinae39,45.
Estimation of divergence time
The Bayesian uncorrelated relaxed clock46 was used to estimate the divergence timescale of the Schizophora. Fossil constraints related to the radiation of Schizophora (S), Oestroidea (O) and the origin of Anthomyiidae (A) were included for molecular clock calibration. Our results (Fig. 3) place the origin of the Schizophora clade within the Late Cretaceous to Paleogene periods (known as K-Pg boundary), about 68.3 Ma (95% credibility interval [CI]: 65.73–70.86 Ma).

mtDNA time tree of Schizophora.
Divergence timescale for the Schizophora clade inferred under Bayesian uncorrelated relaxed clock method from 48 complete mitochondrial genomes. Node values indicate mean estimated divergence times in million years (Ma) and bars indicate 95% credibility intervals. Calibration points for Schizophora crown (S), Anthomyiidae stem (A) and Oestroidea crown (O) fossil constraints are shown in red circles. Pliocene and Quaternary at geological time scale are shown as “Pli.” and “Q.”, respectively.
Schizophora is usually split into two major groups, namely Acalyptratae and Calyptratae. Controversial relationships within the Acalyptratae clade are often reported because of its extreme diversity. The divergence estimation obtained for this group shows that all sampled families have their last common ancestor in the early Paleogene, around 62.0 Ma. This timescale is corroborated by the earliest Agromyzidae specimen found in Baltic amber deposits dated to 64 Ma47,48. Within the family Drosophilidae, the split of obscura and melanogaster groups was dated back to the Oligocene (~24.4 Ma, CI: 14.94–34.76 Ma), after the divergence of Drosophila and Sophophora subgenus in the late Eocene (~34.7 Ma, CI: 23.34–45.43 Ma). The divergence times inferred for the family Drosophilidae are similar to those based on the complete genomes of 12 Drosophila species, which estimated that Drosophila and Sophophora split 40 Ma and obscura and melanogaster groups were split about 26 Ma49.
For the Calyptratae subsection, the most recent common ancestor was inferred to exist in the early Eocene, about 50 Ma (95% CI: 44.95–55.22 Ma). Within Muscoidea, the divergence of Muscidae and Anthomyiidae families was dated to 43.7 Ma (95% CI: 41.0–46.8 Ma). Main radiation of Muscidae was estimated to have occurred about 39.41 Ma (95% CI: 32.5–44.54 Ma), followed by the emergence of the Muscinae subfamily in the late Eocene to early Miocene, about 29.48 Ma (95% CI: 20.3–36.9 Ma). These inferences support recent findings, suggesting that Muscidae radiation is more recent than previous estimations and probably took place near the final stages of the Gondwana breakup50.
The Oestroidea radiation is nested in a paraphyletic Muscoidea grade and was estimated to have diverged in the late Eocene, about 37.65 Ma (95% CI: 35–40 Ma). Those estimations are in agreement with Wiegmann et al.2, who used nuclear and mitochondrial genes from eleven Oestroidea species. However, our estimations and tree topology differ from Zhao et al.51, who used complete mitochondrial genomes from eight Oestroidea species and could not recover their monophyly in ML analyses. The present study used a broader diversity of twenty-five Oestroidea species, including its main families. In particular, the inclusion of a Mesembrinellinae sample was crucial to shed light on controversial phylogenetic relationships within the Oestroidea superfamily, which has been the subject of taxonomical debate38,39,52. Our results support the placement of Mesembrinellinae in a different family, distinct from the core-Calliphoridae (comprised of the subfamilies Calliphorinae, Chrysomyinae, Lucillinae, Toxotarsinae and Melanomyinae41), as proposed by Marinho et al.39 and Guimarães45. Moreover, our results suggest that the diversification of Calliphoridae took place 10–15 million years after the divergence of Mesembrinella sp. (Fig. 3).
The family Calliphoridae (sensu lato) encompasses ~1500 species commonly known as blowflies. The larvae of most calliforids have saprophagous habits, thus playing an important role in recycling organic matter. This family also includes specific parasites of mammals and birds that cause larval infestations known as myiasis53. Obligate parasites feed on live tissues of cattle and warm blooded mammals, causing losses to the agropecuary sector54. The Calliphoridae family is a particular challenge for Oestroidea relationships, since its composition and monophyletic status has been controversial. Our analyses place the last common ancestor of the core-Calliphoridae at the geological border of Oligocene and Miocene (~22.4 Ma), followed by the rapid radiation of the subfamily Chrysomyinae (~17.74 Ma) and Calliphorinae + Luciliinae sister-lineages (~16.32 Ma). These results are similar to those estimated for the carrion-breeding blowflies from Australia55.
Population analysis
We evaluated the population diversity and structure of two species in urban centers on different continents: the Oriental latrine blowfly Chrysomya megacephala (Fabricius, 1974) and the housefly Musca domestica (Linnaeus, 1758). Both species are among the most abundant and important insects around the world and are widely distributed56. Because of their mobility and association with human habits, they can act as mechanical vectors of diseases by transporting microorganisms from feces, garbage and carcasses to humans and animals53. The population analyses of blowflies and houseflies were based on samples collected for whole genome metagenomic studies in urban and rural areas of South and North America, Australia and Singapore (see Supplementary Table S5 and Fig. S2 for location and map). The increasing number of metagenomic studies in insects generates the full microbiome of individuals, but mitogenomes also can be successfully assembled at deep coverage (Supplementary Table S2). In this work, metagenomic datasets generated for 64 specimens of blowflies and houseflies were used as a model to advance mitogenomics of insects at population level.
The analyses of 50 mitogenomes of C. megacephala revealed a low genetic diversity (Fig. 4), with a total of 22 haplotypes and only 34 variable sites (1 indel in non-coding region) along 14,852 sites analyzed. The mean number of pairwise differences over all sequences was 2.87 ± 2.0, while the average nucleotide diversity (p-distance) was 0.00019 ± 0.0001. Samples from Brazil (BR; n = 44), Singapore (SG; n = 3), Australia (AU; n = 2) and India (IN; n = 1) were grouped and the p-distance within groups was 0.00027 for SG, 0.00018 for BR and p = 0.00013 for AU. The average p-distance between groups (p = 0.00031 ± 0.0001) was larger than within groups, but overlapping values between maximum intra-group and minimum inter-group distance is noticeable. The average number of pairwise differences between and within populations, as well as Nei’s distance (d) is shown on Supplementary Fig. S4. The fixation index (FST) is low among the Australian, Singaporean and Brazilian sub-groups ranging from 0.03 to 0.15, indicating admixture among these populations. The Indian sub-group showed isolation from others, but was removed from the pairwise FST analyses due to low sampling.

Blowflies’ mitochondrial variation.
Alignment of complete mtDNA sequences of 50 specimens of the Oriental latrine blowfly C. megacephala (control region, shown in grey in the top linear mtDNA scheme, is excluded). Substitutions are shown as a black bar along the mtDNA for each sample. Consensus tree on the left was inferred through MrBayes. Mitochondrial PCGs coded in the major strand are in blue and those coded in the minor strand are in yellow. rRNA subunits are shown in green and tRNAs in orange. The direction of the arrows indicates the strand location (“+strand” to the right and “− strand” to the left).
The population analyses of 14 samples of M. domestica (Fig. 5) show a total of 13 haplotypes with 100 variable sites (3 indels in non-coding regions) out of 14,820 sites. The overall mean number of pairwise differences was 21.65 ± 17.4 and the average p-distance was 0.0015 ± 0.0012. Houseflies from Brazil (BR; n = 3), United States (US; n = 4) and Singapore (SG; n = 6) were grouped in populations, while the reference mitogenome KM20072357 could not be assigned to any location due to lack of this information in GenBank metadata. The p-distance within groups was 0.0022 for BR, 0.00013 for US and 0.0005 for SG. The average p-distance between groups was p = 0.0014 ± 0.0004. The overlapping values within and between populations are due to the sample DF68 from Brazil, which does not cluster with other Brazilian samples. The average number of pairwise differences between and within populations and the Nei’s distance (d) are shown on Supplementary Fig. S5. Together, these results suggest that the Brazilian population is the most diverse. The FST matrix in Supplementary Fig. S5 shows significant values of FST between the SG group compared to the others, which causes the Asian and American groups (except for DF68) to cluster separately. The FST matrix indicates intermediate to high levels of isolation of M. domestica, forming two major clusters (US + BR groups and SG group).

Houseflies’ mitochondrial variation.
Alignment of complete mtDNA sequences of 14 specimens of the housefly M. domestica (control region, shown in grey in the top linear mtDNA scheme, is excluded). Substitutions are shown as a black bar along the mtDNA for each sample. Consensus tree on the left was inferred through MrBayes. Mitochondrial PCGs coded in the major strand are in blue and those coded in the minor strand are in yellow. rRNA subunits are shown in green and tRNAs in orange. The direction of the arrows indicates the strand orientation (“+strand” to the right and “− strand” to the left).
Discussion
The three strategies used to obtain the complete mitogenomes of a highly diverse group of species (~80 million years of evolution) successfully yield mtDNA sequences from small parts, dried specimens and fresh samples. Although providing high coverage of the mtDNA, the PCR + WGS technique prevents the sequencing of the CR and shows a variable coverage pattern (Fig. 1), depending on the overlap of amplicons. However, this method produced sequences from small parts (wing, muscles and legs) and tiny specimens, proving to be a reliable strategy for mtDNA sequencing. The WGS technique provided a uniform depth of coverage, either with short or long reads (Fig. 1B,C). The advantage of using the short reads is the large number of sequences generated in a highly multiplexed run that currently allows for up to 96 indexed samples. Despite the large amount of DNA required as input, the SMRT platform yields do not allow for more than 10 multiplexed samples, otherwise the depth of coverage is low. However, the long-read assembly was able to generate a full-length mitogenome that included the complex CR sequence. The CR of dipterans is highly biased towards A + T (>90% content) and forms secondary structures that usually hinder its sequencing58.
In a comparative perspective, all techniques can be fully automated with liquid handling systems and analyzed with a high-throughput pipeline. The price range to generate mitogenomes with different strategies and sequencing platforms is in Supplementary Table S6. The WGS using short reads is the cheapest approach to generate nearly complete mitogenomes (USD 103), followed by the combination of long-range PCR and shotgun sequencing (USD107). The use of the SMRT platform was capable of generating full-length mtDNA, but was demonstrated to be the most expensive (USD 358). Therefore, the choice of the mitogenomic approach will largely rely on the needs for complete or near complete genomes and the resources available.
The phylogenetic and molecular dating analyses showed that the main radiation of Schizophora took place in the Paleogene period, particularly in the Paleocene and Eocene epochs, when most schizophoran families diverged. Some authors argue that this clade is more diverse today in the tropics than in other geographic zones because of climate change during the early Eocene, leading to higher temperatures59. The Paleogene period witnessed series of global changes during a cooling period with intervals of global warming that reached its peak in the Paleocene-Eocene Thermal Maximum60. The diversification of insects, in particular phytophagous and mycophagous acalyptrates, also seems to be correlated with the radiation of angiosperms59. Flowering plants dominated environments in the Late Cretaceous61 and are associated with the radiation of pollinators and herbivorous insects during the Paleogene period. The calyptrate diversity was indirectly affected by the angiosperms radiation, as is the case for parasites of phytophagous insects, such as flies from the family Tachinidae. Moreover, low extinction rates also were fundamental for the diversification and survival of Schizophora clade during the K-Pg mass extinction2.
Papavero62 hypothesized that the emergence of early mammals in the Cretaceous also affected the diversification and radiation of the Oestroidea superfamily (Calyptratae), since many lineages present a parasitic relationship with vertebrate hosts63, mainly mammals. An example is the Oestridae family that parasitizes mammals, usually exhibiting a host-specific endoparasitism. Oestrid parasites may have originated from rodent parasitism64 during the K-Pg boundary, but the major diversification of Oestridae began with the wide radiation of mammals during the Paleogene.
The best sampled group in our analyses comprises the monophyletic core of the family Calliphoridae, sensu Rognes 1997. This family contains species known as blowflies and screwworm flies that can impact human and animal health. The branch lengths and diversification of species within Calliphoridae (Figs 2 and 3) suggest that most of its diversity arose in the last 20 million years. The rise and diversification of the core-Calliphoridae in the early Miocene may be related to peculiar geological changes and evolutionary processes that created new niches to be occupied and, therefore, triggered an adaptive radiation of blowflies. The Oligocene-Miocene border was an important period for the diversification and spread of large mammals, after global warming was followed by a drought. The dry conditions led to the replacement of tropical forests with grasslands65,66 that are associated with the radiation of grazing animals. Herbivores became more common starting in the Oligocene, increasing the diversification of carnivores. In particular, artiodactyl species flourished in this grassy landscape and their dispersion became facilitated with geological changes that closed the connection of the Mediterranean Sea and Indian Ocean67, linking Africa and Eurasia. Furthermore, East Asia and North America were connected through the Bering land bridge67, allowing the dispersal of animals and plants to the New World. These conditions also may have created the perfect landscape for calliforid diversification, with an increase in organic matter widely produced by large mammals. To corroborate this hypothesis, we analyzed the pattern of diversification of the family Sarcophagidae (flesh flies), which has feeding and breeding habits similar to Calliphoridae and lays their eggs in decaying organic matter. The flesh flies show a similar pattern of diversification in our analyses with a common ancestor being placed ~23.9 Ma (CI: 16.9 – 30.57 Ma). In-depth analyses of published trees of Oestroidea flies38,39,68,69 revealed that branch lengths and genus diversification are similar between blowflies and flesh flies. Taken together, geological and biological conditions could have increased the availability of manure and organic matter that served as a trigger for the radiation of blowflies and flesh flies during the Miocene epoch. After genus diversification about 15 Ma, flies may have adapted to specific hosts in the family Calliphoridae, ultimately leading to the evolution of ectoparasitic lifestyles dated at 6.95 Ma (CI: 3.77–10.41 Ma) for the obligatory parasite C. hominivorax (screwworm fly) and 2.28 Ma (CI: 0.86–3.96 Ma) for the facultative parasite L. cuprina (Australian sheep blowfly). The ectoparasitic habit within the family Calliphoridae could have emerged in parallel, after continents drifted. This could explain the non-overlapping distribution of parasitic flies species in different continents.
The mitogenomic population analysis indicated low population subdivision (FST < 0.15) and genetic distance among C. megacephala sampled in Australia, Singapore and Brazil (Fig. 4 and Supplementary Fig. S4), suggesting a panmitic population. The average number of differences between all individuals analyzed is 2.9 (4.6 between populations), which is equivalent to the genetic diversity found in extinct animals such as the Tasmanian tiger70. C. megacephala invaded Southern Brazil in the mid-70’s and rapidly spread across South America71. Initially, the introduction was reported to be from Africa72, but a different hypothesis considered the Australasian region as the source of introduction73. Our results show that haplotypes from Singapore and Australia are found in Brazilian samples, thus suggesting that the introduction may originate from the Australasian region. However, the source of the introduction in South America is uncertain and analysis would be improved with samples from ancestral populations from Africa. The Oriental latrine fly can occupy a variety of niches and is found in diverse habitats ranging from savannas and rainforests to urban centers around the world. A previous study suggested that the synanthropic form of C. megacephala has its origin in New Guinea74. However, analyzing the variation among samples from urban areas in Sao Paulo and in the rainforest in Amazon, we could not find exclusive haplotypes in urban and natural environments. Likely, the low mtDNA diversity found in this species is caused by a severe bottleneck during introduction into the New World and subsequent spread of one or a few lineages of mtDNA carried by a few females. Alternatively, potential linkage of mtDNA and insect symbionts was also considered as a cause of the low mtDNA variation75. The Wolbachia symbiont is widely spread in insects76 and was found in all samples of C. megacephala analyzed in this work. Wolbachia can be vertically transmitted by females and affects the sex ratio of offspring through feminization, parthenogenesis, male killing and cytoplasmic incompatibility in invertebrates76. The extent of impact caused by Wolbachia in the genetic variation and structure of C. megacephala is under investigation and early results show a low genetic diversity in Wolbachia infecting different populations (data not shown). Additionally, it was previously described that infected blowflies of the species Protocalliphora sialia (Calliphoridae) carry less mtDNA diversity than uninfected blowflies, suggesting a selective sweep and linkage disequilibrium that may drive geographical and genetic structure77. If the same pattern occurs in C. megacephala, mtDNA haplotypes correlated to the initial infection of the symbiont could hitchhike, resulting in reduced mitochondrial diversity due to indirect selection rather than a bottleneck.
The housefly is a widespread species that is found on all continents and survives in temperate and tropical climates. It is reported that populations from temperate climates undergo multiple bottlenecks because of low temperatures that reduce the population size drastically78. Such bottlenecks might be the cause of the lower diversity found in the US populations than in populations in tropical regions (Brazil and Singapore), where breeding can take place year-round. The presence of different haplotypes in the New World (Neartic and Neotropical zones) is consistent with the hypothesis that M. domestica underwent multiple introductions from Paleartic regions, where the species likely originated79. Also consistent with this hypothesis, we find clustering of haplotypes into two groups, one from the Americas (US + BR samples) and another encompassing the Indomalayan zoogeographical zone, represented by SG samples. Despite the observation of clusters from different regions, M. domestica shows a low mitochondrial genetic diversity, with an average of 21.6 nucleotide differences between two individuals, but a significant population differentiation. This indicates that most genetic variation is among populations of different zoogeographical zones and that the gene flow among them is low. Genetic drift might be an explanation for this pattern, but differential sex-ratio also may influence female fitness. This could lead to the increase of eggs laid by one female genotype and enhance survivorship of the offspring80, affecting the fixation of mitochondrial lineages in different locations. Further investigations are necessary to correlate mitochondrial haplotypes with nuclear genotypes and to address ancestral populations.
Population approaches such as those shown in this work highlights the potential of mitogenomics developed from environmental sequencing projects. The evaluation of the genetic diversity in a population level proved to be reliable, at relatively low costs, from a complex mixture of reads initially generated for a metagenomic study. The development of new high-throughput pipelines to efficiently analyse molecular data from invertebrates also provides insights into species relationship, population structure and evolution of one of the most diverse groups of animals on Earth. Given the impact of flies on environmental and human health, the availability of new molecular data generated in this work will provide a phylogenetic timeframe reference for Schizophora, with estimates based on full mitogenomes and fossil records that can aid future comparative studies in Diptera.
Methods
Sampling, DNA extraction and mtDNA sequencing
Specimens of the suborder Brachycera were collected with an entomological net by sweeping or with decomposing fish as a bait to attract adults. Samples were collected in Sao Paulo and Amazonas states in Brazil, in Pennsylvania in the United States, Western Australia in Australia and in Singapore. Representative sequences for 32 species were used for phylogenetic analysis (Supplementary Tables S1 and S3), while multiple sequences of C. megacephala and M. domestica were used in population-level analyses (Supplementary Table S2 and S5) using datasets generated for a metagenomic study. Specimens were identified through morphological traits and with the cox1 gene as a DNA barcode81.
For most of the samples, two main strategies were adopted to generate mtDNA sequences from short reads: (i) coupling long-range PCR with sequencing on a MiSeq (Illumina Inc.) platform and (ii) WGS sequencing on a MiSeq or HiSeq2000 (Illumina Inc.). A third strategy was also performed using single molecule long reads generated by a PacBio RSII (Pacific Biosciences). Total genomic DNA was extracted from legs or thoracic muscle of dried and ethanol-preserved specimens using an adapted protocol for DNAzol® (Invitrogen)82 or with Spin Tissue Mini-Kit (Invitek), following manufacturer’s instructions. Frozen specimens had DNA extracted with Phenol/chlorophorm83 or using the DNeasy Blood and Tissue Kit (Qiagen), following the manufacturer’s instructions.
Long-range PCR was performed to generate two amplicons of 8 and 9.2 kb. Reactions were carried out following a protocol with optimized primers described previously28. Amplicons were purified with illustra GFX™ Purification Kit (GE Healthcare) and quantified by Qubit® (Life Technologies). Equal amounts of the two amplicons were pooled to a final amount of 1 μg for each sample. For the WGS approach, DNA was quantified by Qubit® and analyzed on an Agilent 2100 Bioanalyzer with the DNA 12000 kit (Agilent Technologies). A total of 1 μg of total DNA was used for sequencing.
Pooled amplicons and genomic DNA were sheared to 300 bp using a Covaris S220 focused-ultrasonicator (Covaris Inc.), according to Illumina’s protocol. Library construction was fully automated using SPRIworks Fragment Library System I (Beckman Coulter) with TruSeq Indexed Adapters (Illumina Inc.). Libraries were size-selected with Pippin Prep (Sage Science) and enriched according to the TruSeq kit protocol. Each library was quantified using Quant-iT™ PicoGreen® (Invitrogen) and qPCR was performed according to KAPA SYBR® FAST qPCR kit instructions (Kapa Biosystems). Equimolar amounts (1 nM) of 24 indexed libraries were pooled for multiplex sequencing. A total of 8 pmol of each pool was added to a flowcell and sequenced on a MiSeq or HiSeq 2500 (Illumina Inc.) platform using 150 × 150 bp paired-end run.
For SMRTbell™ library construction, high molecular weight DNA was quantified by Qubit® and the OD260/OD280 ratio was assessed by a Nanodrop (Thermo Scientific). A total of 10 μg of DNA (OD260/OD280 = 1.8) was pooled from siblings of the same offspring (same mtDNA lineage) and sheared to 9 and 15 kb using a Covaris S220. SMRTbell™ libraries’ yield and size distribution were measured on an Agilent 2100 Bioanalyzer using the High Sensitivity Kit chip. The libraries were fully constructed and sequenced on a PacBio RSII at the Pacific Biosciences headquarter (Menlo Park, USA).
Mitochondrial Genome Assembly and Annotation
The representative mtDNA sequences for each species were generated using a reference-assisted assembly approach. The short Illumina sequences were aligned to the mtDNA of Exorista sorbillans (GenBank accession NC_014704.1) using LASTZ84, requiring an identity of 75% or greater over at least 90% of the sequence. The sequences that aligned uniquely to the reference were input to YASRA85 to generate a consensus sequence. The resulting contigs were then refined by realigning all the short reads to them using a BWA/SAMtools86,87 pipeline with default parameters. The assembled representative sequences of C. megacephala and M. domestica were used as references for individuals of the same species. These alignments were generated using BWA with default parameters and the consensus sequences were generated using SAMtools.
The blowfly genome was assembled with Falcon v 0.2.2 and was polished with Quiver in SMRTanalysis portal v 2.3. The parameters are available upon request. The mitochondrion contig was extracted from the rest of the blowfly contigs based on its similarity with the reference NC_019633.1.
For each of the 32 species, annotation of PCGs was assisted by DOGMA88. Transfer RNAs (tRNA) were annotated based on tRNAscan-SE 1.2189,90 predictions and ribosomal RNAs (rRNA) on sequence and positional homology. For the population analyses, mtDNA sequences of the same species were aligned with MUSCLE91 and the annotation was automated using Geneious® 8.1.3. Annotated files were generated using Sequin 13.70 and submitted to NCBI.
Phylogenetic Analyses
The database MetAmigA92 was used to generate the complete mtDNA dataset used for further analyses (Supplementary Table S3). PCGs were individually aligned using MUSCLE91 in TranslatorX93 server. Alignments were back translated to nucleotide sequences and poorly aligned regions were filtered using Gblocks v. 0.9194. Alignments of the 12 S and 16 S rRNAs were conducted in ClustalX v.2.195 and secondary structures were modeled by homology, based on the structures described for D. melanogaster96, D. virilis96 and other insects97. The variable helix H2077 was predicted in silico using mfold v3.098 with default parameters. Secondary structure information was used to guide adjustments in final alignments and is shown in Supplementary Fig. S6.
Evolutionary divergences among mitochondrial genes were accessed indirectly based on the profile of intra- and inter-group (families) genetic distances, calculated using MEGA 6.0699 and analyzed with R100. Model selection for each gene (considering dsRNA, ssRNA and codon positions) were performed with Mr. AIC 1.4.4101. Different ad hoc partitioning strategies (by genome, gene or codon) were evaluated for phylogenetic inferences. Optimal combinations of partitions, as determined by PartitionFinder 1.1.1102, were also used in phylogenetic inferences. For the BI, partitioning strategies were further improved with the rRNAs secondary structure consensus generated by PHASE 2.0 package103. All partition schemes used and the best-fit model for each mitochondrial gene are shown in the Supplementary Table S7.
ML analyses were conducted using GARLI v. 2.01104 with two independent search replicates for 25 million generations and 50 individuals per generation. Node supports were evaluated with 1,000 bootstrap (BS) resampling. All phylogenetic analyses were performed on the CIPRES gateway105 using the species Ocyptamus sativus (Aschiza: Syrphoidea) as the outgroup. Bayesian analyses were carried out using MrBayes v3.2.2106. Two independent analyses were run for 50 million generations (sample frequency = 1.000), burn-in set to 25% after checking for convergence (standard deviation of split frequencies < 0.01) and effective sample size ESS ≥ 200. Remaining samples were used to generate a 50% majority-rule consensus tree and node supports were analyzed based on posterior probabilities (PP).
In order to evaluate whether the effects of different partitioning strategies were significant, the resultant BI phylogenies were compared using the Bayes Factor statistic107 (Supplementary Table S4), considering the harmonic mean of the likelihood values sampled during the stationary phase of the Markov Chain Monte Carlo (MCMC) run as an estimator for the marginal-likelihoods of the models101.
Estimation of divergence times
There is a growing evidence that substitution rate patterns of Metazoan mtDNA are time-dependent biased108. In order to overcome error estimations in recent evolutionary events of Schizophora, we used fossil constraints close to the divergence events of interest and applied a relaxed clock model that allows for substitution rates to vary among lineages, as previously proposed109. Divergence times were estimated using the Bayesian uncorrelated relaxed clock method46 implemented in BEAST v.2.1.3110. Rate variations among lineages were drawn from a log-normal distribution across branches. The Yule birth-death process was used for the tree prior. The favored BI tree was used as starting topology (not fixed) with the mtDNA11PF partition strategy. No significant changes were observed when using the favored ML tree as a starting topology. Three fossil constraints were included for calibration, comprising the radiations of the Schizophora and Oestroidea lineages and the origin of Anthomyiidae (Supplementary Methods for detailed information). A MCMC sampling size of 60 million was used with samples drawn every 1000 steps. Burn-in was set to 25% after checking for convergence and ESS ≥ 200. The maximum clade credibility tree was annotated using TreeAnnotator v.2.1.2. Three independent analyses excluding each of the fossil constraints were performed in order to evaluate their changing effects in estimation of Schizophora radiation. No significant changes were observed in this analysis.
Population Analyses
Complete mtDNA sequences for multiple samples of C. megacephala and M. domestica were aligned with mitogenomes available on GenBank26,57,111 using MUSCLE91. For C. megacephala, the analyses encompassed 50 mtDNA sequences including 14,929 sites. For M. domestica, 14,820 sites were considered within 14 mitogenomes. Samples were obtained from a WGS metagenomic dataset and used as models to develop the mitogenomic pipeline. Alignment gaps and the complete control region were removed. The nucleotide substitution model was selected based on jModelTest 2112, using the Bayesian information criterion (BIC)113. The variable sites were called using Geneious® 8.0.5. ML phylogenetic trees were generated with PHYML v.3.1114 using 1000 non-parametric bootstrap replicates under the HKY substitution model. Bayesian analyses were performed with MrBayes 3106 using the HKY substitution model with 4 heated chains during 2,000,000 generations, sub-sampling trees every 500 cycles and burn-in of 500,000. Remaining tree samples were used to generate a 50% majority-rule consensus tree.
Estimation of differences over all sequence pairs and p-distance were obtained with MEGA699. The haplotype diversity was calculated using DnaSP v5115. The inter-haplotype distance matrix for each population group and the FST matrix were inferred using Arlequin 3.5116.
Additional Information
How to cite this article: Junqueira, A. C. M. et al. Large-scale mitogenomics enables insights into Schizophora (Diptera) radiation and population diversity. Sci. Rep. 6, 21762; doi: 10.1038/srep21762 (2016).
References
Yeates, D. K. et al. Phylogeny and systematics of Diptera: two decades of progress and prospects. Zootaxa 1668, 565–590 (2007).
Wiegmann, B. M. et al. Episodic radiations in the fly tree of life. Proceedings of the National Academy of Sciences USA 108, 5690–5695 (2011).
Yeates, D. K. & Wiegmann, B. M. Congruence and controversy: toward a higher-level phylogeny of Diptera. Annu. Rev. Entomol. 44, 397–428 (1999).
McAlpine, J. F. & Wood, D. M. Manual of Nearctic Diptera. Volume 3, 1–260 (Research Branch Agriculture Canada, 1990).
Karl, S. A., Toonen, R. J., Grant, W. S. & Bowen, B. W. Common misconceptions in molecular ecology: echoes of the modern synthesis. Molecular Ecology 21, 4171–4189 (2012).
Sorenson, M. D., Ast, J. C., Dimcheff, D. E., Yuri, T. & Mindell, D. P. Primers for a PCR-based approach to mitochondrial genome sequencing in birds and other vertebrates. Mol. Phylogenet. Evol. 12, 105 (1999).
Cooper, A. et al. Complete mitochondrial genome sequences of two extinct moas clarify ratite evolution. Nature 409, 704–707 (2001).
Miller, W. et al. Sequencing the nuclear genome of the extinct woolly mammoth. Nature 456, 387–390 (2008).
Lindqvist, C. et al. Complete mitochondrial genome of a Pleistocene jawbone unveils the origin of polar bear. Proceedings of the National Academy of Sciences USA 107, 5053–5057 (2010).
Paijmans, J. L. A., Gilbert, M. T. P. & Hofreiter, M. Mitogenomic analyses from ancient DNA. Mol. Phylogenet. Evol. (2012), 10.1016/j.ympev.2012.06.002.
Lang, B. F. & Burger, G. Purification of mitochondrial and plastid DNA. Nat Protoc 2, 652–660 (2007).
Sheffield, N. C., Song, H., Cameron, S. L. & Whiting, M. F. A comparative analysis of mitochondrial genomes in Coleoptera (Arthropoda: Insecta) and genome descriptions of six new beetles. Molecular Biology and Evolution 25, 2499–2509 (2008).
Oliveira, M. T. et al. Structure and evolution of the mitochondrial genomes of Haematobia irritans and Stomoxys calcitrans: the Muscidae (Diptera: Calyptratae) perspective. Mol. Phylogenet. Evol. 48, 850–857 (2008).
Gilbert, M. T. P. et al. Whole-genome shotgun sequencing of mitochondria from ancient hair shafts. Science (New York, N.Y.) 317, 1927–1930 (2007).
Morin, P. A. et al. Complete mitochondrial genome phylogeographic analysis of killer whales (Orcinus orca) indicates multiple species. Genome Research 20, 908–916 (2010).
Cameron, S. L. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu. Rev. Entomol. 59, 95–117 (2014).
Junqueira, A. C. M. et al. The mitochondrial genome of the blowfly Chrysomya chloropyga (Diptera: Calliphoridae). Gene 339, 7–15 (2004).
Oliveira, D. C. S. G., Raychoudhury, R., Lavrov, D. V. & Werren, J. H. Rapidly evolving mitochondrial genome and directional selection in mitochondrial genes in the parasitic wasp Nasonia (Hymenoptera: Pteromalidae). Molecular Biology and Evolution 25, 2167–2180 (2008).
Shoemaker, D. D., Dyer, K. A., Ahrens, M., McAbee, K. & Jaenike, J. Decreased diversity but increased substitution rate in host mtDNA as a consequence of Wolbachia endosymbiont infection. Genetics 168, 2049–2058 (2004).
Clary, D. O. & Wolstenholme, D. R. The mitochondrial DNA molecule of Drosophila yakuba: Nucleotide sequence, gene organization and genetic code. J Mol Evol 22, 252–271 (1985).
Beard, C. B., Hamm, D. M. & Collins, F. H. The mitochondrial genome of the mosquito Anopheles gambiae: DNA sequence, genome organization and comparisons with mitochondrial sequences of other insects. Insect Molecular Biology 2, 103–124 (1993).
Crozier, R. H. & Crozier, Y. C. The mitochondrial genome of the honeybee Apis mellifera: complete sequence and genome organization. Genetics 133, 97–117 (1993).
Lessinger, A. C. et al. The mitochondrial genome of the primary screwworm fly Cochliomyia hominivorax (Diptera: Calliphoridae). Insect Molecular Biology 9, 521–529 (2000).
Simon, C., Buckley, T. R., Frati, F., Stewart, J. B. & Beckenbach, A. T. Incorporating Molecular Evolution into Phylogenetic Analysis and a New Compilation of Conserved Polymerase Chain Reaction Primers for Animal Mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 37, 545–579 (2006).
Cameron, S. L., Miller, K. B., D’Haese, C. A., Whiting, M. F. & Barker, S. C. Mitochondrial genome data alone are not enough to unambiguously resolve the relationships of Entognatha, Insecta and Crustacea sensu lato (Arthropoda). Cladistics 20, 534–557 (2004).
Nelson, L. A. et al. Beyond barcoding: A mitochondrial genomics approach to molecular phylogenetics and diagnostics of blowflies (Diptera: Calliphoridae). Gene 511, 131–142 (2012).
Roehrdanz, R. L., Degrugillier, M. E. & Black, W. C. Novel rearrangements of arthropod mitochondrial DNA detected with long-PCR: applications to arthropod phylogeny and evolution. Molecular Biology and Evolution 19, 841–849 (2002).
Barau, J. G., de Azeredo-Espin, A. M. L. & Lessinger, A. C. Conservation and versatility of a new set of primers for long-PCR amplification of complete insect mitochondrial genomes based on Haematobia irritans mtDNA sequences. Mol Ecol Notes 5, 885–887 (2005).
Bourguignon, T. et al. The evolutionary history of termites as inferred from 66 mitochondrial genomes. Molecular Biology and Evolution 32, 406–421 (2015).
Jex, A. R., Hall, R. S., Littlewood, D. T. J. & Gasser, R. B. An integrated pipeline for next-generation sequencing and annotation of mitochondrial genomes. Nucleic Acids Research 38, 522–533 (2010).
Feldmeyer, B., Hoffmeier, K. & Pfenninger, M. The complete mitochondrial genome of Radix balthica (Pulmonata, Basommatophora), obtained by low coverage shot gun next generation sequencing. Mol. Phylogenet. Evol. 57, 1329–1333 (2010).
Webb, K. M. & Rosenthal, B. M. Deep resequencing of Trichinella spiralis reveals previously un-described single nucleotide polymorphisms and intra-isolate variation within the mitochondrial genome. Infect. Genet. Evol. 10, 304–310 (2010).
Webb, K. M. & Rosenthal, B. M. Next-generation sequencing of the Trichinella murrelli mitochondrial genome allows comprehensive comparison of its divergence from the principal agent of human trichinellosis, Trichinella spiralis. Infect. Genet. Evol. 11, 116–123 (2011).
Groenenberg, D. S., Pirovano, W., Gittenberger, E. & Schilthuizen, M. The complete mitogenome of Cylindrus obtusus (Helicidae, Ariantinae) using Illumina Next Generation Sequencing. BMC Genomics 13, 114 (2012).
Mao, M., Valerio, A., Austin, A. D., Dowton, M. & Johnson, N. F. The first mitochondrial genome for the wasp superfamily Platygastroidea: the egg parasitoid Trissolcus basalis. Genome 55, 194–204 (2012).
Johansson, M., Duda, E., Sremba, A., Banks, M. & Peterson, W. Assessing population-level variation in the mitochondrial genome of Euphausia superba using 454 next-generation sequencing. Mol Biol Rep 39, 5755–5760 (2012).
Yeates, D. K. & Wiegmann, B. M. The Evolutionary Biology Of Flies. (2005).
Kutty, S. N., Pape, T., Wiegmann, B. M. & Meier, R. Molecular phylogeny of the Calyptratae (Diptera: Cyclorrhapha) with an emphasis on the superfamily Oestroidea and the position of Mystacinobiidae and McAlpine’s fly. Systematic Entomology 35, 614–635 (2010).
Marinho, M. A. T. et al. Molecular phylogenetics of Oestroidea (Diptera: Calyptratae) with emphasis on Calliphoridae: Insights into the inter-familial relationships and additional evidence for paraphyly among blowflies. Mol. Phylogenet. Evol. 65, 840–854 (2012).
Lambkin, C. L. et al. The phylogenetic relationships among infraorders and superfamilies of Diptera based on morphological evidence. Systematic Entomology 38, 164–179 (2012).
Rognes, K. The Calliphoridae (Blowflies)(Diptera: Oestroidea) are not a monophyletic group. Cladistics 13, 27–66 (1997).
Agriculture, C. D. O. Manual of Nearctic Diptera, Volume 3. (1989).
Pape, T. Phylogeny of Oestridae (Insecta: Diptera). Systematic Entomology 26, 133–171 (2001).
Singh, B. & Wells, J. D. Molecular systematics of the Calliphoridae (Diptera: Oestroidea): evidence from one mitochondrial and three nuclear genes. J. Med. Entomol. 50, 15–23 (2013).
Guimaraes, J. H. A systematic revision of the Mesembrinellidae, stat. nov. (Diptera, Cyclorrhapha). Arquivos de Zoologia 29, 1–109 (1977).
Drummond, A. J., Ho, S. Y. W., Phillips, M. J. & Rambaut, A. Relaxed phylogenetics and dating with confidence. Plos Biol 4, e88 (2006).
Tschirnhaus von, M. & Hoffeins, C. Fossil flies in Baltic amber—Insights in the diversity of Tertiary Acalyptratae (Diptera, Schizophora), with new morphological characters and a key based on 1000 collected inclusions. Denisia 26, 171–212 (2009).
Winkler, I. S., Labandeira, C. C., Wappler, T. & Wilf, P. Distinguishing Agromyzidae (Diptera) leaf mines in the fossil record: new taxa from the Paleogene of North America and Germany and their evolutionary implications. Journal of Paleontology 84, 935–954 (2010).
Clark, A. G. et al. Evolution of genes and genomes on the Drosophila phylogeny. Nature 450, 203–218 (2007).
Haseyama, K. L. F., Wiegmann, B. M., Almeida, E. A. B. & de Carvalho, C. J. B. Say goodbye to tribes in the new house fly classification: A new molecular phylogenetic analysis and an updated biogeographical narrative for the Muscidae (Diptera). Mol. Phylogenet. Evol. 89, 1–12 (2015).
Zhao, Z. et al. The mitochondrial genome of Elodia flavipalpis Aldrich (Diptera: Tachinidae) and the evolutionary timescale of Tachinid flies. PLoS ONE 8, e61814 (2013).
Singh, B., Kurahashi, H. & Wells, J. D. Molecular phylogeny of the blowfly genus Chrysomya. Med Vet Entomol 25, 126–134 (2010).
Zumpt, F. Myiasis in man and animals in the Old World: a textbook for physicians, veterinarians and zoologists. (Butterworth & Co Publishers Ltd, 1965).
Azeredo-Espin, A. M. L. & Lessinger, A. C. Genetic approaches for studying myiasis-causing flies: molecular markers and mitochondrial genomics. Genetica 126, 111–131 (2006).
Wallman, J. F., Leys, R. & Hogendoorn, K. Molecular systematics of Australian carrion-breeding blowflies (Diptera: Calliphoridae) based on mitochondrial DNA. Invertebrate Systematics 19, 1–15 (2005).
Greenberg, B. Flies and disease. Vol. I. Ecology, classification and biotic associations. … Vol I Ecology (1971).
Li, X., Wang, Y., Su, S. & Yang, D. The complete mitochondrial genomes of Musca domestica and Scathophaga stercoraria (Diptera: Muscoidea: Muscidae and Scathophagidae). Mitochondrial DNA 1–2 (2014). 10.3109/19401736.2014.953080
Duarte, G. T., De Azeredo-Espin, A. M. L. & Junqueira, A. C. M. The mitochondrial control region of blowflies (Diptera: Calliphoridae): a hot spot for mitochondrial genome rearrangements. J. Med. Entomol. 45, 667–676 (2008).
Grimaldi, D. A. & Engel, M. S. Evolution of the Insects. (Cambridge University Press, 2005).
Jardine, P. The Paleocene-Eocene Thermal Maximum. Palaeontology Online 1, 1–7 (2011).
Vajda, V. & Bercovici, A. The global vegetation pattern across the Cretaceous–Paleogene mass extinction interval: A template for other extinction events. Global and Planetary Change 122, 29–49 (2014).
Papavero, N. The world Oestridae (Diptera), mammals and continental drift. 14, (Springer Netherlands, 1977).
Stevens, J. R. The evolution of myiasis in blowflies (Calliphoridae). International Journal for Parasitology 33, 1105–1113 (2003).
Pape, T. In The oestrid flies: biology, host-parasite relationships, impact and management (eds. Colwell, D. D., Hall, M. J. R. & Scholl, P. J. ) (CABI Pub, 2006), 10.1079/9780851996844.0020.
Stebbins, G. L. Coevolution of grasses and herbivores. Annals of the Missouri Botanical Garden 68, 75–86 (1981).
Bredenkamp, G. J., Spada, F. & Kazmierczak, E. On the origin of northern and southern hemisphere grasslands. Plant Ecology 163, 209–229 (2002).
Behrensmeyer, A. K. Terrestrial Ecosystems Through Time. (University of Chicago Press, 1992).
Song, Z.-K., Wang, X.-Z. & Liang, G.-Q. Molecular Evolution and Phylogenetic Utility of the Internal Transcribed Spacer 2 (ITS2) in Calyptratae (Diptera: Brachycera). J Mol Evol 67, 448–464 (2008).
Meiklejohn, K. A., Wallman, J. F., Pape, T., Cameron, S. L. & Dowton, M. Utility of COI, CAD and morphological data for resolving relationships within the genus Sarcophaga (sensu lato) (Diptera: Sarcophagidae): a preliminary study. Mol. Phylogenet. Evol. 69, 133–141 (2013).
Miller, W. et al. Genetic diversity and population structure of the endangered marsupial Sarcophilus harrisii (Tasmanian devil). Proc. Natl. Acad. Sci. USA 108, 12348–12353 (2011).
Guimaraes, J. H., Prado, A. P. & Buralli, G. M. Dispersal and distribution of three newly introduced species of Chrysomya Robineau-Desvoidy in Brazil (Diptera, Calliphoridae). Revista Brasileira de Entomologia 23, 245–255 (1979).
Baumgartner, D. L. & Greenberg, B. The Genus Chrysomya (Diptera: Calliphoridae) in the New World. J. Med. Entomol. 21, 105–113 (1984).
Sanford, M. et al. Observations on the Oriental Latrine Fly, Chrysomya megacephala in the McFaddin National Wildlife Refuge, Sabine Pass, Texas. Southwestern Entomologist 35, 109–112 (2010).
Kurahashi, H. In Biogeography and ecology of New Guinea 42, 689–698 (Springer Netherlands, 1982).
Hurst, G. D. D. & Jiggins, F. M. Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: the effects of inherited symbionts. Proc. Biol. Sci. 272, 1525–1534 (2005).
Werren, J. H., Baldo, L. & Clark, M. E. Wolbachia: master manipulators of invertebrate biology. Nat Rev Micro 6, 741–751 (2008).
Baudry, E., Bartos, J., Emerson, K., Whitworth, T. & Werren, J. H. Wolbachia and genetic variability in the birdnest blowfly Protocalliphora sialia. Molecular Ecology 12, 1843–1854 (2003).
Marquez, J. G. & Krafsur, E. S. Gene flow among geographically diverse housefly populations (Musca domestica L.): a worldwide survey of mitochondrial diversity. J. Hered. 93, 254–259 (2002).
Skidmore, P. The Biology of the Muscidae of the World. (Springer Science & Business Media, 1985).
Carrillo, J., Danielson-François, A., Siemann, E. & Meffert, L. Male-biased sex ratio increases female egg laying and fitness in the housefly, Musca domestica. J Ethol 30, 247–254 (2011).
Hebert, P. D. N., Cywinska, A., Ball, S. L. & deWaard, J. R. Biological identifications through DNA barcodes. Proceedings of the Royal Society B: Biological Sciences 270, 313–321 (2003).
Junqueira, A. C. M., Lessinger, A. C. & Azeredo-Espin, A. M. L. Methods for the recovery of mitochondrial DNA sequences from museum specimens of myiasis-causing flies. Med Vet Entomol 16, 39–45 (2002).
Vargas, M. E. & Espin, A. M. Genetic variability in mitochondrial DNA of the screwworm, Cochliomyia hominivorax (Diptera: Calliphoridae), from Brazil. Biochem. Genet. 33, 237–256 (1995).
Harris, R. S. Improved Pairwise Alignment of Genomic DNA. (The Pennsylvania State University, 2007).
Ratan, A. Assembly algorithms for next-generation sequence data. (The Pennsylvania State University, 2009).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Wyman, S. K., Jansen, R. K. & Boore, J. L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255 (2004).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Research 25, 0955–0964 (1997).
Schattner, P., Brooks, A. N. & Lowe, T. M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Research 33, W686–9 (2005).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 32, 1792–1797 (2004).
Feijão, P. C., Neiva, L. S., de Azeredo-Espin, A. M. L. & Lessinger, A. C. AMiGA: the arthropodan mitochondrial genomes accessible database. Bioinformatics 22, 902–903 (2006).
Abascal, F., Zardoya, R. & Telford, M. J. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Research 38, W7–W13 (2010).
Talavera, G. & Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Systematic Biology 56, 564–577 (2007).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007).
Cannone, J. J. et al. The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron and other RNAs. BMC Bioinformatics 3, 2 (2002).
Buckley, T. R., Simon, C., Flook, P. K. & Misof, B. Secondary structure and conserved motifs of the frequently sequenced domains IV and V of the insect mitochondrial large subunit rRNA gene. Insect Molecular Biology 9, 565–580 (2000).
Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Research 31, 3406–3415 (2003).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Molecular Biology and Evolution 30, 2725–2729 (2013).
R. Core Team (2013). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL http://www.R-project.org/.
Nylander, J. A. A. MrAIC.pl. Program distributed by the author. (Evolutionary Biology Centre, Uppsala University, 2004).
Lanfear, R., Calcott, B., Ho, S. Y. W. & Guindon, S. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Molecular Biology and Evolution 29, 1695–1701 (2012).
Jow, H., Gowri-Shankar, V. & Guillard, B. PHASE: a software package for phylogenetics and sequence evolution. University of Manchester (2003).
Zwickl, J. D. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion. (The University of Texas at Austin, 2006).
Miller, M. A., Pfeiffer, W. & Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. 1–8 (2010).
Ronquist, F. et al. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Systematic Biology 61, 539–542 (2012).
Kass, R. E. & Raftery, A. E. Bayes Factors. Journal of the American Statistical Association 90, 773–795 (1995).
Molak, M. & Ho, S. Y. W. Prolonged decay of molecular rate estimates for metazoan mitochondrial DNA. PeerJ 3, e821 (2015).
Ho, S. & Larson, G. Molecular clocks: when times are a-changin’. Trends in Genetics 22, 79–83 (2006).
Bouckaert, R. et al. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 10, e1003537 (2014).
Stevens, J. R., West, H. & Wall, R. Mitochondrial genomes of the sheep blowfly, Lucilia sericata and the secondary blowfly, Chrysomya megacephala. Med Vet Entomol 22, 89–91 (2008).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. jModelTest 2: more models, new heuristics and parallel computing. Nature Methods 9, 772–772 (2012).
Schwarz, G. Estimating the dimension of a model. The annals of statistics 6, 461–464 (1978).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Systematic Biology 59, 307–321 (2010).
Librado, P. & Rozas, J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009).
Excoffier, L. & Lischer, H. E. L. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources 10, 564–567 (2010).
Acknowledgements
The authors would like to thank Vera C. Silva and Dalton S. Amorim for the help with sampling and identification of flies and Jeffrey Wells, Louis B. Klaczko and Silvio S. Nihei for providing samples. Authors are also grateful to Adeline Seah for proofreading the manuscript and to Claudia Augusta de Moraes Russo for helpful comments. This work was funded by a grant from FAPESP to A.M.L.A-E (09/51723-7). A.C.M.J, D.F.P. and M.A.T.M. were supported by a fellowship of Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP; grants 08/56769-2, 10/09961-5 and 12/23200-2, respectively).
Author information
Authors and Affiliations
Contributions
A.C.M.J., A.M.L.A.E., D.F.P. and S.C.S. designed the research and collected samples. A.C.M.J., L.P.T. and D.I.M. conducted library construction and sequencing. A.C.M.J., D.F.P., M.A.T.M., R.W.P., A.R. and S.C.S. analyzed the data. A.C.M.J. drafted the paper with the input of D.F.P., M.A.T.M., A.R. and S.C.S. All authors read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Junqueira, A., Azeredo-Espin, A., Paulo, D. et al. Large-scale mitogenomics enables insights into Schizophora (Diptera) radiation and population diversity. Sci Rep 6, 21762 (2016). https://doi.org/10.1038/srep21762
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep21762
This article is cited by
-
Comparative analysis of four complete mitogenomes from hoverfly genus Eristalinus with phylogenetic implications
Scientific Reports (2022)
-
Regulatory regions in natural transposable element insertions drive interindividual differences in response to immune challenges in Drosophila
Genome Biology (2021)
-
Disruption of the odorant coreceptor Orco impairs foraging and host finding behaviors in the New World screwworm fly
Scientific Reports (2021)
-
Comparative mitogenomics of Drosophilidae and the evolution of the Zygothrica genus group (Diptera, Drosophilidae)
Genetica (2021)
-
The genomes of a monogenic fly: views of primitive sex chromosomes
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
