
Abstract
The extreme environment of the Qinghai-Tibet Plateau (QTP) provides an ideal natural laboratory for studies on adaptive evolution. Few genome/transcriptome based studies have been conducted on how plants adapt to the environments of QTP compared to numerous studies on vertebrates. Crucihimalaya himalaica is a close relative of Arabidopsis with typical QTP distribution, and is hoped to be a new model system to study speciation and ecological adaptation in extreme environment. In this study, we de novo generated a transcriptome sequence of C. himalaica, with a total of 49,438 unigenes. Compared to five relatives, 10,487 orthogroups were shared by all six species, and 4,286 orthogroups contain putative single copy gene. Further analysis identified 487 extremely significantly positively selected genes (PSGs) in C. himalaica transcriptome. Theses PSGs were enriched in functions related to specific adaptation traits, such as response to radiation, DNA repair, nitrogen metabolism, and stabilization of membrane. These functions are responsible for the adaptation of C. himalaica to the high radiation, soil depletion and low temperature environments on QTP. Our findings indicate that C. himalaica has evolved complex strategies for adapting to the extreme environments on QTP and provide novel insights into genetic mechanisms of highland adaptation in plants.
Similar content being viewed by others

Introduction
Extreme environments provide an ideal natural laboratory for studies on adaptive evolution. The Qinghai-Tibet Plateau (QTP) is the highest (above 4,000 meters of average elevation) and largest young plateau in the world, which has low temperature, low oxygen, poor soils, and strong ultraviolet (UV) radiation environments. Understanding how organisms adapt to these various extreme environments can make a significant contribution to evolutionary ecology. Recently, research on the adaptive genetic mechanism of non-model organisms based on genome-wide scale can be conducted using the next generation sequencing technology1. Previous genome-wide studies on adaptive evolution on this region have focused mainly on humans and vertebrates2,3,4,5,6,7,8. These studies revealed that genes involved in hypoxia response, energy metabolism, and skeletal development were under positive selection and rapidly evolving. However, little genome/transcriptome-based research has been devoted to plants in this region.
Relatives of Arabidopsis thaliana are important model systems to study evolutionary ecology and comparative genomics9,10. Several studies have been conducted on the evolution of mating systems in Capsella11,12, life history form in A. lyrata and Capsella rubella13, as well as adaptation to serpentinite soils in A. lyrata14 and external climates in A. halleri15. Notably, there are other close relatives of Arabidopsis, which not only occupy distinctive niche, but also are of ecological importance. Crucihimalaya himalaica (Edgeworth) Al-Shehbaz has been known as A. rupestris Edgeworth, A. himalaica (Edgeworth) O. E. Schulz, Arabis brevicaulis Jafri, and A. brevicaulis (Jafri) Jafri16. This species is a diploid (2n = 16) with a relatively small genome size of 319 Mb17. It usually grows in rocky hillsides, sandy slopes, alpine meadows, and screes in areas around Himalayas, e.g. QTP.
Previous reviews suggested that Crucihimalaya is a specialized group in Himalayas derived from the Arabidopsis genus in very late geological history18,19. However, recent molecular phylogenetic result showed that Crucihimalaya, Capsella, Arabidopsis and several genera constitute a paraphyly20. Dated phylogenies also indicated that the genus Crucihimalaya origin in about 5.2 Mya, and that C. himalaica split from C. lasiocarpa in about 3.56 Mya20, which is in accordance with the time of QTP rapid uplift, 3.6 Mya21. Conceivably, this species must have undergone significant genetic changes to adapt to stress factors following the rapid uplift of QTP. Therefore, C. himalaica is hoped to be a new model system to study speciation and ecological adaptation in extreme environments.
Transcriptome analysis (RNA-seq) provides a rapid and effective approach to obtain massive protein-coding genes, which can be used for understanding ecological, comparative and evolutionary genomics questions for non-model organisms22,23. In this study, we performed RNA-seq to obtain most transcript sequences of C. himalaica. Subsequently, positively selected genes (PSGs) in C. himalaica related to environmental adaptation were identified by comparative genomics with closely related species whose genome have been sequenced. We aim to reveal how this Arabidopsis relative adapts to the complicated extreme environments on QTP at genome/transcriptome level.
Results
Transcriptome assembly and annotation
In this study, we generated 132.29 million clean reads and 16.52 Gb of RNA-seq data after quality filtering (Supplementary Table S1). The clean data were submitted to the NCBI Sequence Reads Archive (SRA) database (no. SRR3138110). De novo assembly of these high-quality reads generated 66,084 transcripts and 49,438 unigenes (the longest transcript in one gene) (Table 1). The unigenes are 896 bp on average with an N50 length of 1,641 bp.
There are 29,760 (60.19%) and 33,667 (68.09%) unigenes identified homologs in Nr and Nt databases on the basis of similarity, respectively. A total of 39,189 (79.26%) unigenes were successfully annotated using at least one database (a total of seven databases, see methods) with a significant match (e-value < 10−5) (Supplementary Table S2). The top species classification hits for C. himalaica in the Nr database are Capsella rubella, A. lyrata, and A. thaliana (Fig. 1A), which all belong to Brassicaceae. The e-values are very significant, which mostly close to zero (32.1%) and 0∼1e–100 (16.5%) (Fig. 1B), suggesting that most unigenes of C. himalaica have very similar homologs in above relatives.

Species classification (A) and e-value distribution (B) of the unigenes of C. himalaica annotated to NCBI Nr database.
Orthogroups identified and positive selection analysis
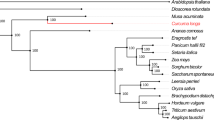
In this study, a total of 22,572 orthogroups were detected, including 152,769 genes. Among these, 10,487 orthogroups were shared by all six species, and 4,286 orthogroups contained putative one-to-one single copy genes. To further confirm the phylogenetic position of C. himalaica, we generated a phylogenetic tree based on 1,506,379 amino acid sequences of the trimmed and concatenated 4,286 single copy gene alignments from six species (Fig. 2A). The phylogenic tree showed that C. himalaica was closely related to Capsella rubella, and then clustered with the clade of A. lyrata and A. thaliana. The placement of L. stylosa was outside of these two clades with B. rapa as outgroup.

(A) Phylogenetic tree derived from concatenated all orthologs (1,506,379 amino acids) of six species. (B) Boxplots of dN/dS ratios for each species. The median dN/dS ratio and significances of the deviations using Wilcoxon rank sum test are also showed in the boxplots. (C) Number of orthologs with given dN/dS ratios for the six species.
We also estimated the substitution rates for each orthogroups by free-ratio model in PAML, which allows an independent dN/dS ratio for each branch24. For each species, the dN/dS ratio major concentrated in 0.1–0.2 (Fig. 2B,C). The median of dN/dS ratio in C. himalaica (0.1873) across all the orthologs was significantly larger than that of other relatives (0.1442–0.1572) (Fig. 2B, Wilcoxon rank sum test, p < 0.0001). The frequency distribution of dN/dS ratios evidently showed that C. himalaica (1172 genes) has more genes with elevated dN/dS ratios (dN/dS > 0.3) than others (592–815 genes) (Fig. 2C).
For 4,286 single copy orthogroups, the branch-site model of the PAML 4 package24 was used to detect genes with signals of positive selection. As a result, 1,444 genes possibly under positive selection were identified in the C. himalaica genome (ω > 1); of these genes, 598 showed significant evidence of positive selection (P-value < 0.05), and 487 were undergo extremely significant positive selection (P-value < 0.01) (Supplementary Table S3).
We also conducted KEGG (Fig. 3, Supplementary Table S4) and GO (Fig. 4, Supplementary Table S5) functional classification for these PSGs (P-value < 0.01) in C. himalaica. The distribution of KEGG classification of PSGs showed that in all annotated categories, metabolic processes have the most hits, such as amino acid metabolism (10 PSGs), cofactors and vitamins metabolism (7 PSGs), lipid metabolism (7 PSGs), and energy metabolism (5 PSGs) (Fig. 3). Beyond that, another predominant hit was DNA repair and recombination, which included 21 PSGs. It contains several DNA repair pathways, such as mismatch repair (6 PSGs), nucleotide excision repair (5 PSGs), base excision repair (3 PSGs) and homologous recombination (2 PSGs) (Supplementary Table S4).

(A) Cellular Processes; (B) Environmental Information Processing; (C) Genetic Information Processing; (D) Metabolism; (E) Organismal Systems.

The Arabic numbers show the enriched number of PSGs in each term.
In addition, GO annotation also showed that many PSGs are related to specific adaptation traits, including nitrogen compound metabolic process (150 PSGs), regulation of nitrogen metabolic (73 PSGs), response to radiation (48 PSGs), DNA repair (21 PSGs), and photoperiodism, flowering (13 PSGs) (Fig. 4). In the following paragraphs, we will discuss the important roles of these PSGs in the adaptation of C. himalaica to the various extreme environments in QTP, respectively.
Discussion
Our phylogenetic analysis based on genomic level highly supported that C. himalaica was most closed to Capsella rather than Arabidopsis. The relationship among these species agrees with previous study20. Although previous reviews suggested that Crucihimalaya is a specialized group in Himalayas derived from the Arabidopsis genus18,19. Our result indicates that it should be derived from the common ancestor of the Arabidopsis lineage and Crucihimalaya lineage. In fact, C. himalaica grouped with C. rubella, but not A. thaliana, is conflict with morphological data, especially the principal character for taxonomy (dehiscent siliques, linear fruits in Crucihimalaya & Arabidopsis vs. dehiscent silicles, obdeltoid fruits in Capsella). Maybe that is why Crucihimalaya was classified into Arabidopsis in previous taxonomic system. This conflict between the morphological and molecular result highlights the need for further studies on the intrinsic mechanism of speciation.
Orthologs are genes that have evolved from a common ancestral gene via speciation25. To investigate the selective pressures at the branch level in C. himalaica and its relatives, we estimated the substitution rates for each orthogroups. The median of dN/dS ratio in C. himalaica was significantly larger than that of other relatives, which strongly supported the accelerated evolution in C. himalaica after splitting from its ancestor lineage. Furthermore, the frequency distribution of dN/dS ratios evidently showed that C. himalaica has more genes with elevated dN/dS ratios than other five species. These accelerated evolution of genes is often driven by positive selection or relaxed selection pressure. Combined with results of branch-site model, which revealed that many functional genes in C. himalaica undergo positive selection in extreme environment of QTP, we speculated that the evolutionary rate increased in C. himalaica are due to positive selection rather than relaxation of select.
There are very high light radiation on QTP, especially the UV-B radiation during the summer (approximately 65 kJ/m2) is among the highest worldwide26. The light radiation can influence plant growth and development, such as photoperiodism, flowering, and DNA damage. Our results showed that as many as 43 PSGs in C. himalaica were annotated as response to light stimulus according to GO category, among which, 13 PSGs in the C. himalaica genome were involved in photoperiodism, flowering process (Fig. 4, Supplementary Table S5). Among these, the gene encoding early flowering 6 protein (lysine-specific demethylase, ELF6) is a repressor in the photoperiodic flowering pathway and its loss-of-function mutation causes early flowering27. Similarly, another gene encoding sensitive to freezing 6 protein (mediator of RNA polymerase II transcription subunit 16), plays an important role in regulation of the circadian clock and in the control of flowering time28. In the short growing season condition of QTP, flowering time is particularly critical and affect both the life cycles and reproductive success of alpine flora29,30. C. himalaica starts flowering relatively early (April), and has a very long duration of the flowering period (from April to September). This pattern suggests that C. himalaica has evolved specific reproductive strategies via positively selected more than a dozen genes associated with photoperiodism and flowering process as adaptation to the short growing season in the harsh environments of QTP.
In addition, highly energetic UV radiation causes direct damage to DNA, RNA, and proteins31. Our results showed that 25 PSGs were involved in response to DNA damage stimulus (Fig. 4, Supplementary Table S5). This result was consistent with 21 PSGs enriched in DNA repair pathway (Fig. 4, Table 2). As an essential system for correcting UV-induced DNA damage, nucleotide excision repair (NER) play more important roles in UV-resistance for plants32. It is worth to mention that several essential genes participating in NER process showed positive selection. Such as gene encoding DNA excision repair protein ERCC-1 (or ultraviolet hypersensitive 7), which has been reported to function as a part of a structure-specific endonuclease that cleaves 5′ to UV photoproducts in DNA33. And gene encoding DNA damage-binding protein 2, which forms a complex with DDB1 to recognize damaged DNA and initiation of NER process after exposure to UV light34. Another gene, encoding TFIIH protein, is an essential transcription initiation factor that is also pivotal for NER35. Besides NER, other DNA repair mechanism, such as base excision repair (BER), mismatch excision repair (MMR) and homologous recombination (HR) also participated in response to light radiation and other external environment stimulation in QTP (Table 2). Accordingly, we found PSGs encoding DNA repair protein XRCC4, DNA mismatch repair protein MUTS2, MUTS protein homolog 2/5 (MSH2/5), MUTL protein homolog 3 (MLH3), DNA repair and recombination protein RAD54, and DNA damage repair/toleration protein DRT111 (Supplementary Table S3). KEGG enrichment showed that these genes covered almost all aspects of the DNA repair mechanism (Supplementary Table S4), suggesting that C. himalaica has evolved an integrated DNA repair mechanism to adapt to the harsh habitat following the uplift of QTP.
Nitrogen compound metabolism is the basic physiological processes, which can generate components of cells (e.g. nucleic acids and proteins) as well as convert sugars and proteins into energy. Moreover, nitrogen compound metabolism is even more important for C. himalaica, because nutrient deficiency, especially nitrogen deficiency is a typical feature of the sandy soil habitats for its living36,37. It means nitrogen assimilation under low-nitrogen condition is also a challenge for C. himalaica living in QTP. From our results, it was evident that as many as 150 PSGs involved in nitrogen compound metabolism in C. himalaica, which included GO categories associated with DNA metabolic process (41genes) and regulation of nitrogen compound metabolic process (73 genes) (Fig. 4, Supplementary Table S5). The latter included many transcription factors involved in the nitrogen assimilation process, including the primary assimilation of ammonia to carbon skeletons to biosynthesize amino acids and other organic compounds. Remarkably, three members of DOF (DNA BINDING WITH ONE FINGER) family, a class of zinc finger domain TFs, showed significant positive selection. DOF factors could play an important role in nitrogen regulation, which was supported by the fact that Dof1 over-expressed in Arabidopsis promote the nitrogen assimilation, thus improved plant growth under low-nitrogen conditions38. Moreover, a PSG encoding GATA zinc finger protein homolog NTL10, also has been reported activates the expression of nitrogen-catabolic enzymes during conditions of nitrogen limitation39. The functions of these PSGs suggested that C. himalaica may have adaptively sped up evolutionary rates of genes involved in nitrogen metabolic regulation to better adapt to the nitrogen limitation in its habitat, which seems to be the driving force behind rapid evolution of these genes.
Low temperatures and rapid changes of temperature are also prevalent features of QTP. Organisms that live in this alpine ecosystem face various growth-related challenges from low temperatures, such as reduced fluidity of lipid membranes40. The lipid bilayers transmit external signals to its interior as well as protect the integrality of various organelle membranes in unfavorable environments. Our GO category results indicated that 22 genes related to lipid biosynthesis were under significant positive selection, including 14 PSGs genes involved in phospholipid biosynthetic process and 5 PSGs in glycerolipid biosynthetic process (Supplementary Table S3). Among these, gene encoding lipid-A-disaccharide synthase with function of condense the lipid A, could play a structural role by stabilizing the plasma membrane lipid bilayer in plants, and involved in signal transduction or plant defense responses41. Other PSGs, such as gene encoding Phosphatidyl-N-methylethanolamine N-methyltransferase, was reported to produce the abundant membrane lipid phosphatidylcholine42. Previous findings also showed that the degree of unsaturated lipids remains unchanged in C. himalaica under rapid temperature changes43, suggesting that the remodeling of membrane lipids might protect membranes against frequent temperature changes. Based on above results, we speculate that these membranes lipids and proteins might play major roles in maintaining membrane integrity under low temperatures and rapid changes of temperature conditions.
Conclusion
Organisms that live in QTP face a variety of external stress from their harsh environments. As such, it is often believed that these organisms have undergone a series of adaptive evolutionary changes. Low oxygen was reported as the most challenge for animals living in high-altitude, thus several genes response to hypoxia showed signature of positive selection in different species2,3,4,5,6,7,8. Unlike animals, plants as sessile organisms are constantly exposed to these stresses in QTP. It is conceivable that they have developed more sophisticate mechanisms to protect themselves. Our results of transcriptome sequence of C. himalaica and evolutionary genomic analysis provided evidence for this belief. In this study, we identified 487 significantly PSGs in transcriptome sequences of C. himalaica. Functions of these PSGs elucidate that they potentially possess specific traits of adaptive significance, such as response to radiation, DNA repair, membrane stabilization and organic metabolism. These functions are likely responsible for the adaptation of C. himalaica to the high radiation, low temperature and soil depletion environments on QTP. Due to the RNA-seq technology is difficult to obtain the entire and full length transcripts, further studies are required. Nevertheless, our findings indicate that sophisticated genetic mechanisms have evolved in C. himalaica to survive the harsh conditions in QTP.
Materials and Methods
Sample collection and transcriptome sequencing
The original seedlings of C. himalaica were sampled in August 24, 2013 in Xiangcheng County (alt. 4,104 m, 29°02′47″ N, 99°42′55″ E) of QTP. These seedlings from the same individual were cultivated in greenhouse in Kunming Institute of Botany. In order to obtain more expression transcripts, two developmental stages (15 days, 30 days) of different organs (leaves, stems, and roots) were sampled and stored at −80 °C.
High quality total RNA was extracted using the TRIZOL reagent (Sigma Aldrich) following the manufacturer’s instructions. A total amount of 3 μg RNA per developmental stage was used as input material for the RNA sample preparations. Sequencing libraries were generated using NEBNext Ultra™ RNA Library Prep Kit for Illumina Inc. (NEB, USA) following manufacturer’s recommendations. The clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumina Inc.). After cluster generation, the library preparations were sequenced on an Illumina Hiseq 2500 platform and paired-end reads were generated. The whole step of library construction and Illumina sequencing was performed at Novogene Bioinformatics Technology Co., Ltd (Beijing, China).
De novo assembly and functional annotation
Clean data (clean reads) were obtained by removing reads containing adapter, reads containing ploy-N and low quality reads from raw data. At the same time, Q20, Q30, GC-content and sequence duplication level of the clean data were calculated. The sequenced left files (read1 files) from two samples were pooled into one big left.fq file, and right files (read2 files) into one big right.fq file. Transcriptome assembly was accomplished based on the left.fq and right.fq using Trinity program (trinityrnaseq_r20140413) with minimum k-mer coverage set to 2 and all other parameters set by default44.
Functional annotations of all assembled unigenes were conducted by searching against the following databases: NCBI non-redundant protein (Nr), NCBI non-redundant nucleotide (Nt), Protein family (Pfam), Clusters of Orthologous Groups of proteins (KOG), Swiss-Prot protein (Swiss-Prot), KEGG Ortholog database (KO), and Gene Ontology database (GO).
Orthologous genes identified and phylogenetic analysis
Based on previous studies in Brassicaceae10,20, we selected genomes of five relatives (A. thaliana, A. lyrata, Capsella rubella, Leavenworthia alabamica and Brassica rapa) and C. himalaica to identify orthologs. Furthermore, to define a set of conserved genes for cross-taxa comparison, we used OrthoMCL software45 to identify homologous gene clusters (orthogroups) among the six genomes. Genes with lengths less than 50 amino acids were excluded. OrthoMCL was run with an e-value cut-off of 1e-15 and an inflation parameter of 2.0 due to the close genetic relationship between six relatives.
Orthologroups with only single copy genes (one-to-one orthologs) that were shared by all six genomes were retained for further analysis. Each orthogroups was aligned using MUSCLE v3.8.3146 with default parameters. The poorly aligned regions were further strictly trimmed by using the trimAl v1.4 software47 with the parameter “-gt 0.8 -st 0.001”. Alignments of all orthogroups were concatenated by our python script. Then maximum likelihood (ML) trees were generated using RAxML v7.0.448 with PROTCATJTT model, the maximum likelihood criteria.
Positive selection analysis
In the positive selection analysis, only above single copy genes were considered. To calculate the nonsynonymous (Ka) and synonymous (Ks) substitution rates between pairs of orthogroups, above amino acid alignments were reverse-translated to the corresponding codon-based nucleotide alignments by PAL2NAL49. For each alignment, a gene tree was constructed by RAxML48 using GTR + GAMMA model.
To estimate lineage-specific evolutionary rates for each branch of the six species, the codeml program in the PAML 4 package24 with the free-ratio model (model = 1) was run on each orthogroups. We conducted the boxplot analysis using the dN/dS ratio derived from free-ratio model results and filtered dS >3 or dN/dS >3. Significances of the deviations from the median dN/dS ratio between six species branches were detected using Wilcoxon rank sum test. We also established frequency distribution plots of all dN/dS ratios of six species.
To increase the power of our tests for positive selection, we applied the improved branch-site model50 implemented in codeml program24 to estimate the dN/dS substitution rates (ω value). We also deleted all gaps (clean data = 1) from the alignments to lower the effect of ambiguous bases on the inference of positive selection. A foreground branch was specified as the clade of C. himalaica. A significant likelihood ratio test (LRT) was conducted to determine whether positive selection is operating in the foreground branch. In this study, the extremely significant positively selected genes (PSGs) were inferred if the P-value was less than 0.01.
For each PSG in C. himalaica, functional information was inferred based on its ortholog in A. thaliana. Gene Ontology (GO) enrichment analyses of PSGs were conducted using web-based agriGO (bioinfo.cau.edu.cn/agriGO)51 with singular enrichment analysis (SEA) method and TAIR10 database. The KOBAS software52 was also used to test the statistical enrichment of PSGs in Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways53.
Additional Information
How to cite this article: Qiao, Q. et al. Transcriptome sequencing of Crucihimalaya himalaica (Brassicaceae) reveals how Arabidopsis close relative adapt to the Qinghai-Tibet Plateau. Sci. Rep. 6, 21729; doi: 10.1038/srep21729 (2016).
References
Ekblom, R. & Galindo, J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity 107, 1–15 (2011).
Cheviron, Z. A. & Brumfield, R. T. Genomic insights into adaptation to high-altitude environments. Heredity 108, 354–361 (2012).
Qiu, Q. et al. The yak genome and adaptation to life at high altitude. Nature Genetics 44, 946–949 (2012).
Qu, Y. et al. Ground tit genome reveals avian adaptation to living at high altitudes in the Tibetan plateau. Nature Communications 4 (2013).
Li, M. et al. Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nature Genetics 45, 1431–1438 (2013).
Ge, R.-L. et al. Draft genome sequence of the Tibetan antelope. Nature Communications 4, 1858 (2013).
Gou, X. et al. Whole-genome sequencing of six dog breeds from continuous altitudes reveals adaptation to high-altitude hypoxia. Genome Research 24, 1308–1315 (2014).
Li, Y. et al. Population variation revealed high-altitude adaptation of Tibetan mastiffs. Molecular Biology & Evolution 31, 1200–1205 (2014).
Koenig, D. & Weigel, D. Beyond the thale: comparative genomics and genetics of Arabidopsis relatives. Nat Rev Genet 16, 285–298 (2015).
Mitchell-Olds, T. Arabidopsis thaliana and its wild relatives: a model system for ecology and evolution. Trends in Ecology & Evolution 16, 693–700 (2001).
Guo, Y.-L. et al. Recent speciation of Capsella rubella from Capsella grandiflora, associated with loss of self-incompatibility and an extreme bottleneck. Proceedings of the National Academy of Science 106, 5246–5251 (2009).
John Paul, F. et al. Recent speciation associated with the evolution of selfing in Capsella. Proceedings of the National Academy of Sciences of the United States of America 106, 5241–5245 (2009).
Guo, Y. L., Todesco, M., Hagmann, J., Das, S. & Weigel, D. Independent FLC Mutations as Causes of Flowering-Time Variation in Arabidopsis thaliana and Capsella rubella. Genetics 192, 729–739 (2012).
Turner, T. L., Bourne, E. C., Von Wettberg, E. J., Hu, T. T. & Nuzhdin, S. V. Population resequencing reveals local adaptation of Arabidopsis lyrata to serpentine soils. Nat Genet 42, 260–263 (2010).
Fischer, M. C. et al. Population genomic footprints of selection and associations with climate in natural populations of Arabidopsis halleri from the Alps. Molecular Ecology 22, 5594–5607 (2013).
Al-Shehbaz, I. A. & Price, R. A. Generic Placement of Species Excluded from Arabidopsis (Brassicaceae). Novon 9, 296–307 (1999).
Johnston, J. S. et al. Evolution of Genome Size in Brassicaceae. Annals of Botany 95, 229–235 (2005).
Wu, Z., Lu, A., Tang, Y., Chen, Z. & Li, D. The families and genera of angiosperms in China. (Science Press, 2003).
Wu, Z., Sun, H., Zhou, Z., Li, D. & Peng, H. Floristics of seed plants from China. (Science Press, 2011).
Beilstein, M. A., Nagalingum, N. S., Clements, M. D., Manchester, S. R. & Sarah, M. Dated Molecular Phylogenies Indicate A Miocene Origin For Arabidopsis Thaliana. Proc Natl Acad Sci USA 107, 18724–18728 (2010).
Li, J. & Fang, X. Uplift of the Tibetan Plateau and environmental changes. Chin. Sci. Bull. 44, 2117–2124 (1999).
Ward, J. A., Ponnala, L. & Weber, C. A. Strategies for transcriptome analysis in nonmodel plants. American Journal of Botany 99, 267–276 (2012).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protocols 8, 1494–1512 (2013).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution 24, 1586–1591 (2007).
Fitch, W. M. Distinguishing homologous from analogous proteins. Systematic Biology 19, 99–113 (1970).
Norsang, G. et al. Ground-based measurements and modeling of solar UV-B radiation in Lhasa, Tibet. Atmospheric Environment 43, 1498–1502 (2009).
Yu, X. et al. Modulation of brassinosteroid-regulated gene expression by jumonji domain-containing proteins ELF6 and REF6 in Arabidopsis. Proceedings of the National Academy of Sciences 105, 7618–7623 (2008).
Knight, H., Thomson, A. & Mcwatters, H. Sensitive to freezing6 integrates cellular and environmental inputs to the plant circadian clock. Plant Physiology 148, 293–303 (2008).
Zhang, L., Turkington & Tang . Flowering and Fruiting Phenology of 24 Plant Species on the North Slope of Mt. Qomolangma (Mt. Everest). Journal of Mountain Science 7, 45–54 (2010).
Thórhallsdóttir, T. E. Flowering phenology in the central highland of Iceland and implications for climatic warming in the Arctic. Oecologia 114, 43–49 (1998).
Frohnmeyer, H. & Staiger, D. Ultraviolet-B Radiation-Mediated Responses in Plants. Balancing Damage and Protection. Plant Physiology 133, 1420–1428 (2003).
Singh, S. K., Choudhury, S. R., Roy, S. & Sengupta, D. N. Understanding DNA repair and recombination in higher plant genome: information from genome-wide screens in Arabidopsis and rice. Plant Signaling & Behavior 6, 120–122 (2011).
Reed, E., Larkins, T., Chau, C. & Figg, W. In Handbook of Anticancer Pharmacokinetics and Pharmacodynamics Cancer Drug Discovery and Development (eds Rudek, Michelle A, Chau, Cindy H., Figg, William D. & McLeod, Howard L. ) Ch. 18, 333–349 (Springer: New York,, 2014).
Biedermann, S. & Hellmann, H. The DDB1a interacting proteins ATCSA-1 and DDB2 are critical factors for UV-B tolerance and genomic integrity in Arabidopsis thaliana. Plant Journal for Cell & Molecular Biology 62, 404–415 (2010).
Theil, A. F., Julie, N., Nils, W., Wim, V. & Giuseppina, G. M. Slowly progressing nucleotide excision repair in trichothiodystrophy group A patient fibroblasts. Molecular & Cellular Biology 31, 3630–3638 (2011).
Chen, J. et al. Soil nutrient availability determines the facilitative effects of cushion plants on other plant species at high elevations in the south-eastern Himalayas. Plant Ecology & Diversity 8, 199–210 (2014).
Wang, D. D., Zheng, G. W. & Wei-Qi, L. I. Plants Adapt to Long-Term Potassium Deficiency by Accumulation of Membrane Lipids in Leaves and Maintenance of Lipid Composition in Roots. Plant Diversity 36, 163–176 (2014).
Shuichi, Y., Ai, A., Hiroaki, K., Hirofumi, U. & Tetuya, M. Metabolic engineering with Dof1 transcription factor in plants: Improved nitrogen assimilation and growth under low-nitrogen conditions. Proceedings of the National Academy of Sciences 101, 7833–7838 (2004).
Daniel-Vedele, F. & Caboche, M. A tobacco cDNA clone encoding a GATA-1 zinc finger protein homologous to regulators of nitrogen metabolism in fungi. Molecular & General Genetics Mgg 240, 365–373 (1993).
Cavicchioli, R. Cold-adapted archaea. Nature Reviews Microbiology 4, 331–343 (2006).
Chijun, L., Ziqiang, G., Dan, L. & Raetz, C. R. H. Pathway for lipid A biosynthesis in Arabidopsis thaliana resembling that of Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America 108, 11387–11392 (2011).
Keogh, M. R., Courtney, P. D., Kinney, A. J. & Dewey, R. E. Functional Characterization of Phospholipid N -Methyltransferases from Arabidopsis and Soybean. Journal of Biological Chemistry 284, 15439–15447 (2009).
Zheng, G., Tian, B., Zhang, F., Tao, F. & Li, W. Plant adaptation to frequent alterations between high and low temperatures: remodelling of membrane lipids and maintenance of unsaturation levels. Plant Cell & Environment 34, 1431–1442 (2011).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology 29, 644–652 (2011).
Li, L., Stoeckert, C. J. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Research 13, 2178–2189 (2003).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 32, 1792–1797 (2004).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Stamatakis, A., Ludwig, T. & Meier, H. RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics 21, 456–463 (2005).
Suyama, M., Torrents, D. & Bork, P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Research 34, W609–W612 (2006).
Zhang, J., Nielsen, R. & Yang, Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Molecular Biology and Evolution 22, 2472–2479 (2005).
Du, Z., Zhou, X., Ling, Y., Zhang, Z. & Su, Z. agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Research 38, W64–W70 (2010).
Xie, C. et al. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Research 39, W316–W322 (2011).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Research 28, 27–30 (2000).
Acknowledgements
We thank David Boufford, Yang Niu, Xiangguang Ma, Deli Peng, Lie Yang, Zhiwei Wang, and Hongliang Chen for help with the field survey. We also appreciate Prof. Xiangyang Hu for giving some suggestions to improve the manuscript. This work is supported by grants from National Natural Science Foundation of China (31590820, 31590823, 31300201), National High Technology Research and Development Program of China (2014AA020528).
Author information
Authors and Affiliations
Contributions
T.Z., Y.Z., J.H. and H.S. conceived and designed the project. Q.Q., Q.W., T.Z. and X.H. prepared samples. Q.Q., T.Z. and Y.G. contributed to sequencing experiment. T.Z., Q.Q. and Q.W. performed data analyses. T.Z., Q.Q. and J.H. wrote the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Qiao, Q., Wang, Q., Han, X. et al. Transcriptome sequencing of Crucihimalaya himalaica (Brassicaceae) reveals how Arabidopsis close relative adapt to the Qinghai-Tibet Plateau. Sci Rep 6, 21729 (2016). https://doi.org/10.1038/srep21729
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep21729
This article is cited by
-
Molecular footprints of selection effects and whole genome duplication (WGD) events in three blueberry species: detected by transcriptome dataset
BMC Plant Biology (2020)
-
Physiological and transcriptome analyses of photosynthesis and chlorophyll metabolism in variegated Citrus (Shiranuhi and Huangguogan) seedlings
Scientific Reports (2019)
-
De novo transcriptome provides insights into the growth behaviour and resveratrol and trans-stilbenes biosynthesis in Dactylorhiza hatagirea - An endangered alpine terrestrial orchid of western Himalaya
Scientific Reports (2019)
-
Comparative transcriptomics provides insight into the molecular basis of species diversification of section Trigonopedia (Cypripedium) on the Qinghai-Tibetan Plateau
Scientific Reports (2018)
-
Recent Perspective of Next Generation Sequencing: Applications in Molecular Plant Biology and Crop Improvement
Proceedings of the National Academy of Sciences, India Section B: Biological Sciences (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
